COVID-19 Related Coagulopathy: A Distinct Entity?

 , , , ,
, , , , {kind=link}
Abstract
:1. Introduction
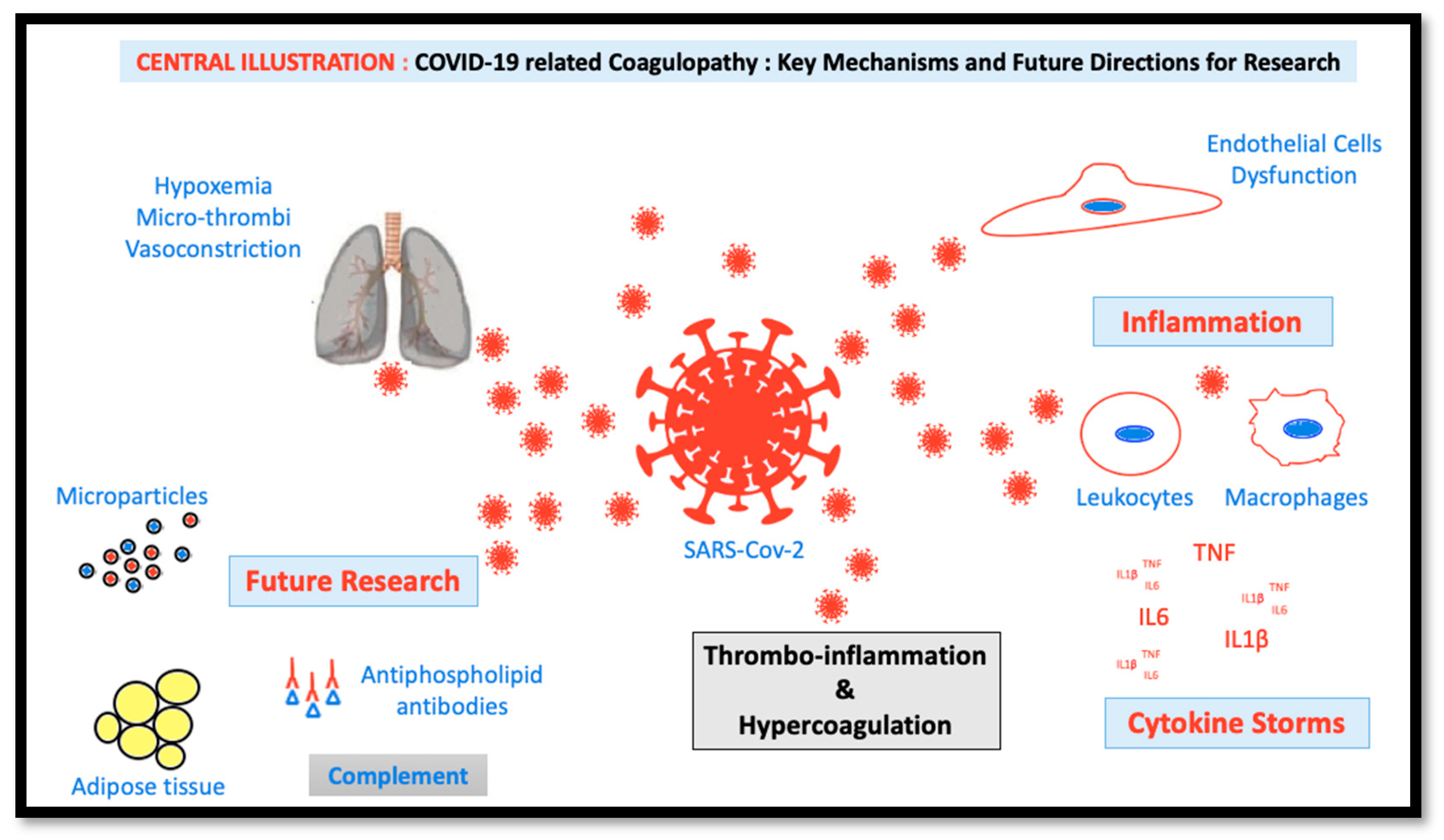
2. Pathogenesis of COVID-19
2.1. Hemostasis Parameters in COVID-19
2.2. Disseminated Intravascular Coagulation in COVID-19
2.3. D-Dimer Generation in COVID-19: Different Pathways?
2.4. Gaps in Evidence
3. Pathophysiology of COVID-19-Induced Coagulopathy
3.1. Inflammation
3.2. Endothelial Activation
3.3. Severe Hypoxemia
3.4. Pulmonary Microvascular Thrombosis
4. Coagulopathy Disorders Disease in COVID-19 Patients
Prevalence of Venous Thromboembolic Disease in COVID-19
5. Antithrombotic Therapy and COVID-19-Related Coagulopathy
5.1. Current Recommendations
5.2. Heparin and COVID-19
5.3. Gaps in Evidence
6. Future Research
6.1. A Role for Obesity
6.2. Possible Role of Microparticles in the Development of COVID-19-Induced Coagulopathy
6.3. Antiphospholipid Antibodies in COVID-19 Patients
6.4. Complement Activation in COVID-19 Patients
7. Conclusions
Author Contributions
Conflicts of Interest
References
- Chan, J.F.; Yuan, S.; Kok, K.H.; To, K.K.; Chu, H.; Yang, J.; Xing, F.; Liu, J.; Yip, C.C.; Poon, R.; et al. A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: A study of a family cluster. Lancet 2020, 395, 514–523. [Google Scholar] [CrossRef] [Green Version]
- Dong, E.; Du, H.; Gardner, L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect. Dis. 2020, 20, 533–534. [Google Scholar] [CrossRef]
- COVID-19 Dashboard by the Center for Systems Science and Engineering (CSSE) at Johns Hopkins University (JHU). Available online: https://www.arcgis.com/apps/opsdashboard/index.html#/bda7594740fd40299423467b48e9ecf6 (accessed on 28 April 2020).
- Matsushita, K.; Marchandot, B.; Jesel, L.; Ohlmann, P.; Morel, O. Impact of COVID-19 on Cardiovascular System: A Review. J. Clin. Med. 2020, 9, 1407. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Wang, Y.; Shao, C.; Huang, J.; Gan, J.; Huang, X.; Bucci, E.; Piacentini, M.; Ippolito, G.; Melino, G. COVID-19 infection: The perspectives on immune responses. Cell Death Differ. 2020, 27, 1451–1454. [Google Scholar] [CrossRef] [Green Version]
- Guan, W.J.; Ni, Z.Y.; Hu, Y.; Liang, W.H.; Ou, C.Q.; He, J.X.; Liu, L.; Shan, H.; Lei, C.L.; Hui, D.S.; et al. China Medical Treatment Expert Group for Covid-19. Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef]
- Tang, N.; Li, D.; Wang, X.; Sun, Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J. Thromb. Haemost. 2020, 18, 844–847. [Google Scholar] [CrossRef] [Green Version]
- Susen, S.; Tacquard, C.A.; Godon, A.; Mansour, A.; Garrigue, D.; Nguyen, P.; Godier, A.; Testa, S.; Albaladejo, P.; Gruel, Y.; on behalf of the GIHP and GFHT Group. Diagnosis and Management of Heparin-Induced Thrombocytopenia: Proposals from the French Working Group on Perioperative Haemostasis (GIHP) and the French Study Group on Thrombosis and Haemostasis (GFHT), in Collaboration with the French Society for Anesthesia and Intensive Care (SFAR). Available online: https://sfar.org/propositions-tih-gihp-gfht-sfar (accessed on 28 April 2020).
- Shanghai Clinical Treatment Expert Group for COVID-19. Comprehensive treatment and management of coronavirus disease 2019: Expert consensus statement from Shanghai (in Chinese). Chin. J. Infect. 2020, 38. published online ahead of print. [Google Scholar]
- Hunt, B.; Retter, A.; McClintock, C. Practical Guidance for the Prevention of Thrombosis and Management of Coagulopathy and Disseminated Intravascular Coagulation of Patients Infected with COVID-19. Available online: https://thrombosisuk.org/downloads/T&H%20and%20COVID.pdf (accessed on 28 April 2020).
- Marietta, M.; Ageno, W.; Artoni, A.; De Candia, E.; Gresele, P.; Marchetti, M.; Marcucci, R.; Tripodi, A. COVID-19 and haemostasis: A position paper from Italian Society on Thrombosis and Haemostasis (SISET). Blood Transfus 2020. [Google Scholar] [CrossRef]
- Casini, A.; Alberio, L.; Angelillo-Scherrer, A.; Fontana, P.; Gerber, B.; Graf, L.; Hegemann, I.; Korte, W.; Kremer Hovinga, J.; Lecompte, T.; et al. Thromboprophylaxis and laboratory monitoring for in-hospital patients with Covid-19-a Swiss consensus statement by the Working Party Hemostasis. Swiss Med. Wkly. 2020, 150, w20247. [Google Scholar] [CrossRef] [Green Version]
- Thachil, J.; Tang, N.; Gando, S.; Falanga, A.; Cattaneo, M.; Levi, M.; Clark, C.; Iba, T. ISTH interim guidance on recognition and management of coagulopathy in COVID-19. J. Thromb. Haemost. 2020, 18, 1023–1026. [Google Scholar] [CrossRef]
- Bikdeli, B.; Madhavan, M.V.; Jimenez, D.; Chuich, T.; Dreyfus, I.; Driggin, E.; Nigoghossian, C.D.; Ageno, W.; Madjid, M.; Guo, Y.; et al. COVID-19 and Thrombotic or Thromboembolic Disease: Implications for Prevention, Antithrombotic Therapy, and Follow-up. J. Am. Coll. Cardiol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Driggin, E.; Madhavan, M.V.; Bikdeli, B.; Chuich, T.; Laracy, J.; Bondi-Zoccai, G.; Brown, T.S.; Nigoghossian, C.D.; Zidar, D.A.; Haythe, J.; et al. Cardiovascular Considerations for Patients, Health Care Workers, and Health Systems During the Coronavirus Disease 2019 (COVID-19) Pandemic. J. Am. Coll. Cardiol. 2020, 75, 2352–2371. [Google Scholar] [CrossRef]
- Ge, X.Y.; Li, J.L.; Yang, X.; Chmura, A.A.; Zhu, G.; Epstein, J.H.; Mazet, J.K.; Hu, B.; Zhang, W.; Peng, C.; et al. Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature 2013, 503, 535–538. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Zhang, H.; Penninger, J.M.; Li, Y.; Zhong, N.; Slutsky, A.S. Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: Molecular mechanisms and potential therapeutic target. Intensive Care Med. 2020. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Zhong, L.; Deng, J.; Peng, J.; Dan, H.; Zeng, X.; Li, T.; Chen, Q. High expression of ACE2 receptor of 2019-nCoV on the epithelial cells of oral mucosa. Int. J. Oral Sci. 2020, 12, 1–5. [Google Scholar] [CrossRef]
- Chen, L.; Li, X.; Chen, M.; Feng, Y.; Xiong, C. The ACE2 expression in human heart indicates new potential mechanism of heart injury among patients infected with SARS-CoV-2. Cardiovasc. Res. 2020, 116, 1097–1100. [Google Scholar] [CrossRef] [Green Version]
- Centers for Disease Control and Prevention. Interim, U.S. Guidance for Risk Assessment and Public Health Management of Healthcare Personnel with Potential Exposure in a Healthcare Setting to Patients with Coronavirus Disease (COVID-19). Available online: https://www.cdc.gov/coronavirus/2019-ncov/hcp/guidance-risk-assesment-hcp.html (accessed on 9 April 2020).
- Zou, L.; Ruan, F.; Huang, M.; Liang, L.; Huang, H.; Hong, Z.; Yu, J.; Kang, M.; Song, Y.; Xia, J.; et al. SARS-CoV-2 Viral Load in Upper Respiratory Specimens of Infected Patients. N. Engl. J. Med. 2020, 382, 1177–1179. [Google Scholar] [CrossRef] [PubMed]
- Peiris, J.S.; Chu, C.M.; Cheng, V.C.; Chan, K.S.; Hung, I.F.; Poon, L.L.; Law, K.l.; Tang, B.S.; Hon, T.Y.; Chan, C.S.; et al. Clinical progression and viral load in a community outbreak of coronavirus-associated SARS pneumonia: A prospective study. Lancet 2003, 361, 1767–1772. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; McGoogan, J.M. Characteristics of and Important Lessons from the Coronavirus Disease 2019 (COVID-19) Outbreak in China: Summary of a Report of 72314 Cases from the Chinese Center for Disease Control and Prevention. JAMA 2020, 323, 1239. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Wu, C.; Chen, X.; Cai, Y.; Xia, J.; Zhou, X.; Xu, S.; Huang, H.; Zhang, L.; Zhou, X.; Du, C.; et al. Risk Factors Associated with Acute Respiratory Distress Syndrome and Death in Patients With Coronavirus Disease 2019 Pneumonia in Wuhan, China. JAMA Intern. Med. 2020. [Google Scholar] [CrossRef] [Green Version]
- Henry, B.M.; De Oliveira, M.H.S.; Benoit, S.; Plebani, M.; Lippi, G. Hematologic, biochemical and immune biomarker abnormalities associated with severe illness and mortality in coronavirus disease 2019 (COVID-19): A meta-analysis. Clin. Chem. Lab. Med. 2020. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.; Liu, W.; Liu, K.; Fang, Y.-Y.; Shang, J.; Zhou, L.; Wang, K.; Leng, F.; Wei, S.; Chen, L.; et al. Clinical characteristics of fatal and recovered cases of coronavirus disease 2019 (COVID-19) in Wuhan, China: A retrospective study. Chin. Med. J. 2020. [Google Scholar] [CrossRef]
- Guo, T.; Fan, Y.; Chen, M.; Wu, X.; Zhang, L.; He, T.; Wang, H.; Wan, J.; Wang, X.; Lu, Z. Cardiovascular Implications of Fatal Outcomes of Patients with Coronavirus Disease 2019 (COVID-19). JAMA Cardiol. 2020. [Google Scholar] [CrossRef] [Green Version]
- Terpos, E.; Ntanasis-Stathopoulos, I.; Elalamy, I.; Kastritis, E.; Sergentanis, T.N.; Politou, M.; Psaltopoulou, T.; Gerotziafas, G.; Dimopoulos, M.A. Hematological findings and complications of COVID -19. Am. J. Hematol. 2020. [Google Scholar] [CrossRef] [Green Version]
- Gando, S.; Levi, M.; Toh, C.-H. Disseminated intravascular coagulation. Nat. Rev. Dis. Prim. 2016, 2, 16038. [Google Scholar] [CrossRef] [Green Version]
- Wada, H.; Matsumoto, T.; Yamashita, Y. Diagnosis and treatment of disseminated intravascular coagulation (DIC) according to four DIC guidelines. J. Intensiv. Care 2014, 2, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bick, R.L. Disseminated intravascular coagulation: A review of etiology, pathophysiology, diagnosis, and management: Guidelines for care. Clin. Appl. Thromb. Hemost. 2002, 8, 1–31. [Google Scholar] [CrossRef] [PubMed]
- Lillicrap, D. Disseminated intravascular coagulation in patients with 2019-nCoV pneumonia. J. Thromb. Haemost. 2020, 18, 786–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, R.S.; Wu, A.; To, K.F.; Lee, N.; Lam, C.W.; Wong, C.K.; Chan, P.K.; Ng, M.H.; Yu, L.M.; Hui, D.S.; et al. Haematological manifestations in patients with severe acute respiratory syndrome: Retrospective analysis. BMJ 2003, 326, 1358–1362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, S.; Huang, M.; Li, D.; Tang, N. Difference of coagulation features between severe pneumonia induced by SARS-CoV2 and non-SARS-CoV2. J. Thromb. Thrombolysis 2020, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Levi, M.; Van Der Poll, T. Coagulation and sepsis. Thromb. Res. 2017, 149, 38–44. [Google Scholar] [CrossRef]
- Iba, T.; Levy, J.H.; Raj, A.; Warkentin, T.E. Advance in the Management of Sepsis-Induced Coagulopathy and Disseminated Intravascular Coagulation. J. Clin. Med. 2019, 8, 728. [Google Scholar] [CrossRef] [Green Version]
- Iba, T.; Levy, J.H.; Warkentin, T.E.; Thachil, J.; van der Poll, T.; Levi, M. Scientific and Standardization Committee on DIC, and the Scientific and Standardization Committee on Perioperative and Critical Care of the International Society on Thrombosis and Haemostasis Diagnosis and management of sepsis-induced coagulopathy and disseminated intravascular coagulation. J. Thromb. Haemost. 2019, 17, 1989–1994. [Google Scholar] [CrossRef] [Green Version]
- Gupta, N.; Zhao, Y.-Y.; Evans, C.E. The stimulation of thrombosis by hypoxia. Thromb. Res. 2019, 181, 77–83. [Google Scholar] [CrossRef]
- Luo, W.; Yu, H.; Gou, J.; Li, X.; Sun, Y.; Li, J.; Liu, L. Clinical Pathology of Critical Patient with Novel Coronavirus Pneumonia (COVID-19). Preprints 2020, 2020, 2020020407. [Google Scholar]
- Dolhnikoff, M.; Duarte-Neto, A.N.; de Almeida Monteiro, R.A.; Ferraz Da Silva, L.F.; Pierre de Oliveira, E.; Nascimento Saldiva, P.H.; Mauad, T.; Marcia Negri, E. Pathological evidence of pulmonary thrombotic phenomena in severe COVID-19. J. Thromb. Haemost. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marongiu, F.; Grandone, E.; Barcellona, D. Pulmonary thrombosis in 2019-nCoV pneumonia? J. Thromb. Haemost. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Li, Y.; Yang, Q. Antiplatelet Therapy Following Percutaneous Coronary Intervention in Patients Complicated by COVID-19: Implications from Clinical Features to Pathological Findings. Circulation 2020. [Google Scholar] [CrossRef] [PubMed]
- Koupenova, M.; Clancy, L.; Corkrey, H.A.; Freedman, J.E. Circulating Platelets as Mediators of Immunity, Inflammation, and Thrombosis. Circ. Res. 2018, 122, 337–351. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Shi, L.; Wang, F.S. Liver injury in COVID-19: Management and challenges. Lancet Gastroenterol. Hepatol. 2020, 5, 428–430. [Google Scholar] [CrossRef]
- Zhang, Y.; Xiao, M.; Zhang, S.; Xia, P.; Cao, W.; Jiang, W.; Chen, H.; Ding, X.; Zhao, H.; Zhang, H.; et al. Coagulopathy and Antiphospholipid Antibodies in Patients with Covid-19. N. Engl. J. Med. 2020, 382, e38. [Google Scholar] [CrossRef]
- Zulfiqar, A.-A.; Lorenzo-Villalba, N.; Hassler, P.; Andres, E. Immune Thrombocytopenic Purpura in a Patient with Covid-19. N. Engl. J. Med. 2020, 382, e43. [Google Scholar] [CrossRef]
- Fox, E.A.; Kahn, S.R. The relationship between inflammation and venous thrombosis. A systematic review of clinical studies. Thromb. Haemost. 2005, 94, 362–365. [Google Scholar] [CrossRef]
- Libby, P.; Loscalzo, J.; Ridker, P.M.; Farkouh, M.E.; Hsue, P.Y.; Fuster, V.; Hasan, A.A.; Amar, S. Inflammation, Immunity, and Infection in Atherothrombosis: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2018, 72, 2071–2081. [Google Scholar] [CrossRef]
- Wong, C.K.; Lam, C.W.; Wu, A.K.L.; Ip, W.K.; Lee, N.; Chan, I.H.S.; Lit, L.C.W.; Hui, D.S.; Chan, M.H.M.; Chung, S.S.C.; et al. Plasma inflammatory cytokines and chemokines in severe acute respiratory syndrome. Clin. Exp. Immunol. 2004, 136, 95–103. [Google Scholar] [CrossRef] [Green Version]
- Min, C.-K.; Cheon, S.; Ha, N.-Y.; Sohn, K.M.; Kim, Y.; Aigerim, A.; Shin, H.M.; Choi, J.-Y.; Inn, K.-S.; Kim, J.H.; et al. Comparative and kinetic analysis of viral shedding and immunological responses in MERS patients representing a broad spectrum of disease severity. Sci. Rep. 2016, 6, 25359. [Google Scholar] [CrossRef] [PubMed]
- Channappanavar, R.; Perlman, S. Pathogenic human coronavirus infections: Causes and consequences of cytokine storm and immunopathology. Semin. Immunopathol. 2017, 39, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Darbousset, R.; Schoenwaelder, S.M. Thromboinflammation: Challenges of therapeutically targeting coagulation and other host defense mechanisms. Blood 2019, 133, 906–918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Libby, P. The Heart in COVID19: Primary Target or Secondary Bystander? JACC Basic Transl. Sci. 2020. [Google Scholar] [CrossRef]
- Qin, C.; Zhou, L.; Hu, Z.; Zhang, S.; Yang, S.; Tao, Y.; Xie, C.; Ma, K.; Shang, K.; Wang, W.; et al. Dysregulation of immune response in patients with COVID-19 in Wuhan, China. Clin. Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Liu, J.; Li, S.; Liu, J.; Liang, B.; Wang, X.; Wang, H.; Li, W.; Tong, Q.; Yi, J.; Zhao, L.; et al. Longitudinal characteristics of lymphocyte responses and cytokine profiles in the peripheral blood of SARS-CoV-2 infected patients. medRxiv 2020. preprint. [Google Scholar] [CrossRef]
- Clerkin, K.J.; Fried, J.A.; Raikhelkar, J.; Sayer, G.; Griffin, J.M.; Masoumi, A.; Jain, S.S.; Burkhoff, D.; Kumaraiah, D.; Rabbani, L.; et al. Coronavirus Disease 2019 (COVID-19) and Cardiovascular Disease. Circulation 2020, 141, 1648–1655. [Google Scholar] [CrossRef] [Green Version]
- Xiong, T.-Y.; Redwood, S.; Prendergast, B.; Chen, M. Coronaviruses and the cardiovascular system: Acute and long-term implications. Eur. Hear J. 2020, 41, 1798–1800. [Google Scholar] [CrossRef] [Green Version]
- Mehta, P.; Mc Auley, D.F.; Brown, M.; Sanchez, E.; Tattersall, R.S.; Manson, J.J. COVID-19: Considercytokinestorm syndromes and immunosuppression. Lancet 2020. [Google Scholar] [CrossRef]
- Siddiqi, H.K.; Mehra, M.R. COVID-19 illness in native and immunosuppressed states: A clinical–therapeutic staging proposal. J. Hear Lung Transpl. 2020, 39, 405–407. [Google Scholar] [CrossRef] [Green Version]
- Ramacciotti, E.; Agati, L.B.; Aguiar, V.C.R.; Wolosker, N.; Guerra, J.C.; De Almeida, R.P.; Alves, J.C.; Lopes, R.D.; Wakefield, T.W.; Comerota, A.J.; et al. Zika and Chikungunya Virus and Risk for Venous Thromboembolism. Clin. Appl. Thromb. Hemost. 2019, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smither, S.J.; O’Brien, L.M.; Eastaugh, L.; Woolley, T.; Lever, M.; Fletcher, T.; Parmar, K.; Hunt, B.J.; Watts, S.; Kirkman, E.; et al. Haemostatic Changes in Five Patients Infected with Ebola Virus. Viruses 2019, 11, 647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tisoncik, J.R.; Korth, M.J.; Simmons, C.P.; Farrar, J.; Martin, T.R.; Katze, M.G. Into the eye of the cytokine storm. Microbiol. Mol. Biol. Rev. 2012, 76, 16–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sardu, C.; Gambardella, J.; Morelli, M.B.; Wang, X.; Marfella, R.; Santulli, G. Is COVID-19 an Endothelial Disease? Clinical and Basic Evidence. Preprints 2020, 2020040204. [Google Scholar] [CrossRef]
- Escher, R.; Breakey, N.; Lämmle, B. Severe COVID-19 infection associated with endothelial activation. Thromb. Res. 2020, 190, 62. [Google Scholar] [CrossRef] [PubMed]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Hamming, I.; Timens, W.; Bulthuis, M.; Lely, A.T.; Navis, G.; Van Goor, H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 2004, 203, 631–637. [Google Scholar] [CrossRef]
- Song, J.; Hu, B.; Qu, H.; Wang, L.; Huang, X.; Li, M.; Zhang, M. Upregulation of angiotensin converting enzyme 2 by shear stress reduced inflammation and proliferation in vascular endothelial cells. Biochem. Biophys. Res. Commun. 2020. [Google Scholar] [CrossRef]
- Takei, Y.; Yamada, M.; Saito, K.; Kameyama, Y.; Sugiura, H.; Makiguchi, T.; Fujino, N.; Koarai, A.; Toyama, H.; Saito, K.; et al. Increase in circulating ACE-positive endothelial microparticles during acute lung injury. Eur. Respir. J. 2019, 54, 1801188. [Google Scholar] [CrossRef]
- Grasselli, G.; Zangrillo, A.; Zanella, A.; Antonelli, M.; Cabrini, L.; Castelli, A.; Cereda, D.; Coluccello, A.; Foti, G.; Fumagalli, R.; et al. COVID-19 Lombardy ICU Network. Baseline Characteristics and Outcomes of 1591 Patients Infected With SARS-CoV-2 Admitted to ICUs of the Lombardy Region, Italy. JAMA 2020, 323, 1574. [Google Scholar] [CrossRef] [Green Version]
- Xie, J.; Covassin, N.; Fan, Z.; Singh, P.; Gao, W.; Li, G.; Kara, T.; Somers, V.K. Association Between Hypoxemia and Mortality in Patients With COVID-19. Mayo Clin. Proc. 2020. [Google Scholar] [CrossRef] [PubMed]
- Mo, P.; Xing, Y.; Xiao, Y.; Deng, L.; Zhao, Q.; Wang, H.; Xiong, Y.; Cheng, Z.; Gao, S.; Liang, K.; et al. Clinical characteristics of refractory COVID-19 pneumonia in Wuhan, China. Clin. Infect. Dis. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gattinoni, L.; Chiumello, D.; Rossi, S. COVID-19 pneumonia: ARDS or not? Crit. Care 2020, 24, 154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Z.; Shi, L.; Wang, Y.; Zhang, J.; Huang, L.; Zhang, C.; Liu, S.; Zhao, P.; Liu, H.; Zhu, L.; et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir. Med. 2020, 8, 420–422. [Google Scholar] [CrossRef]
- Fox, S.E.; Akmatbekov, A.; Harbert, J.L.; Li, G.; Quincy Brown, J.; Vander Heide, R.S. Pulmonary and Cardiac Pathology in Covid-19: The First Autopsy Series from New Orleans. medRxiv 2020. [Google Scholar] [CrossRef]
- Ranucci, M.; Ballotta, A.; Di Dedda, U.; Bayshnikova, E.; Dei Poli, M.; Resta, M.; Falco, M.; Albano, M.; Albano, G.; Menicanti, L. The procoagulant pattern of patients with COVID-19 acute respiratory distress syndrome. J. Thromb. Haemost. 2020. [Google Scholar] [CrossRef]
- Chong, P.Y.; Chui, P.; Ling, A.E.; Franks, T.J.; Tai, D.Y.; Leo, Y.S.; Kaw, G.J.; Wansaicheong, G.; Chan, K.P.; Ean Oon, L.L.; et al. Analysis of deaths during the severe acute respiratory syndrome (SARS) epidemic in Singapore: Challenges in determining a SARS diagnosis. Arch. Pathol. Lab. Med. 2004, 128, 195–204. [Google Scholar]
- Obi, A.T.; Tignanelli, C.J.; Jacobs, B.N.; Arya, S.; Park, P.K.; Wakefield, T.W.; Henke, P.K.; Napolitano, L.M. Empirical systemic anticoagulation is associated with decreased venous thromboembolism in critically ill influenza A H1N1 acute respiratory distress syndrome patients. J. Vasc. Surg. Venous Lymphat. Disord. 2019, 7, 317–324. [Google Scholar] [CrossRef]
- Wang, T.; Chen, R.; Liu, C.; Liang, W.; Guan, W.; Tang, R.; Tang, C.; Zhang, N.; Zhong, N.; Li, S. Attention should be paid to venous thromboembolism prophylaxis in the management of COVID-19. Lancet Haematol. 2020, 7, e362–e363. [Google Scholar] [CrossRef]
- Zhang, L.; Zhu, F.; Xie, L.; Wang, C.; Wang, J.; Chen, R.; Jia, P.; Guan, H.Q.; Peng, L.; Chen, Y.; et al. Clinical characteristics of COVID-19-infected cancer patients: A retrospective case study in three hospitals within Wuhan, China. Ann. Oncol. 2020. [Google Scholar] [CrossRef]
- Danzi, G.B.; Loffi, M.; Galeazzi, G.; Gherbesi, E. Acute pulmonary embolism and COVID-19 pneumonia: A random association? Eur. Hear. J. 2020, 41, 1858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Y.; Wang, X.; Yang, P.; Zhang, S. COVID-19 Complicated by Acute Pulmonary Embolism. Radiol. Cardiothorac. Imaging 2020, 2, e200067. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Wang, X.; Zhang, S.; Liu, B.; Wu, X.; Wang, Y.; Wang, X.; Yang, M.; Sun, J.; Xie, Y. Findings of Acute Pulmonary Embolism in COVID-19 Patients. SSRN Electron. J. 2020. [Google Scholar] [CrossRef]
- Cui, S.; Chen, S.; Li, X.; Liu, S.; Wang, F. Prevalence of venous thromboembolism in patients with severe novel coronavirus pneumonia. J. Thromb. Haemost. 2020. [Google Scholar] [CrossRef] [PubMed]
- Klok, F.A.; Kruip, M.; Van Der Meer, N.; Arbous, M.S.; Gommers, D.; Kant, K.; Kaptein, F.; Van Paassen, J.; Stals, M.; Huisman, M.; et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb. Res. 2020. [Google Scholar] [CrossRef]
- Helms, J.; Tacquard, C.; Severac, F.; Leonard-Lorant, I.; Ohana, M.; Delabranche, X.; Merdji, H.; Clere-Jehl, R.; Schenck, M.; Fagot Gandet, F.; et al. High risk of thrombosis in patients in severe SARS-CoV-2 infection: A multicenter prospective cohort study. Intensiv. Care Med. 2020, 1–10. [Google Scholar] [CrossRef]
- Wada, H.; Thachil, J.; Di Nisio, M.; Mathew, P.; Kurosawa, S.; Gando, S.; Kim, H.; Nielsen, J.; Dempfle, C.-E.; Levi, M.; et al. Guidance for diagnosis and treatment of DIC from harmonization of the recommendations from three guidelines. The Scientific Standardization Committee on DIC of the International Society on Thrombosis Haemostasis. J. Thromb. Haemost. 2013, 11, 761–767. [Google Scholar] [CrossRef]
- Tang, N.; Bai, H.; Chen, X.; Gong, J.; Li, D.; Sun, Z. Anticoagulant treatment is associated with decreased mortality in severe coronavirus disease 2019 patients with coagulopathy. J. Thromb. Haemost. 2020, 18, 1094–1099. [Google Scholar] [CrossRef]
- Alexander, G.C.; Qato, D.M. Ensuring Access to Medications in the US During the COVID-19 Pandemic. JAMA 2020. [Google Scholar] [CrossRef] [Green Version]
- Poterucha, T.J.; Libby, P.; Goldhaber, S.Z. More than an anticoagulant: Do heparins have direct anti-inflammatory effects? Thromb. Haemost. 2017, 117, 437–444. [Google Scholar] [CrossRef]
- Shi, C.; Wang, C.; Wang, H.; Yang, C.; Cai, F.; Zeng, F.; Cheng, F.; Liu, Y.; Zhou, T.; Deng, B.; et al. The potential of low molecular weight heparin to mitigate cytokine storm in severe COVID-19 patients: A retrospective clinical study. medRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Shukla, D.; Spear, P.G. Herpesviruses and heparan sulfate: An intimate relationship in aid of viral entry. J. Clin. Investig. 2001, 108, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Ghezzi, S.; Cooper, L.; Rubio, A.; Pagani, I.; Capobianchi, M.R.; Ippolito, G.; Pelletier, J.; Meneghetti, M.C.Z.; Lima, M.A.; Skidmore, M.A.; et al. Heparin prevents Zika virus induced-cytopathic effects in human neural progenitor cells. Antivir. Res. 2017, 140, 13–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanders, J.M.; Monogue, M.L.; Jodlowski, T.Z.; Cutrell, J.B. Pharmacologic Treatments for Coronavirus Disease 2019 (COVID-19) A Review. JAMA 2020. [Google Scholar] [CrossRef]
- Thachil, J. The versatile heparin in COVID-19. J. Thromb. Haemost. 2020, 18, 1020–1022. [Google Scholar] [CrossRef] [Green Version]
- Gibson, C.M.; Spyropoulos, A.C.; Cohen, A.T.; Hull, R.D.; Goldhaber, S.Z.; Yusen, R.D.; Hernandez, A.F.; Korjian, S.; Daaboul, Y.; Gold, A.; et al. The IMPROVEDD VTE risk score: Incorporation of D-dimer into the IMPROVE score to improve venous thromboembolism risk stratification. TH Open 2017, 1, e56–e65. [Google Scholar] [CrossRef] [Green Version]
- Cohen, A.T.; Alikhan, R.; Arcelus, J.; Bergmann, J.-F.; Haas, S.; Merli, G.J.; Spyropoulos, A.C.; Tapson, V.F.; Turpie, A.G.G. Assessment of venous thromboembolism risk and the benefits of thromboprophylaxis in medical patients. Thromb. Haemost. 2005, 94, 750–759. [Google Scholar] [CrossRef]
- Cohen, A.T.; Harrington, R.A.; Goldhaber, S.Z.; Hull, R.D.; Wiens, B.L.; Gold, A.; Hernandez, A.F.; Gibson, C.M. Extended Thromboprophylaxis with Betrixaban in Acutely Ill Medical Patients. N. Engl. J. Med. 2016, 375, 534–544. [Google Scholar] [CrossRef]
- Goldhaber, S.Z.; Leizorovicz, A.; Kakkar, A.K.; Haas, S.; Merli, G.; Knabb, R.M.; Weitz, J.I. Apixaban versus enoxaparin for thromboprophylaxis in medically ill patients. N. Engl. J. Med. 2011, 365, 2167–2177. [Google Scholar] [CrossRef] [Green Version]
- Spyropoulos, A.C.; Ageno, W.; Albers, G.W.; Elliott, C.G.; Halperin, J.L.; Hiatt, W.R.; Maynard, G.A.; Steg, P.G.; Weitz, J.I.; Suh, E.; et al. Rivaroxaban for Thromboprophylaxis after Hospitalization for Medical Illness. N. Engl. J. Med. 2018, 379, 1118–1127. [Google Scholar] [CrossRef]
- Wang, J.; Hajizadeh, N.; Moore, E.E.; McIntyre, R.C.; Moore, P.K.; Veress, L.A.; Yaffe, M.B.; Moore, H.B.; Barrett, C.D. Tissue Plasminogen Activator (tPA) Treatment for COVID-19 Associated Acute Respiratory Distress Syndrome (ARDS): A Case Series. J. Thromb. Haemost. 2020. [Google Scholar] [CrossRef] [PubMed]
- Lighter, J.; Phillips, M.; Hochman, S.; Sterling, S.; Johnson, D.; Francois, F.; Stachel, A. Obesity in patients younger than 60 years is a risk factor for Covid-19 hospital admission. Clin. Infect. Dis. 2020, ciaa415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simonnet, A.; Chetboun, M.; Poissy, J.; Raverdy, V.; Noulette, J.; Duhamel, A.; Labreuche, J.; Mathieu, D.; Pattou, F.; Jourdain, M.; et al. High prevalence of obesity in severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) requiring invasive mechanical ventilation. Obsity 2020. [Google Scholar] [CrossRef] [PubMed]
- Barrasa, H.; Rello, J.; Tejada, S.; Martín, A.; Balziskueta, G.; Vinuesa, C.; Fernández-Miret, B.; Villagra, A.; Vallejo, A.; Sebastián, A.S.; et al. Alava COVID19 Study Investigators. SARS-Cov-2 in Spanish Intensive Care: Early Experience with 15-day Survival in Vitoria. Anaesth. Crit. Care Pain Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Venkata, C.; Sampathkumar, P.; Afessa, B. Hospitalized patients with 2009 H1N1 influenza infection: The Mayo Clinic experience. Mayo Clin. Proc. 2010, 85, 798–805. [Google Scholar] [CrossRef]
- Morel, O.; Luca, F.; Grunebaum, L.; Jesel, L.; Meyer, N.; Desprez, D.; Robert, S.; Dignat-George, F.; Toti, F.; Simon, C.; et al. Short-term very low-calorie diet in obese females improves the haemostatic balance through the reduction of leptin levels, PAI-1 concentrations and a diminished release of platelet and leukocyte-derived microparticles. Int. J. Obes. 2011, 35, 1479–1486. [Google Scholar] [CrossRef] [Green Version]
- Badimon, L.; Bugiardini, R.; Cenko, E.; Cubedo, J.; Dorobantu, M.; Duncker, D.J.; Estruch, R.; Miličić, D.; Tousoulis, D.; Vasiljevic, Z.; et al. Position Paper of the European Society of Cardiology working group of coronary pathophysiology and microcirculation: Obesity and heart disease. Eur. Hear J. 2017, 38, 1951–1958. [Google Scholar] [CrossRef]
- Caër, C.; Rouault, C.; Le Roy, T.; Poitou, C.; Aron-Wisnewsky, J.; Torcivia, A.; Bichet, J.-C.; Clément, K.; Guerre-Millo, M.; André, S. Immune cell-derived cytokines contribute to obesity related inflammation, fibrogenesis and metabolic deregulation in human adipose tissue. Sci. Rep. 2017, 7, 3000. [Google Scholar] [CrossRef]
- Morel, O.; Jesel, L.; Freyssinet, J.-M.; Toti, F. Cellular mechanisms underlying the formation of circulating microparticles. Arter. Thromb. Vasc. Biol. 2011, 31, 15–26. [Google Scholar] [CrossRef] [Green Version]
- Amabile, N.; Cheng, S.; Renard, J.M.; Larson, M.G.; Ghorbani, A.; McCabe, E.; Griffin, G.; Guerin, C.; Ho, J.E.; Shaw, S.Y.; et al. Association of circulating endothelial microparticles with cardiometabolic risk factors in the Framingham Heart Study. Eur. Hear J. 2014, 35, 2972–2979. [Google Scholar] [CrossRef]
- Abbas, M.; Jesel, L.; Auger, C.; Amoura, L.K.; Messas, N.; Manin, G.; Rumig, C.; León-González, A.J.; Ribeiro, T.P.; Silva, G.C.; et al. Endothelial Microparticles From Acute Coronary Syndrome Patients Induce Premature Coronary Artery Endothelial Cell Aging and Thrombogenicity: Role of the Ang II/AT1 Receptor/NADPH Oxidase-Mediated Activation of MAPKs and PI3-Kinase Pathways. Circulation 2017, 135, 280–296. [Google Scholar] [CrossRef] [PubMed]
- Mack, M.; Kleinschmidt, A.; Brühl, H.; Klier, C.; Nelson, P.J.; Cihak, J.; Plachý, J.; Stangassinger, M.; Erfle, V.; Schlöndorff, D. Transfer of the chemokine receptor CCR5 between cells by membrane-derived microparticles: A mechanism for cellular human immunodeficiency virus 1 infection. Nat. Med. 2000, 6, 769–775. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chen, S.; Bihl, J. Exosome-Mediated Transfer of ACE2 (Angiotensin-Converting Enzyme 2) from Endothelial Progenitor Cells Promotes Survival and Function of Endothelial Cell. Oxidative Med. Cell. Longev. 2020, 2020, 4213541–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lóczi, L.; Kappelmayer, J.; Tarr, T.; Bagoly, Z. Antiphospholipid syndrome and the risk of myocardial infarction: Current evidence and uncertainties. Kardiol. Polska 2019, 78, 6–14. [Google Scholar] [CrossRef] [Green Version]
- Mobarrez, F.; Gunnarsson, I.; Svenungsson, E. Altered β2 glycoprotein I expression on microparticles in the presence of antiphospholipid antibodies. J. Thromb. Haemost. 2017, 15, 1799–1806. [Google Scholar] [CrossRef]
- Nomijra, S.; Komiyama, Y.; Kokawa, T.; Takahashi, H.; Koike, T.; Matsuura, E. Binding of beta2-glycoprotein I to platelet-derived microparticles. Br. J. Haematol. 2008, 85, 639. [Google Scholar] [CrossRef]
- Gralinski, L.E.; Sheahan, T.P.; Morrison, T.E.; Menachery, V.D.; Jensen, K.; Leist, S.R.; Whitmore, A.; Heise, M.T.; Baric, R.S.; Enjuanes, L.; et al. Complement Activation Contributes to Severe Acute Respiratory Syndrome Coronavirus Pathogenesis. mBio 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Campbell, C.M.; Kahwash, R. Will Complement Inhibition be the New Target in Treating COVID-19 Related Systemic Thrombosis? Circulation 2020. [Google Scholar] [CrossRef] [Green Version]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marchandot, B.; Sattler, L.; Jesel, L.; Matsushita, K.; Schini-Kerth, V.; Grunebaum, L.; Morel, O. COVID-19 Related Coagulopathy: A Distinct Entity? J. Clin. Med. 2020, 9, 1651. https://doi.org/10.3390/jcm9061651
Marchandot B, Sattler L, Jesel L, Matsushita K, Schini-Kerth V, Grunebaum L, Morel O. COVID-19 Related Coagulopathy: A Distinct Entity? Journal of Clinical Medicine. 2020; 9(6):1651. https://doi.org/10.3390/jcm9061651
Chicago/Turabian StyleMarchandot, Benjamin, Laurent Sattler, Laurence Jesel, Kensuke Matsushita, Valerie Schini-Kerth, Lelia Grunebaum, and Olivier Morel. 2020. "COVID-19 Related Coagulopathy: A Distinct Entity?" Journal of Clinical Medicine 9, no. 6: 1651. https://doi.org/10.3390/jcm9061651
APA StyleMarchandot, B., Sattler, L., Jesel, L., Matsushita, K., Schini-Kerth, V., Grunebaum, L., & Morel, O. (2020). COVID-19 Related Coagulopathy: A Distinct Entity? Journal of Clinical Medicine, 9(6), 1651. https://doi.org/10.3390/jcm9061651