Has Drug Repurposing Fulfilled Its Promise in Acute Myeloid Leukaemia?


Abstract
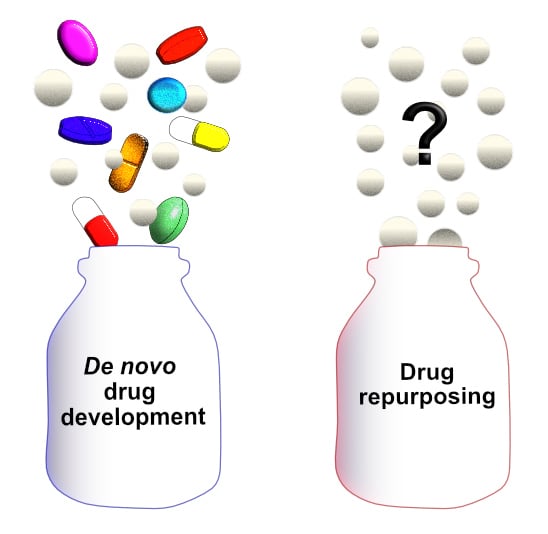
:
1. Introduction: What Is Drug Repurposing?
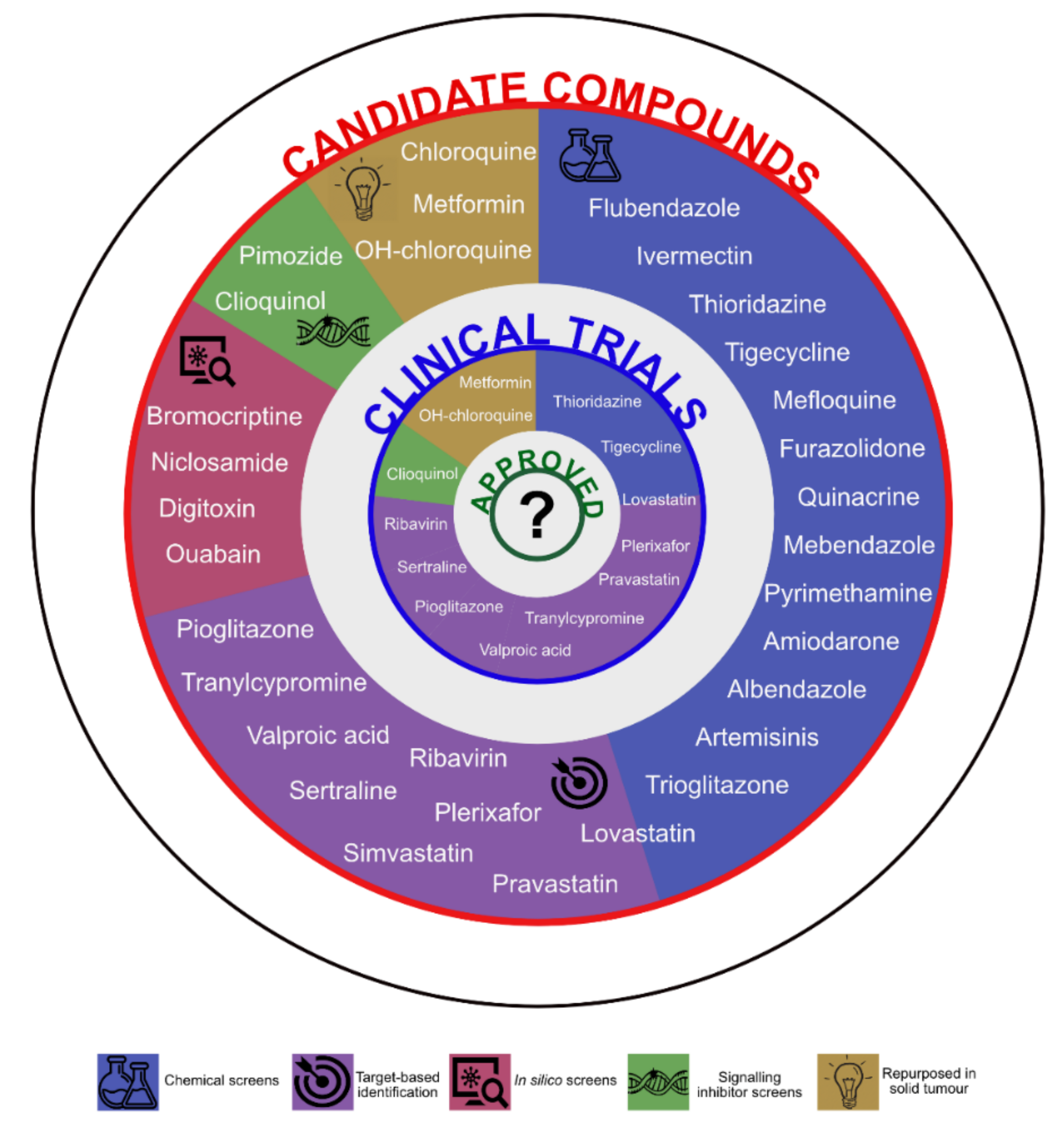
2. Drug Repurposing in AML
2.1. Drug Repositioning (or Soft Drug Repurposing)
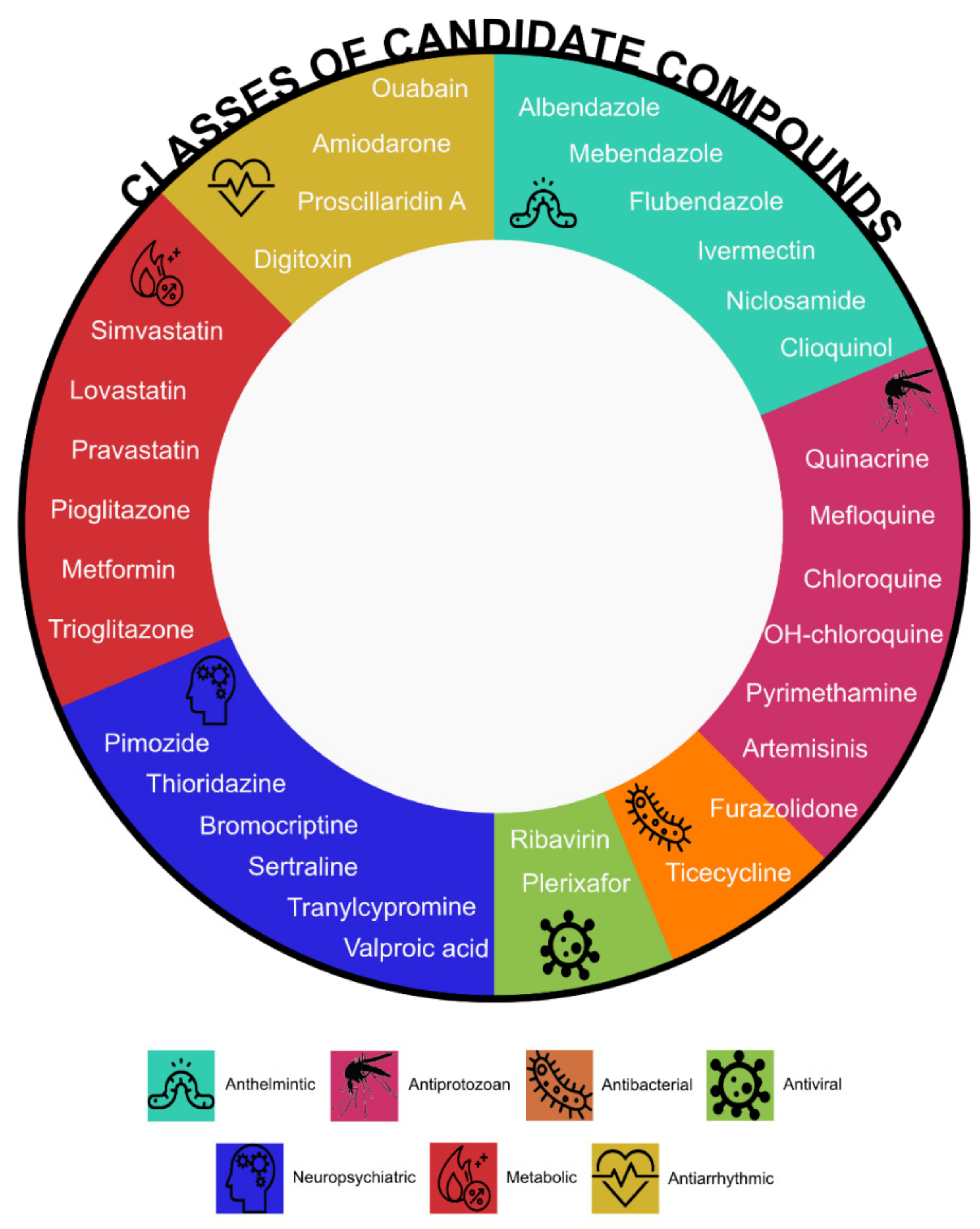
2.2. (Hard) Drug Repurposing
2.2.1. Screening and Hits
2.2.2. Preclinical Validation
2.2.3. Clinical Trials
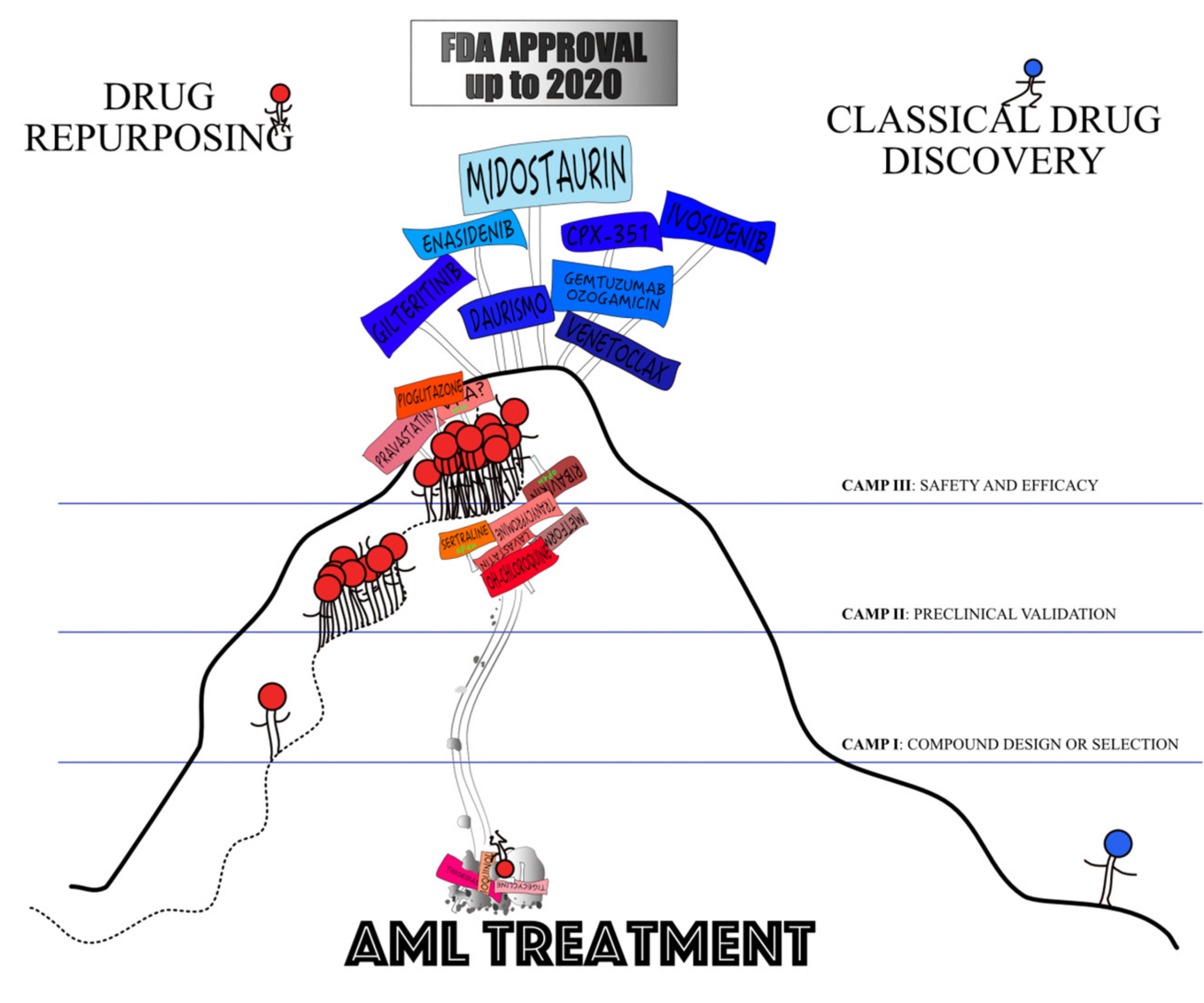
3. Where Are We Now?
4. Does the Promise Hold?
- Dogma 1: Drug repurposing saves time
- Dogma 2: Phase I clinical trials can be skipped.
- Dogma 3: Repurposed drugs are safe as their toxicity profile is known
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sleire, L.; Førde-Tislevoll, H.E.; Netland, I.A.; Leiss, L.; Skeie, B.S.; Enger, P.Ø. Drug repurposing in cancer. Pharmacol. Res. 2017, 124, 74–91. [Google Scholar] [CrossRef] [PubMed]
- Pantziarka, P.; Bouche, G.; Meheus, L.; Sukhatme, V.; Sukhatme, V.P. Repurposing drugs in your medicine cabinet: Untapped opportunities for cancer therapy? Futur. Oncol. 2015, 11, 181–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, S.C.; Sung, B.; Prasad, S.; Webb, L.J.; Aggarwal, B.B. Cancer drug discovery by repurposing: Teaching new tricks to old dogs. Trends Pharmacol. Sci. 2013, 34, 508–517. [Google Scholar] [CrossRef] [PubMed]
- Sukhai, M.A.; Spagnuolo, P.A.; Weir, S.; Kasper, J.; Patton, L.; Schimmer, A.D. New sources of drugs for hematologic malignancies. Blood 2011, 117, 6747–6755. [Google Scholar] [CrossRef] [Green Version]
- Bertolini, F.; Sukhatme, V.P.; Bouche, G. Drug repurposing in oncology—Patient and health systems opportunities. Nat. Publ. Gr. 2015, 12. [Google Scholar] [CrossRef]
- Talevi, A. Drug repositioning: Current approaches and their implications in the precision medicine era. Expert Rev. Precis. Med. Drug Dev. 2018, 3, 49–61. [Google Scholar] [CrossRef]
- Cha, Y.; Erez, T.; Reynolds, I.J.; Kumar, D.; Ross, J.; Koytiger, G.; Kusko, R.; Zeskind, B.; Risso, S.; Kagan, E.; et al. Drug repurposing from the perspective of pharmaceutical companies. Br. J. Pharmacol. 2018, 175, 168–180. [Google Scholar] [CrossRef] [Green Version]
- Swinney, D.C.; Anthony, J. How were new medicines discovered? Nat. Rev. Drug Discov. 2011, 10, 507–519. [Google Scholar] [CrossRef]
- Lamb, J.; Crawford, E.D.; Peck, D.; Modell, J.W.; Blat, I.C.; Wrobel, M.J.; Lerner, J.; Brunet, J.P.; Subramanian, A.; Ross, K.N.; et al. The connectivity map: Using gene-expression signatures to connect small molecules, genes, and disease. Science 2006, 313, 1929–1935. [Google Scholar] [CrossRef] [Green Version]
- Iorio, F.; Shrestha, R.L.; Levin, N.; Boilot, V.; Garnett, M.J.; Saez-Rodriguez, J.; Draviam, V.M. A semi-supervised approach for refining transcriptional signatures of drug response and repositioning predictions. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oprea, T.I.; Tropsha, A.; Faulon, J.L.; Rintoul, M.D. Systems chemical biology. Nat. Chem. Biol. 2007, 3, 447–450. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Agarwal, P. Systematic drug repositioning based on clinical side-effects. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Campillos, M.; Kuhn, M.; Gavin, A.C.; Jensen, L.J.; Bork, P. Drug target identification using side-effect similarity. Science 2008, 321, 263–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitchen, D.B.; Decornez, H.; Furr, J.R.; Bajorath, J. Docking and scoring in virtual screening for drug discovery: Methods and applications. Nat. Rev. Drug Discov. 2004, 3, 935–949. [Google Scholar] [CrossRef] [PubMed]
- Sanseau, P.; Agarwal, P.; Barnes, M.R.; Pastinen, T.; Richards, J.B.; Cardon, L.R.; Mooser, V. Use of genome-wide association studies for drug repositioning. Nat. Biotechnol. 2012, 30, 317–320. [Google Scholar] [CrossRef]
- Jensen, P.B.; Jensen, L.J.; Brunak, S. Mining electronic health records: Towards better research applications and clinical care. Nat. Rev. Genet. 2012, 13, 395–405. [Google Scholar] [CrossRef]
- Nosengo, N. Can you teach old drugs new tricks? Nature 2016, 534, 314–316. [Google Scholar] [CrossRef]
- FDA. The FDA’s Drug Review Process: Ensuring Drugs Are Safe and Effective. Available online: https://www.fda.gov/drugs/drug-information-consumers/fdas-drug-review-process-ensuring-drugs-are-safe-and-effective (accessed on 27 May 2020).
- Gurova, K. New hopes from old drugs: Revisiting DNA-binding small molecules as anticancer agents. Futur. Oncol. 2009, 5, 1685–1704. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Zhao, C.; Wang, L. Molecular-targeted agents combination therapy for cancer: Developments and potentials. Int. J. Cancer 2014, 134, 1257–1269. [Google Scholar] [CrossRef]
- Pantziarka, P.; Pirmohamed, M.; Mirza, N. New uses for old drugs. BMJ 2018, 361, k2701. [Google Scholar] [CrossRef]
- Saultz, J.; Garzon, R. Acute myeloid leukemia: A concise review. J. Clin. Med. 2016, 5, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Döhner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute myeloid leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breitenbuecher, F.; Schnittger, S.; Grundler, R.; Markova, B.; Carius, B.; Brecht, A.; Duyster, J.; Haferlach, T.; Huber, C.; Fischer, T. Identification of a novel type of ITD mutations located in nonjuxtamembrane domains of the FLT3 tyrosine kinase receptor. Blood 2009, 113, 4074–4077. [Google Scholar] [CrossRef] [PubMed]
- Schröder, M.; Kaufman, R.J. The mammalian unfolded protein response. Annu. Rev. Biochem. 2005, 74, 739–789. [Google Scholar] [CrossRef]
- Lai, C.; Doucette, K.; Norsworthy, K. Recent drug approvals for acute myeloid leukemia. J. Hematol. Oncol. 2019, 12, 100. [Google Scholar] [CrossRef] [PubMed]
- Pantziarka, P.; Bouche, G.; Meheus, L.; Sukhatme, V.; Sukhatme, V.P. The repurposing drugs in oncology (ReDO) project. Ecancermedicalscience 2014, 8. [Google Scholar] [CrossRef]
- Pantziarka, P.; Bouche, G.; André, N. “Hard” drug repurposing for precision oncology: The missing link? Front. Pharmacol. 2018, 9. [Google Scholar] [CrossRef]
- Ghanem, H.; Jabbour, E.; Faderl, S.; Ghandhi, V.; Plunkett, W.; Kantarjian, H. Clofarabine in leukemia. Expert Rev. Hematol. 2010, 3, 15–22. [Google Scholar] [CrossRef]
- Ho, K.V.; Solimando, D.A.; Waddell, J.A. Clofarabine and cytarabine regimen for acute myeloid leukemia. Hosp. Pharm. 2015, 50, 969–974. [Google Scholar] [CrossRef] [Green Version]
- Zhou, A.; Han, Q.; Song, H.; Zi, J.; Ma, J.; Ge, Z. Efficacy and toxicity of cladribine for the treatment of refractory acute myeloid leukemia: A meta-analysis. Drug Des. Devel. Ther. 2019, 13, 1867–1878. [Google Scholar] [CrossRef] [Green Version]
- Seligson, N.D.; Hobbs, A.L.V.; Leonard, J.M.; Mills, E.L.; Evans, A.G.; Goorha, S. Evaluating the impact of the addition of cladribine to standard acute myeloid leukemia induction therapy. Ann. Pharmacother. 2018, 52, 439–445. [Google Scholar] [CrossRef]
- Falini, B.; Brunetti, L.; Martelli, M.P. Dactinomycin in NPM1-Mutated acute myeloid leukemia. N. Engl. J. Med. 2015, 373, 1180–1182. [Google Scholar] [CrossRef]
- Pleyer, L.; Döhner, H.; Dombret, H.; Seymour, J.F.; Schuh, A.C.; Beach, C.L.; Swern, A.S.; Burgstaller, S.; Stauder, R.; Girschikofsky, M.; et al. Azacitidine for front-line therapy of patients with AML: Reproducible efficacy established by direct comparison of international phase 3 trial data with registry data from the austrian azacitidine registry of the AGMT study group. Int. J. Mol. Sci. 2017, 18, 415. [Google Scholar] [CrossRef] [PubMed]
- Kerr, R.; Cunningham, J.; Bowen, D.T. Low-dose melphalan in elderly acute myeloid leukemia: Complete remissions but resistant relapse with therapy-related karyotypes. Leukemia 2000, 14, 953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stratmann, J.; van Kann, E.; Rummelt, C.; Koschade, S.; Röllig, C.; Lübbert, M.; Schaich, M.; Parmentier, S.; Sebastian, M.; Chromik, J.; et al. Low-dose melphalan in elderly patients with relapsed or refractory acute myeloid leukemia: A well-tolerated and effective treatment after hypomethylating-agent failure. Leuk. Res. 2019, 85, 106192. [Google Scholar] [CrossRef] [PubMed]
- Rudd, S.G.; Tsesmetzis, N.; Sanjiv, K.; Paulin, C.B.; Sandhow, L.; Kutzner, J.; Hed Myrberg, I.; Bunten, S.S.; Axelsson, H.; Zhang, S.M.; et al. Ribonucleotide reductase inhibitors suppress SAMHD1 ara- CTPase activity enhancing cytarabine efficacy. EMBO Mol. Med. 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Sekeres, M.A.; MacIejewski, J.P.; Erba, H.P.; Afable, M.; Englehaupt, R.; Sobecks, R.; Advani, A.; Seel, S.; Chan, J.; Kalaycio, M.E. A Phase 2 study of combination therapy with arsenic trioxide and gemtuzumab ozogamicin in patients with myelodysplastic syndromes or secondary acute myeloid leukemia. Cancer 2011, 117, 1253–1261. [Google Scholar] [CrossRef] [Green Version]
- Wetzler, M.; Andrews, C.; Ford, L.A.; Tighe, S.; Barcos, M.; Sait, S.N.J.; Block, A.W.; Nowak, N.J.; Baer, M.R.; Wang, E.S.; et al. Phase 1 study of arsenic trioxide, high-dose cytarabine, and idarubicin to down-regulate constitutive signal transducer and activator of transcription 3 activity in patients aged <60 years with acute myeloid leukemia. Cancer 2011, 117, 4831–4868. [Google Scholar] [CrossRef]
- Burnett, A.K.; Hills, R.K.; Hunter, A.; Milligan, D.; Kell, J.; Wheatley, K.; Yin, J.; McMullin, M.F.; Cahalin, P.; Craig, J.; et al. The addition of arsenic trioxide to low-dose Ara-C in older patients with AML does not improve outcome. Leukemia 2011, 25, 1122–1127. [Google Scholar] [CrossRef]
- Douer, D.; Watkins, K.; Louie, R.; Weitz, I.; Mohrbacher, A.; Levine, A.M. Treatment of acute myelogenous leukemia (non-APL) with intravenous Trisenox® (arsenic trioxide) and ascorbic acid: Preliminary results. Blood 2004, 104, 1815. [Google Scholar] [CrossRef]
- Malani, D.; Murumägi, A.; Yadav, B.; Kontro, M.; Eldfors, S.; Kumar, A.; Karjalainen, R.; Majumder, M.M.; Ojamies, P.; Pemovska, T.; et al. Enhanced sensitivity to glucocorticoids in cytarabine-resistant AML. Leukemia 2017, 31, 1187–1195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, L.; Wen, Y.; Yao, Y.; Chen, F.; Wang, G.; Wu, F.; Wu, J.; Narayanan, P.; Redell, M.; Mo, Q.; et al. Glucocorticoids inhibit oncogenic RUNX1-ETO in acute myeloid leukemia with chromosome translocation t(8;21). Theranostics 2018, 8, 2189–2201. [Google Scholar] [CrossRef] [Green Version]
- Roulston, G.D.R.; Burt, C.L.; Kettyle, L.M.J.; Matchett, K.B.; Keenan, H.L.; Mulgrew, N.M.; Ramsey, J.M.; Dougan, C.; McKiernan, J.; Grishagin, I.V.; et al. Low-dose salinomycin induces anti-leukemic responses in AML and MLL. Oncotarget 2016, 7, 73448–73461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Remsing Rix, L.L.; Kuenzi, B.M.; Luo, Y.; Remily-Wood, E.; Kinose, F.; Wright, G.; Li, J.; Koomen, J.M.; Haura, E.B.; Lawrence, H.R.; et al. GSK3 alpha and beta are new functionally relevant targets of tivantinib in lung cancer cells. ACS Chem. Biol. 2014, 9, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Banerji, V.; Frumm, S.M.; Ross, K.N.; Li, L.S.; Schinzel, A.C.; Hahn, C.K.; Kakoza, R.M.; Chow, K.T.; Ross, L.; Alexe, G.; et al. The intersection of genetic and chemical genomic screens identifies GSK-3α as a target in human acute myeloid leukemia. J. Clin. Investig. 2012, 122, 935–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuenzi, B.M.; Rix, L.L.R.; Kinose, F.; Kroeger, J.L.; Lancet, J.E.; Padron, E.; Rix, U. Off-target based drug repurposing opportunities for tivantinib in acute myeloid leukemia OPEN. Sci. Rep. 2019, 9, 606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diggle, G.E. Thalidomide: 40 years on. Int. J. Clin. Pract. 2001, 55, 627–631. [Google Scholar]
- Mao, X.; Stewart, A.K.; Hurren, R.; Datti, A.; Zhu, X.; Zhu, Y.; Shi, C.; Lee, K.; Tiedemann, R.; Eberhard, Y.; et al. A chemical biology screen identifies glucocorticoids that regulate c-maf expression by increasing its proteasomal degradation through up-regulation of ubiquitin. Blood 2007, 110, 4047–4054. [Google Scholar] [CrossRef] [Green Version]
- Mao, X.; Li, X.; Hurren, R.; Venugopal, A.; Trudel, S.; Stewart, A.K.; Schimmer, A.D. The oral off-patent antimicrobial clioquinol inhibits the proteasome and induces cell death in AML and myeloma cells through a copper-dependent mechanism. Blood 2007, 110, 1595. [Google Scholar] [CrossRef]
- Spagnuolo, P.A.; Hu, J.; Hurren, R.; Wang, X.; Gronda, M.; Sukhai, M.A.; Di Meo, A.; Boss, J.; Ashali, I.; Zavareh, R.B.; et al. The antihelmintic flubendazole inhibits microtubule function through a mechanism distinct from Vinca alkaloids and displays preclinical activity in leukemia and myeloma. Blood 2010, 115, 4824–4833. [Google Scholar] [CrossRef] [Green Version]
- Sharmeen, S.; Skrtic, M.; Sukhai, M.A.; Hurren, R.; Gronda, M.; Wang, X.; Fonseca, S.B.; Sun, H.; Wood, T.E.; Ward, R.; et al. The antiparasitic agent ivermectin induces chloride-dependent membrane hyperpolarization and cell death in leukemia cells. Blood 2010, 116, 3593–3603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Škrtić, M.; Sriskanthadevan, S.; Jhas, B.; Gebbia, M.; Wang, X.; Wang, Z.; Hurren, R.; Jitkova, Y.; Gronda, M.; Maclean, N.; et al. Inhibition of mitochondrial translation as a therapeutic strategy for human acute myeloid leukemia. Cancer Cell 2011, 20, 674–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, E.A.; Walker, S.R.; Weisberg, E.; Bar-Natan, M.; Barrett, R.; Gashin, L.B.; Terrell, S.; Klitgaard, J.L.; Santo, L.; Addorio, M.R.; et al. The STAT5 inhibitor pimozide decreases survival of chronic myelogenous leukemia cells resistant to kinase inhibitors. Blood 2011, 117, 3421–3429. [Google Scholar] [CrossRef]
- Nelson, E.A.; Walker, S.R.; Xiang, M.; Weisberg, E.; Bar-Natan, M.; Barrett, R.; Liu, S.; Kharbanda, S.; Christie, A.L.; Nicolais, M.; et al. The STAT5 inhibitor pimozide displays efficacy in models of acute myelogenous leukemia driven by FLT3 mutations. Genes Cancer 2012, 3, 503–511. [Google Scholar] [CrossRef] [Green Version]
- Sachlos, E.; Risueño, R.M.; Laronde, S.; Shapovalova, Z.; Lee, J.H.; Russell, J.; Malig, M.; McNicol, J.D.; Fiebig-Comyn, A.; Graham, M.; et al. Identification of drugs including a dopamine receptor antagonist that selectively target cancer stem cells. Cell 2012, 149, 1284–1297. [Google Scholar] [CrossRef] [Green Version]
- Sukhai, M.A.; Prabha, S.; Hurren, R.; Rutledge, A.C.; Lee, A.Y.; Sriskanthadevan, S.; Sun, H.; Wang, X.; Skrtic, M.; Seneviratne, A.; et al. Lysosomal disruption preferentially targets acute myeloid leukemia cells and progenitors. J. Clin. Investig. 2013, 123, 315–328. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Sun, L.; Qiu, J.J.; Sun, X.; Li, S.; Wang, X.; So, C.W.E.; Dong, S. A novel application of furazolidone: Anti-leukemic activity in acute myeloid leukemia. PLoS ONE 2013, 8, e72335. [Google Scholar] [CrossRef] [Green Version]
- Hartwell, K.A.; Miller, P.G.; Mukherjee, S.; Kahn, A.R.; Stewart, A.L.; Logan, D.J.; Negri, J.M.; Duvet, M.; Järås, M.; Puram, R.; et al. Niche-based screening identifies small-molecule inhibitors of leukemia stem cells. Nat. Chem. Biol. 2013, 9, 840–848. [Google Scholar] [CrossRef] [Green Version]
- Eriksson, A.; Österroos, A.; Hassan, S.; Gullbo, J.; Rickardson, L.; Jarvius, M.; Nygren, P.; Fryknäs, M.; Höglund, M.; Larsson, R. Drug screen in patient cells suggests quinacrine to be repositioned for treatment of acute myeloid leukemia. Blood Cancer J. 2015, 5, e307. [Google Scholar] [CrossRef] [Green Version]
- Matchett, K.B.; Grishagin, I.V.; Kettyle, L.M.; Gavory, G.; Harrison, T.; Mills, K.; Thompson, A. Mebendazole: A candidate FDA approved drug for repurposing in leukaemia. Br. J. Haematol. 2016, 173, 121–122. [Google Scholar]
- Sharma, A.; Jyotsana, N.K.; Lai, C.; Chaturvedi, A.; Gabdoulline, R.; Görlich, K.; Murphy, C.E.; Blanchard, J.; Ganser, A.; Brown, E.; et al. Pyrimethamine as a Potent and Selective Inhibitor of Acute Myeloid Leukemia Identified by High-throughput Drug Screening. Curr. Cancer Drug Targets 2016, 16, 818–828. [Google Scholar] [CrossRef] [PubMed]
- Lara-Castillo, M.C.; Cornet-Masana, J.M.; Etxabe, A.; Banús-Mulet, A.; Torrente, M.Á.; Nomdedeu, M.; Díaz-Beyá, M.; Esteve, J.; Risueño, R.M. Repositioning of bromocriptine for treatment of acute myeloid leukemia. J. Transl. Med. 2016, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matchett, K.B.; Grishagin, I.V.; Kettyle, L.; Dowling, C.; Ni Chonghaile, T.; Mills, K.I.; Thompson, A. High-throughput screen identification of albendazole as a novel repurposed drug in acute myeloid leukaemia. Blood 2017, 130. [Google Scholar] [CrossRef]
- Chae, H.D.; Cox, N.; Dahl, G.V.; Lacayo, N.J.; Davis, K.L.; Capolicchio, S.; Smith, M.; Sakamoto, K.M. Niclosamide suppresses acute myeloid leukemia cell proliferation through inhibition of CREB-dependent signaling pathways. Oncotarget 2018, 9, 4301–4317. [Google Scholar] [CrossRef]
- Ketchem, C.J.; Kucera, C.; Barve, A.; Beverly, L.J. The antiarrhythmic drug, amiodarone, decreases akt activity and sensitizes human acute myeloid leukemia cells to apoptosis by ABT-263. Am. J. Med. Sci. 2018, 355, 488–496. [Google Scholar] [CrossRef] [Green Version]
- Casson, L.; Howell, L.; Mathews, L.A.; Ferrer, M.; Southall, N.; Guha, R.; Keller, J.M.; Thomas, C.; Siskind, L.J.; Beverly, L.J. Inhibition of ceramide metabolism sensitizes human leukemia cells to inhibition of BCL2-like proteins. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Laverdière, I.; Boileau, M.; Neumann, A.L.; Frison, H.; Mitchell, A.; Ng, S.W.K.; Wang, J.C.Y.; Minden, M.D.; Eppert, K. Leukemic stem cell signatures identify novel therapeutics targeting acute myeloid leukemia. Blood Cancer J. 2018, 8, 52. [Google Scholar] [CrossRef]
- Jin, Y.; Lu, Z.; Ding, K.; Li, J.; Du, X.; Chen, C.; Sun, X.; Wu, Y.; Zhou, J.; Pan, J. Antineoplastic mechanisms of niclosamide in acute myelogenous leukemia stem cells: Inactivation of the NF-κB pathway and generation of reactive oxygen species. Cancer Res. 2010, 70, 2516–2527. [Google Scholar] [CrossRef] [Green Version]
- Balgi, A.D.; Fonseca, B.D.; Donohue, E.; Tsang, T.C.F.; Lajoie, P.; Proud, C.G.; Nabi, I.R.; Roberge, M. Screen for chemical modulators of autophagy reveals novel therapeutic inhibitors of mTORC1 signaling. PLoS ONE 2009, 4, e7124. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Wang, J.; Lu, J.; Bond, M.C.; Ren, X.R.; Lyerly, H.K.; Barak, L.S.; Chen, W. The anti-helminthic niclosamide inhibits Wnt/Frizzled1 signaling. Biochemistry 2009, 48, 10267–10274. [Google Scholar] [CrossRef] [Green Version]
- Mao, X.; Li, X.; Sprangers, R.; Wang, X.; Venugopal, A.; Wood, T.; Zhang, Y.; Kuntz, D.A.; Coe, E.; Trudel, S.; et al. Clioquinol inhibits the proteasome and displays preclinical activity in leukemia and myeloma. Leukemia 2009, 23, 585–590. [Google Scholar] [CrossRef]
- Verbaanderd, C.; Maes, H.; Schaaf, M.B.; Sukhatme, V.P.; Pantziarka, P.; Sukhatme, V.; Agostinis, P.; Bouche, G. Repurposing drugs in oncology (ReDO)-Chloroquine and hydroxychloroquine as anti-cancer agents. Ecancermedicalscience 2017, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiraki, K.; Kimura, I. Studies on the treatment of malignant tumors with fibroblast-inhibiting agent. II. Effects of chloroquine on animal tumors. Acta Med. Okayama 1963, 17, 239–252. [Google Scholar]
- Kawaguchi-Ihara, N.; Zhao, Y.; Nakamura, S.; Suzuki, K.; Zhang, Y.; Tohda, S.; Murohashi, I. Chloroquine inhibits self-renewal of blast progenitors synergistically with phytochemicals or nonsteroidal anti-inflammatory drugs in hematological malignant cell lines. Anticancer Res. 2019, 39, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Clark, J.; Wunderlich, M.; Fan, C.; Davis, A.; Chen, S.; Guan, J.L.; Mulloy, J.C.; Kumar, A.; Zheng, Y. Autophagy is dispensable for Kmt2a/Mll-Mllt3/Af9 AML maintenance and anti-leukemic effect of chloroquine. Autophagy 2017, 13, 955–966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.; Eom, J.I.; Jeung, H.K.; Jang, J.E.; Kim, J.S.; Cheong, J.W.; Kim, Y.S.; Min, Y.H. Induction of cytosine arabinoside-resistant human myeloid leukemia cell death through autophagy regulation by hydroxychloroquine. Biomed. Pharmacother. 2015, 73, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Clark, J.; Guan, J.; Kumar, A.R.; Zheng, Y. Susceptibility of AML to chloroquine therapy is independent of autophagy. Blood 2015, 126, 1262. [Google Scholar] [CrossRef]
- Drenberg, C.D.; Buaboonnam, J.; Orwick, S.J.; Hu, S.; Li, L.; Fan, Y.; Shelat, A.A.; Guy, R.K.; Rubnitz, J.; Baker, S.D. Evaluation of artemisinins for the treatment of acute myeloid leukemia. Cancer Chemother. Pharmacol. 2016, 77, 1231–1243. [Google Scholar] [CrossRef] [PubMed]
- Lam, N.S.; Long, X.; Wong, J.W.; Griffin, R.C.; Doery, J.C.G. Artemisinin and its derivatives: A potential treatment for leukemia. Anticancer Drugs 2019, 30, 1–18. [Google Scholar] [CrossRef]
- Topisirovic, I.; Guzman, M.L.; McConnell, M.J.; Licht, J.D.; Culjkovic, B.; Neering, S.J.; Jordan, C.T.; Borden, K.L.B. Aberrant eukaryotic translation initiation factor 4E-dependent mRNA transport impedes hematopoietic differentiation and contributes to leukemogenesis. Mol. Cell. Biol. 2003, 23, 8992–9002. [Google Scholar] [CrossRef] [Green Version]
- Borden, K.L.B.; Culjkovic-Kraljacic, B. Ribavirin as an anti-cancer therapy: Acute myeloid leukemia and beyond? Leuk. Lymphoma 2010, 51, 1805–1815. [Google Scholar] [CrossRef] [Green Version]
- Hendrix, C.W.; Collier, A.C.; Lederman, M.M.; Schols, D.; Pollard, R.B.; Brown, S.; Jackson, J.B.; Coombs, R.W.; Glesby, M.J.; Flexner, C.W.; et al. Safety, pharmacokinetics, and antiviral activity of AMD3100, a selective CXCR4 receptor inhibitor, in HIV-1 infection. J. Acquir. Immune Defic. Syndr. 2004, 37, 1253–1262. [Google Scholar] [CrossRef]
- Gruszka, A.; Valli, D.; Restelli, C.; Alcalay, M. Adhesion deregulation in acute myeloid leukaemia. Cells 2019, 8, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, B.S.; Kim, H.J.; Konopleva, M. Targeting the CXCL12/CXCR4 axis in acute myeloid leukemia: From bench to bedside. Korean J. Intern. Med. 2017, 32, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Nordenberg, J.; Fenig, E.; Landau, M.; Weizman, R.; Weizman, A. Effects of psychotropic drugs on cell proliferation and differentiation. Biochem. Pharmacol. 1999, 58, 1229–1236. [Google Scholar] [CrossRef]
- Tickenbrock, L.; Klein, H.U.; Trento, C.; Hascher, A.; Göllner, S.; Bäumer, N.; Kuss, R.; Agrawal, S.; Bug, G.; Serve, H.; et al. Increased HDAC1 deposition at hematopoietic promoters in AML and its association with patient survival. Leuk. Res. 2011, 35, 620–625. [Google Scholar] [CrossRef]
- Blaheta, R.; Nau, H.; Michaelis, M.; Cinatl, J., Jr. Valproate and valproate-analogues: Potent tools to fight against cancer. Curr. Med. Chem. 2012, 9, 1417–1433. [Google Scholar] [CrossRef]
- Göttlicher, M.; Minucci, S.; Zhu, P.; Krämer, O.H.; Schimpf, A.; Giavara, S.; Sleeman, J.P.; Lo Coco, F.; Nervi, C.; Pelicci, P.G.; et al. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J. 2001, 20, 6969–6978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schenk, T.; Chen, W.C.; Göllner, S.; Howell, L.; Jin, L.; Hebestreit, K.; Klein, H.U.; Popescu, A.C.; Burnett, A.; Mills, K.; et al. Inhibition of the LSD1 (KDM1A) demethylase reactivates the all-trans-retinoic acid differentiation pathway in acute myeloid leukemia. Nat. Med. 2012, 18, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Barth, J.; Abou-El-Ardat, K.; Dalic, D.; Kurrle, N.; Maier, A.M.; Mohr, S.; Schütte, J.; Vassen, L.; Greve, G.; Schulz-Fincke, J.; et al. LSD1 inhibition by tranylcypromine derivatives interferes with GFI1-mediated repression of PU.1 target genes and induces differentiation in AML. Leukemia 2019, 33, 1411–1426. [Google Scholar] [CrossRef]
- Tuynder, M.; Fiucci, G.; Prieur, S.; Lespagnol, A.; Géant, A.; Beaucourt, S.; Duflaut, D.; Besse, S.; Susini, L.; Cavarelli, J.; et al. Translationally controlled tumor protein is a target of tumor reversion. Proc. Natl. Acad. Sci. USA 2004, 101, 15364–15369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, D.; Zhang, Y.T.; Xu, G.P.; Yan, W.W.; Pan, X.R.; Tong, J.H. Sertraline exerts its antitumor functions through both apoptosis and autophagy pathways in acute myeloid leukemia cells. Leuk. Lymphoma 2017, 58, 2208–2217. [Google Scholar] [CrossRef] [PubMed]
- Liberante, F.G.; Pouryahya, T.; McMullin, M.F.; Zhang, S.D.; Mills, K.I. Identification and validation of the dopamine agonist bromocriptine as a novel therapy for high-risk myelodysplastic syndromes and secondary acute myeloid leukemia. Oncotarget 2016, 7, 6609–6619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franciosi, M.; Lucisano, G.; Lapice, E.; Strippoli, G.F.M.; Pellegrini, F.; Nicolucci, A. Metformin therapy and risk of cancer in patients with type 2 diabetes: Systematic review. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Liu, Z.; Zeng, J.; Zhu, H.; Li, J.; Cheng, X.; Jiang, T.; Zhang, L.; Zhang, C.; Chen, T.; et al. Metformin synergistically sensitizes FLT3-ITD-positive acute myeloid leukemia to sorafenib by promoting mTOR-mediated apoptosis and autophagy. Leuk. Res. 2015, 39, 1421–1427. [Google Scholar] [CrossRef] [PubMed]
- Sabnis, H.S.; Bradley, H.L.; Tripathi, S.; Yu, W.M.; Tse, W.; Qu, C.K.; Bunting, K.D. Synergistic cell death in FLT3-ITD positive acute myeloid leukemia by combined treatment with metformin and 6-benzylthioinosine. Leuk. Res. 2016, 50, 132–140. [Google Scholar] [CrossRef] [Green Version]
- Yuan, F.; Cheng, C.; Xiao, F.; Liu, H.; Cao, S.; Zhou, G. Inhibition of mTORC1/P70S6K pathway by Metformin synergistically sensitizes Acute Myeloid Leukemia to Ara-C. Life Sci. 2020, 243, 117276. [Google Scholar] [CrossRef]
- Ghadiany, M.; Tabarraee, M.; Salari, S.; Haghighi, S.; Rezvani, H.; Ghasemi, S.N.; Karimi-Sari, H. Adding oral pioglitazone to standard induction chemotherapy of acute myeloid leukemia: A randomized clinical trial. Clin. Lymphoma Myeloma Leuk. 2019, 19, 206–212. [Google Scholar] [CrossRef]
- Thomas, S.; Schelker, R.; Klobuch, S.; Zaiss, S.; Troppmann, M.; Rehli, M.; Haferlach, T.; Herr, W.; Reichle, A. Biomodulatory therapy induces complete molecular remission in chemorefractory acute myeloid leukemia. Haematologica 2015, 100, e4–e6. [Google Scholar] [CrossRef] [Green Version]
- Klobuch, S.; Steinberg, T.; Bruni, E.; Mirbeth, C.; Heilmeier, B.; Ghibelli, L.; Herr, W.; Reichle, A.; Thomas, S. Biomodulatory treatment with azacitidine, all-trans retinoic acid and pioglitazone induces differentiation of primary AML blasts into neutrophil like cells capable of ROS production and phagocytosis. Front. Pharmacol. 2018, 9. [Google Scholar] [CrossRef]
- Pradelli, D.; Soranna, D.; Zambon, A.; Catapano, A.; Mancia, G.; La Vecchia, C.; Corrao, G. Statins use and the risk of all and subtype hematological malignancies: A meta-analysis of observational studies. Cancer Med. 2015, 4, 770–780. [Google Scholar] [CrossRef] [PubMed]
- Harwood, H.J., Jr.; Alvarez, I.M.; Noyes, W.D.; Stacpoole, P.W. In vivo regulation of human leukocyte 3-hydroxy-3-methylglutaryl coenzyme A reductase: Increased enzyme protein concentration and catalytic efficiency in human leukemia and lymphoma. J. Lipid Res. 1991, 32, 1237–1252. [Google Scholar]
- Newman, A.; Clutterbuck, R.D.; Powles, R.L.; Catovsky, D.; Millar, J.L. A comparison of the effect of the 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors simvastatin, lovastatin and pravastatin on leukaemic and normal bone marrow progenitors. Leuk. Lymphoma 1997, 24, 533–537. [Google Scholar] [CrossRef] [PubMed]
- Perne, A.; Muellner, M.K.; Steinrueck, M.; Craig-Mueller, N.; Mayerhofer, J.; Schwarzinger, I.; Sloane, M.; Uras, I.Z.; Hoermann, G.; Nijman, S.M.B.; et al. Cardiac glycosides induce cell death in human cells by inhibiting general protein synthesis. PLoS ONE 2009, 4, e8292. [Google Scholar] [CrossRef] [PubMed]
- Da Costa, E.M.; Armaos, G.; McInnes, G.; Beaudry, A.; Moquin-Beaudry, G.; Bertrand-Lehouillier, V.; Caron, M.; Richer, C.; St-Onge, P.; Johnson, J.R.; et al. Heart failure drug proscillaridin A targets MYC overexpressing leukemia through global loss of lysine acetylation. J. Exp. Clin. Cancer Res. 2019, 38, 251. [Google Scholar] [CrossRef]
- Taylor, S.J.; Duyvestyn, J.M.; Dagger, S.A.; Dishington, E.J.; Rinaldi, C.A.; Dovey, O.M.; Vassiliou, G.S.; Grove, C.S.; Langdon, W.Y. Preventing chemotherapy-induced myelosuppression by repurposing the FLT3 inhibitor quizartinib. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [Green Version]
- Schimmer, A.D.; Jitkova, Y.; Gronda, M.; Wang, Z.; Brandwein, J.; Chen, C.; Gupta, V.; Schuh, A.; Yee, K.; Chen, J.; et al. A phase i study of the metal ionophore clioquinol in patients with advanced hematologic malignancies. Clin. Lymphoma Myeloma Leuk. 2012, 12, 330–336. [Google Scholar] [CrossRef]
- Phase I Study of Mitoxantrone and Etoposide Combined With Hydroxychloroquine, for Relapsed Acute Myelogenous Leukemia-Full Text View-ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02631252?term=NCT02631252&draw=2&rank=1 (accessed on 30 March 2020).
- Reed, G.A.; Schiller, G.J.; Kambhampati, S.; Tallman, M.S.; Douer, D.; Minden, M.D.; Yee, K.W.; Gupta, V.; Brandwein, J.; Jitkova, Y.; et al. A Phase 1 study of intravenous infusions of tigecycline in patients with acute myeloid leukemia. Cancer Med. 2016, 5, 3031–3040. [Google Scholar] [CrossRef]
- Kentsis, A.; Topisirovic, I.; Culjkovic, B.; Shao, L.; Borden, K.L.B. Ribavirin suppresses elF4E-mediated oncogenic transformation by physical mimicry of the 7-methyl guanosine mRNA cap. Proc. Natl. Acad. Sci. USA 2004, 101, 18105–18110. [Google Scholar] [CrossRef] [Green Version]
- Assouline, S.; Culjkovic, B.; Cocolakis, E.; Rousseau, C.; Beslu, N.; Amri, A.; Caplan, S.; Leber, B.; Roy, D.C.; Miller, W.H.; et al. Molecular targeting of the oncogene eIF4E in acute myeloid leukemia (AML): A proof-of-principle clinical trial with ribavirin. Blood 2009, 114, 257–260. [Google Scholar] [CrossRef]
- Tavor, S.; Petit, I.; Porozov, S.; Avigdor, A.; Dar, A.; Leider-Trejo, L.; Shemtov, N.; Deutsch, V.; Naparstek, E.; Nagler, A.; et al. CXCR4 regulates migration and development of human acute myelogenous leukemia stem cells in transplanted NOD/SCID mice. Cancer Res. 2004, 64, 2817–2824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uy, G.L.; Rettig, M.P.; Motabi, I.H.; McFarland, K.; Trinkaus, K.M.; Hladnik, L.M.; Kulkarni, S.; Abboud, C.N.; Cashen, A.F.; Stockerl-Goldstein, K.E.; et al. A phase 1/2 study of chemosensitization with the CXCR4 antagonist plerixafor in relapsed or refractory acute myeloid leukemia. Blood 2012, 119, 3917–3924. [Google Scholar] [CrossRef] [PubMed]
- Blum, W.; Klisovic, R.B.; Hackanson, B.; Liu, Z.; Liu, S.; Devine, H.; Vukosavljevic, T.; Huynh, L.; Lozanski, G.; Kefauver, C.; et al. Phase I study of decitabine alone or in combination with valproic acid in acute myeloid leukemia. J. Clin. Oncol. 2007, 25, 3884–3891. [Google Scholar] [CrossRef] [PubMed]
- Aslostovar, L.; Boyd, A.L.; Almakadi, M.; Collins, T.J.; Leong, D.P.; Tirona, R.G.; Kim, R.B.; Julian, J.A.; Xenocostas, A.; Leber, B.; et al. A phase 1 trial evaluating thioridazine in combination with cytarabine in patients with acute myeloid leukemia. Blood Adv. 2018, 2, 1935–1945. [Google Scholar] [CrossRef]
- Sertraline and Cytosine Arabinoside in Adults with Relapsed and Refractory AML-Full Text View-ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02891278 (accessed on 30 March 2020).
- Scotland, S.; Micklow, E.; Wang, Z.; Boutzen, H.; Récher, C.; Danet-Desnoyers, G.; Selak, M.; Carroll, M.; Sarry, J.-E. Metformin for therapeutic intervention in acute myeloid leukemia. Blood 2010, 116, 4351. [Google Scholar] [CrossRef]
- Metformin+Cytarabine for the Treatment of Relapsed/Refractory AML-Full Text View-ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT01849276?term=NCT01849276&draw=2&rank=1 (accessed on 30 March 2020).
- Hatta, Y.; Saiki, M.; Enomoto, Y.; Aizawa, S.; Sawada, U.; Horie, T. Pioglitazone inhibits the growth of human leukemic cell lines and primary leukemic cells in vitro. Blood 2004, 104, 4493. [Google Scholar] [CrossRef]
- Heudobler, D.; Klobuch, S.; Lüke, F.; Hahn, J.; Grube, M.; Kremers, S.; Südhoff, T.; Westermann, J.; Luise Hütter-Krönke, M.; Paschka, P.; et al. Low-dose azacitidine, pioglitazone and all-trans retinoic acid versus standard-dose azacitidine in patients ≥ 60 years with acute myeloid leukemia refractory to standard induction chemotherapy (AMLSG 26-16/AML-ViVA): Results of the safety run-in phase I. Blood 2019, 134, 1382. [Google Scholar] [CrossRef]
- Kornblau, S.M.; Banker, D.E.; Stirewalt, D.; Shen, D.; Lemker, E.; Verstovsek, S.; Estrov, Z.; Faderl, S.; Cortes, J.; Beran, M.; et al. Blockade of adaptive defensive changes in cholesterol uptake and synthesis in AML by the addition of pravastatin to idarubicin + high-dose Ara-C: A phase 1 study. Blood 2007, 109, 2999–3006. [Google Scholar] [CrossRef]
- Dose Escalation Phase I/II Study of Lovastatin with High-Dose Cytarabine for Refractory or Relapsed AML-Full Text View-ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/study/NCT00583102?term=NCT00583102&draw=2&rank=1 (accessed on 30 March 2020).
- Sassano, A.; Katsoulidis, E.; Antico, G.; Altman, J.K.; Redig, A.J.; Minucci, S.; Tallman, M.S.; Platanias, L.C. Suppressive effects of statins on acute promyelocytic leukemia cells. Cancer Res. 2007, 67, 4524–4532. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Li, J.; Chen, B.; Zhou, M.; Zeng, Y.; Zhang, Q.; Guo, Y.; Chen, J.; Ouyang, J. Cardiac glycosides inhibit proliferation and induce apoptosis of human hematological malignant cells. Int. J. Clin. Exp. Pathol. 2016, 9, 9268–9275. [Google Scholar]
- Safety and Activity of Digoxin with Decitabine in Adult AML and MDS-Full Text View-ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03113071?term=NCT03113071&draw=1&rank=1 (accessed on 30 March 2020).
- Assouline, S.; Culjkovic-Kraljacic, B.; Bergeron, J.; Caplan, S.; Cocolakis, E.; Lambert, C.; Lau, C.J.; Zahreddine, H.A.; Miller, W.H.; Borden, K.L.B. A phase i trial of ribavirin and low-dose cytarabine for the treatment of relapsed and refractory acute myeloid leukemia with elevated eIF4E. Haematologica 2015, 100, e7–e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uy, G.L.; Rettig, M.P.; Stone, R.M.; Konopleva, M.Y.; Andreeff, M.; McFarland, K.; Shannon, W.; Fletcher, T.R.; Reineck, T.; Eades, W.; et al. A phase 1/2 study of chemosensitization with plerixafor plus G-CSF in relapsed or refractory acute myeloid leukemia. Blood Cancer J. 2017, 7, e542. [Google Scholar] [CrossRef] [PubMed]
- Cancer Research UK. A Study Looking at Treatment for Acute Myeloid Leukaemia and High Risk Myelodysplastic Syndrome (AML18 Pilot). Available online: https://www.cancerresearchuk.org/about-cancer/find-a-clinical-trial/a-study-ac220-or-plerixafor-alongside-chemotheray-for-aml-or-high-risk-mds-aml18-pilot#undefined (accessed on 31 May 2020).
- Allan, J.N.; Roboz, G.J.; Askin, G.; Ritchie, E.; Scandura, J.; Christos, P.; Hassane, D.C.; Guzman, M.L. CD25 expression and outcomes in older patients with acute myelogenous leukemia treated with plerixafor and decitabine. Leuk. Lymphoma 2018, 59, 821–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez-Cuadrón, D.; Boluda, B.; Martínez, P.; Bergua, J.; Rodríguez-Veiga, R.; Esteve, J.; Vives, S.; Serrano, J.; Vidriales, B.; Salamero, O.; et al. A phase I–II study of plerixafor in combination with fludarabine, idarubicin, cytarabine, and G-CSF (PLERIFLAG regimen) for the treatment of patients with the first early-relapsed or refractory acute myeloid leukemia. Ann. Hematol. 2018, 97, 763–772. [Google Scholar] [CrossRef]
- Garcia-Manero, G.; Kantarjian, H.M.; Sanchez-Gonzalez, B.; Yang, H.; Rosner, G.; Verstovsek, S.; Rytting, M.; Wierda, W.G.; Ravandi, F.; Koller, C.; et al. Phase 1/2 study of the combination of 5-aza-2′-deoxycytidine with valproic acid in patients with leukemia. Blood 2006, 108, 3271–3279. [Google Scholar] [CrossRef] [Green Version]
- Lübbert, M.; Rüter, B.H.; Claus, R.; Schmoor, C.; Schmid, M.; Germing, U.; Kuendgen, A.; Rethwisch, V.; Ganser, A.; Platzbecker, U.; et al. A multicenter phase II trial of decitabine as first-line treatment for older patients with acute myeloid leukemia judged unfit for induction chemotherapy. Haematologica 2012, 97, 393–401. [Google Scholar] [CrossRef]
- Lübbert, M.; Grishina, O.; Schmoor, C.; Schlenk, R.F.; Jost, E.; Krauter, J.; Heuser, M.; Thol, F.; Schittenhelm, M.M.; Salih, H.R.; et al. Results of the randomized phase II study decider (AMLSG 14-09) comparing decitabine (DAC) with or without valproic acid (VPA) and with or without All-Trans retinoic acid (ATRA) add-on in newly diagnosed elderly non-fit AML patients. Blood 2016, 128, 589. [Google Scholar] [CrossRef]
- Ryningen, A.; Stapnes, C.; Lassalle, P.; Corbascio, M.; Gjertsen, B.T.; Bruserud, Ø. A subset of patients with high-risk acute myelogenous leukemia shows improved peripheral blood cell counts when treated with the combination of valproic acid, theophylline and all-trans retinoic acid. Leuk. Res. 2009, 33, 779–787. [Google Scholar] [CrossRef]
- Fredly, H.; Ersvær, E.; Kittang, A.O.; Tsykunova, G.; Gjertsen, B.T.; Bruserud, Ø. The combination of valproic acid, all-trans retinoic acid and low-dose cytarabine as disease-stabilizing treatment in acute myeloid leukemia. Clin. Epigenet. 2013, 5, 13. [Google Scholar] [CrossRef] [Green Version]
- Tassara, M.; Döhner, K.; Brossart, P.; Held, G.; Götze, K.; Horst, H.A.; Ringhoffer, M.; Köhne, C.H.; Kremers, S.; Raghavachar, A.; et al. Valproic acid in combination with all-trans retinoic acid and intensive therapy for acute myeloid leukemia in older patients. Blood 2014, 123, 4027–4036. [Google Scholar] [CrossRef] [Green Version]
- Soriano, A.O.; Yang, H.; Faderl, S.; Estrov, Z.; Giles, F.; Ravandi, F.; Cortes, J.; Wierda, W.G.; Ouzounian, S.; Quezada, A.; et al. Safety and clinical activity of the combination of 5-azacytidine, valproic acid, and all-trans retinoic acid in acute myeloid leukemia and myelodysplastic syndrome. Blood 2007, 110, 2302–2308. [Google Scholar] [CrossRef] [PubMed]
- Raffoux, E.; Cras, A.; Recher, C.; Boëlle, P.I.; de Labarthe, A.; Turlure, P.; Marolleau, J.E.; Reman, O.; Gardin, C.; Victor, M.; et al. Phase 2 clinical trial of 5-azacitidine, valproic acid, and all-trans retinoic acid in patients with high-risk acute myeloid leukemia or myelodysplastic syndrome. Oncotarget 2010, 1, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Advani, A.S.; Mcdonough, S.; Copelan, E.; Willman, C.; Mulford, D.A.; List, A.F.; Sekeres, M.A.; Othus, M.; Appelbaum, F.R. SWOG0919: A Phase 2 study of idarubicin and cytarabine in combination with pravastatin for relapsed acute myeloid leukaemia. Br. J. Haematol. 2014, 167, 233–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Advani, A.S.; Li, H.; Michaelis, L.C.; Medeiros, B.C.; Liedtke, M.; List, A.F.; O’Dwyer, K.; Othus, M.; Erba, H.P.; Appelbaum, F.R. Report of the relapsed/refractory cohort of SWOG S0919: A phase 2 study of idarubicin and cytarabine in combination with pravastatin for acute myelogenous leukemia (AML). Leuk. Res. 2018, 67, 17–20. [Google Scholar] [CrossRef]
- Shadman, M.; Mawad, R.; Dean, C.; Chen, T.L.; Shannon-Dorcy, K.; Sandhu, V.; Hendrie, P.C.; Scott, B.L.; Walter, R.B.; Becker, P.S.; et al. Idarubicin, cytarabine, and pravastatin as induction therapy for untreated acute myeloid leukemia and high-risk myelodysplastic syndrome. Am. J. Hematol. 2015, 90, 483–486. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.L.; Estey, E.H.; Othus, M.; Gardner, K.M.; Markle, L.J.; Walter, R.B. Cyclosporine modulation of multidrug resistance in combination with pravastatin, mitoxantrone and etoposide for adult patients with relapsed/refractory acute myeloid leukemia: A phase 1/2 study. Leuk. Lymphoma 2013, 54, 2534–2536. [Google Scholar] [CrossRef] [Green Version]
- ReDO Project. Available online: http://www.redo-project.org/db/ (accessed on 7 March 2020).
- Pantziarka, P.; Verbaanderd, C.; Sukhatme, V.; Rica Capistrano, I.; Crispino, S.; Gyawali, B.; Rooman, I.; Van Nuffel, A.M.T.; Meheus, L.; Sukhatme, V.P.; et al. Redo_DB: The repurposing drugs in oncology database. Ecancermedicalscience 2018, 12. [Google Scholar] [CrossRef] [Green Version]
- Pantziarka, P.; Sukhtame, V.; Meheus, L.; Sukhatme, V.P.V.V.; Bouche, G.; Meheus, L.; Sukhatme, V.P.V.V.; Bouche, G. Repurposing non-cancer Drugs in Oncology—How many drugs are out there? bioRxiv 2017, 1. [Google Scholar] [CrossRef] [Green Version]
- Caravatti, G.; Meyer, T.; Fredenhagen, A.; Trinks, U.; Mett, H.; Fabbro, D. Inhibitory activity and selectivity of staurosporine derivatives towards protein kinase C. Bioorganic Med. Chem. Lett. 1994, 4, 399–404. [Google Scholar] [CrossRef]
- Weisberg, E.; Boulton, C.; Kelly, L.M.; Manley, P.; Fabbro, D.; Meyer, T.; Gilliland, D.G.; Griffin, J.D. Inhibition of mutant FLT3 receptors in leukemia cells by the small molecule tyrosine kinase inhibitor PKC412. Cancer Cell 2002, 1, 433–443. [Google Scholar] [CrossRef] [Green Version]
- Stone, R.M.; DeAngelo, D.J.; Klimek, V.; Galinsky, I.; Estey, E.; Nimer, S.D.; Grandin, W.; Lebwohl, D.; Wang, Y.; Cohen, P.; et al. Patients with acute myeloid leukemia and an activating mutation in FLT3 respond to a small-molecule FLT3 tyrosine kinase inhibitor, PKC412. Blood 2005, 105, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Rydapt Prescribing Information. 2017. Available online: https://www.fda.gov/medwatch (accessed on 27 April 2020).
- Mori, M.; Kaneko, N.; Ueno, Y.; Tanaka, R.; Cho, K.; Saito, R.; Kondoh, Y.; Shimada, I.; Kuromitsu, S. ASP2215, a novel FLT3/AXL inhibitor: Preclinical evaluation in acute myeloid leukemia (AML). J. Clin. Oncol. 2014, 32, 7070. [Google Scholar] [CrossRef]
- Perl, A.E.; Altman, J.K.; Cortes, J.E.; Smith, C.C.; Litzow, M.; Baer, M.R.; Claxton, D.F.; Erba, H.P.; Gill, S.C.; Goldberg, S.L.; et al. Final results of the chrysalis trial: A first-in-human phase 1/2 Dose-Escalation, dose-expansion study of gilteritinib (ASP2215) in patients with relapsed/refractory acute myeloid leukemia (R/R AML). Blood 2016, 128, 1069. [Google Scholar] [CrossRef]
- Xospata Prescribing Information. 2019. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/211349s001lbl.pdf (accessed on 27 April 2020).
- Agios Pharmaceuticals, Inc. Agios Pharmaceuticals Announces that Celgene Exercised its Option to License AG-221 Under Global Strategic Collaboration. Available online: https://investor.agios.com/news-releases/news-release-details/agios-pharmaceuticals-announces-celgene-exercised-its-option (accessed on 28 April 2020).
- Yen, K.; Wang, F.; Travins, J.; Chen, Y.; Yang, H.; Straley, K.; Choe, S.; Dorsch, M.; Schenkein, D.P.; Agresta, S.; et al. AG-221 offers a survival advantage in a primary human IDH2 mutant AML xenograft model. Blood 2013, 122, 240. [Google Scholar] [CrossRef]
- Stein, E.M.; DiNardo, C.D.; Pollyea, D.A.; Fathi, A.T.; Roboz, G.J.; Altman, J.K.; Stone, R.M.; Deangelo, D.J.; Levine, R.L.; Flinn, I.W.; et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood 2017, 130, 722–731. [Google Scholar] [CrossRef]
- IDHIFA Prescribing Information. 2017. Available online: www.fda.gov/medwatch (accessed on 27 April 2020).
- Popovici-Muller, J.; Saunders, J.O.; Salituro, F.G.; Travins, J.M.; Yan, S.; Zhao, F.; Gross, S.; Dang, L.; Yen, K.E.; Yang, H.; et al. Discovery of the first potent inhibitors of mutant IDH1 that lower tumor 2-HG in vivo. ACS Med. Chem. Lett. 2012, 3, 850–855. [Google Scholar] [CrossRef] [Green Version]
- Popovici-Muller, J.; Lemieux, R.M.; Artin, E.; Saunders, J.O.; Salituro, F.G.; Travins, J.; Cianchetta, G.; Cai, Z.; Zhou, D.; Cui, D.; et al. Discovery of AG-120 (Ivosidenib): A first-in-class mutant IDH1 inhibitor for the treatment of IDH1 mutant cancers. ACS Med. Chem. Lett. 2018, 9, 300–305. [Google Scholar] [CrossRef] [Green Version]
- Hansen, E.; Quivoron, C.; Straley, K.; Lemieux, R.; Popovici-Muller, J.; Sadrzadeh, H.; Fathi, A.; Gliser, C.; David, M.; Saada, V.; et al. AG-120, an oral, selective, first-in-class, potent inhibitor of mutant IDH1, reduces intracellular 2HG and induces cellular differentiation in TF-1 R132H cells and primary human IDH1 mutant AML patient samples treated Ex Vivo. Blood 2014, 124, 3734. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Stein, E.M.; de Botton, S.; Roboz, G.J.; Altman, J.K.; Mims, A.S.; Swords, R.; Collins, R.H.; Mannis, G.N.; Pollyea, D.A.; et al. Durable remissions with ivosidenib in IDH1 -mutated relapsed or refractory AML. N. Engl. J. Med. 2018, 378, 2386–2398. [Google Scholar] [CrossRef]
- Tibsovo Prescribing Information. 2019. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/211192s001lbl.pdf (accessed on 28 April 2020).
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef]
- Pan, R.; Hogdal, L.J.; Benito, J.M.; Bucci, D.; Han, L.; Borthakur, G.; Cortes, J.; Deangelo, D.J.; Debose, L.; Mu, H.; et al. Selective BCL-2 inhibition by ABT-199 causes on-target cell death in acute myeloid Leukemia. Cancer Discov. 2014, 4, 362–675. [Google Scholar] [CrossRef] [Green Version]
- Konopleva, M.; Pollyea, D.A.; Potluri, J.; Chyla, B.; Hogdal, L.; Busman, T.; McKeegan, E.; Salem, A.H.; Zhu, M.; Ricker, J.L.; et al. Efficacy and biological correlates of response in a phase II study of venetoclax monotherapy in patients with acute Myelogenous Leukemia. Cancer Discov. 2016, 6, 1106–1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venclexta Prescribing Information. 2018. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/208573s009lbl.pdf (accessed on 28 April 2020).
- Munchhof, M.J.; Li, Q.; Shavnya, A.; Borzillo, G.V.; Boyden, T.L.; Jones, C.S.; Lagreca, S.D.; Martinez-Alsina, L.; Patel, N.; Pelletier, K.; et al. Discovery of PF-04449913, a potent and orally bioavailable inhibitor of smoothened. ACS Med. Chem. Lett. 2012, 3, 106–111. [Google Scholar] [CrossRef] [Green Version]
- Fukushima, N.; Minami, Y.; Kakiuchi, S.; Kuwatsuka, Y.; Hayakawa, F.; Jamieson, C.; Kiyoi, H.; Naoe, T. Small-molecule Hedgehog inhibitor attenuates the leukemia-initiation potential of acute myeloid leukemia cells. Cancer Sci. 2016, 107, 1422–1429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinelli, G.; Oehler, V.G.; Papayannidis, C.; Courtney, R.; Shaik, M.N.; Zhang, X.; O’Connell, A.; McLachlan, K.R.; Zheng, X.; Radich, J.; et al. Treatment with PF-04449913, an oral smoothened antagonist, in patients with myeloid malignancies: A phase 1 safety and pharmacokinetics study. Lancet Haematol. 2015, 2, e339–e346. [Google Scholar] [CrossRef]
- Daurismo Prescribing Information. 2018. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/210656s000lbl.pdf (accessed on 28 April 2020).
- Mayer, L.D.; Harasym, T.O.; Tardi, P.G.; Harasym, N.L.; Shew, C.R.; Johnstone, S.A.; Ramsay, E.C.; Bally, M.B.; Janoff, A.S. Ratiometric dosing of anticancer drug combinations: Controlling drug ratios after systemic administration regulates therapeutic activity in tumor-bearing mice. Mol. Cancer Ther. 2006, 5, 1854–1863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feldman, E.J.; Lancet, J.E.; Kolitz, J.E.; Ritchie, E.K.; Roboz, G.J.; List, A.F.; Allen, S.L.; Asatiani, E.; Mayer, L.D.; Swenson, C.; et al. First-in-man study of CPX-351: A liposomal carrier containing cytarabine and daunorubicin in a fixed 5:1 molar ratio for the treatment of relapsed and refractory acute myeloid leukemia. J. Clin. Oncol. 2011, 29, 979–985. [Google Scholar] [CrossRef] [Green Version]
- Vyxeos Prescribing Information. 2017. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/209401s000lbl.pdf (accessed on 28 April 2020).
- Wojcicki, A.V.; Kadapakkam, M.; Frymoyer, A.; Lacayo, N.; Chae, H.D.; Sakamoto, K.M. Repurposing drugs for acute myeloid leukemia: A worthy cause or a futile pursuit? Cancers (Basel) 2020, 12, 441. [Google Scholar] [CrossRef] [Green Version]
- Regnault, C.; Dheeman, D.S.; Hochstetter, A. Microfluidic devices for drug assays. High-Throughput 2018, 7, 18. [Google Scholar] [CrossRef] [Green Version]
- Zhai, J.; Yi, S.; Jia, Y.; Mak, P.I.; Martins, R.P. Cell-based drug screening on microfluidics. Trends Anal. Chem. 2019, 117, 231–241. [Google Scholar] [CrossRef]
- Qin, Y.; Wu, L.; Schneider, T.; Yen, G.S.; Wang, J.; Xu, S.; Li, M.; Paguirigan, A.L.; Smith, J.L.; Radich, J.P.; et al. A self-digitization dielectrophoretic (SD-DEP) chip for high-efficiency single-cell capture, on-demand compartmentalization, and downstream nucleic acid analysis. Angew. Chem. Int. Ed. 2018, 57, 11378–11383. [Google Scholar] [CrossRef] [PubMed]
- Verbaanderd, C.; Meheus, L.; Huys, I.; Pantziarka, P. Repurposing drugs in oncology: Next steps. Trends Cancer 2017, 3, 543–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, S.K.; Nayak, R.P. Off-label use of medicine: Perspective of physicians, patients, pharmaceutical companies and regulatory authorities. J. Pharmacol. Pharmacother. 2014, 5, 88–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Year | Screen | Library (Compound Number) | Model System | Readout | Concentration | Assay Time | Hits (%) | Candidate | Reference |
|---|---|---|---|---|---|---|---|---|---|
| 2007 | Cyclin D2 transactivation inhibitor screen | Prestwick (1120) and LOPAC (1280) | NIH3T3 expressing c-Maf and the cyclin D2 promoter-driven luciferase reporter | Luminescence and MTS reduction viability assay | 5 µM | 20 h | 39 (1.6) | Clioquinol | [49,50] |
| 2010 | Chemical screen | Custom * (110) | OCI-AML2, HL60, KG1a | MTS reduction viability assay | 5–50 µM | 72 h | Several | Flubendazole | [51] |
| 2010 | Chemical screen | Custom *,$ (100) | OCI-AML2, HL60, KG1a | MTS reduction viability assay | 5–50 µM | 72 h | NR | Ivermectin | [52] |
| 2011 | Chemical screen | Custom * (312) | TEX and M9-ENL1 with LSC features | MTS reduction viability assay | 1–10 µM | 72 h | 5 (1.6) | Tigecycline | [53] |
| 2012 | STAT5 transcriptional activity inhibitor screen | Prestwick (1120) | U3A cell line expressing STAT5 promoter-driven luciferase reporter + cytokine induction | Luminescence | 0–10 µM | 2 + 6 h | NR | Pimozide | [54,55] |
| 2012 | Chemical screen | NIH Clinical Collection (446) and Canadian Compound Collection (144) | Neoplastic and normal hPSCs transduced with EOS-GFP reporter indicative of Oct4 and Sox2 expression levels | Differentiation induction assessment by automated microscopy | 10 µM | 72 h | 11 (1.8) | Thioridazine | [56] |
| Secondary high content analysis | 11 | 3 (27) | |||||||
| 2013 | Chemical screen | Custom *,$ (100) | OCI-AML2, HL60, KG1a | MTS reduction viability assay | 5–50 µM | 72 h | Several | Mefloquine | [57] |
| 2013 | Chemical screen | Prestwick (1120) | Primary murine BM cells transformed with AML1-ETO, cells from second plating | Methylcellulose colony formation assay | 50 µM | 5–7 days | 95 (8.4) | Furazolidone | [58] |
| 2013 | Chemical screen | Broad Institute Compound Collection (14718) ** | Primary murine LSCs from quaternary MLL-AF9 AML grown on primary or OP9 stroma cells mimicking the niche | Leukemic cobblestone area-forming cell assay | 5 µM | 5 days | 415 (2.8) | Lovastatin Trioglitazone | [59] |
| 2015 | Chemical screen | LOPAC (1280) | Primary AML and PBMNC samples (4+4; also, ALL and CLL) | Fluorometric Microculture Cytotoxicity Assay | 10 µM | 72 h | 25 (1.9) | Quinacrine | [60] |
| 2016 | Chemical screen | Screen Well FDA-approved drugs (760) ^ | Primary murine cell lines representative of AML and MLL | <50% viability | NR | NR | 38 (5) | Mebendazole | [61] |
| 2016 | Chemical screen in the presence of ATRA | Biomol (36), MicroSource (1214), Prestwick (1120), Sigma (885) | MN1-transformed murine bone marrow progenitors, known to be resistant to ATRA | Alamar blue viability assay, <80% viability | 2.5 µM + 1 µM ATRA | 45 h | 117 (3.2) | Pyrimethamine | [62] |
| 2016 | In silico: CMap (Build 1.0) | 164 perturbagens | Published expression profile of the HL60 cell line treated with PMA; CMap database | PMA-differentiation signature crossed with CMap; p value <0.05 and a connectivity score >0.75 in HL60 at a concentration <10 µM | N/A | N/A | NR | Bromocriptine | [63] |
| 2017 | Chemical screen | Screen Well FDA-approved drugs (760) ^ | Primary murine cell lines representative of AML with a t(9;11) (MLL-AF9 translocation) and AML with a normal karyotype (HOXA9-Meis 1 -driven). | <50% viability | NR | NR | 38 (5) | Albendazole | [64] |
| 2018 | In silico: 2D chemical similarity analysis | NR | N/A | Structural similarity to XX-650-23 CREB inhibitor | N/A | N/A | NR | Niclosamide | [65] |
| 2018 | Chemical screen in the presence of ABT-737 BCL2 inhibitor. | LOPAC (1280) | RPMI 8226, U937, HL60 | CellTiterGlo viability assay | 1.8, 9, 45 µM + IC30 or IC70 of ABT-737 | 48 h | NR | Amiodarone | [66,67] |
| 2018 | In silico: CMap (Build 2.0) | Small bioactive molecules and tools (1310) | CMap database | First query with two LSC signatures to identify compounds that inhibit LSC gene expression programmes. Second query of the CMap to exclude compounds that inhibit HSCs | N/A | N/A | 151 (11.5) | Digitoxin Ouabain | [68] |
| Secondary chemical screen | 84/151 compounds from the in silico screen. | AML 8227 | Phenotype screen by flow cytometry | 2.5, 5, 10 µM | 6 days | 48 |
| Drug Group | Candidate Compound | First Preclinical Study (Year) | First Clinical Trial (Year) | NCT Number | Status | Phase | Years between Preclinical and Clinical Study | |
|---|---|---|---|---|---|---|---|---|
| Anti-microbial | Clioquinol | 2007 [50] | 2009 [108] | NCT00963495 | Terminated | I | 2 | |
| Hydroxychloroquine | 2015 [77] | 2016 [109] | NCT02631252 | Terminated | I | 1 | ||
| Tigecycline | 2011 [53] | 2011 [110] | NCT01332786 | Completed | I | 0 | ||
| Ribavirin | 2004 [111] | 2007 [112] | NCT00559091 | Completed | II | 3 | ||
| Plerixafor | 2004 [113] | 2007 [114] | NCT00512252 | Completed | I/II | 3 | ||
| Neuro-psychiatric | Valproic acid | 2001 [89] | 2004 [115] | NCT00075010 | Completed | I/II | 3 | |
| Thioridazine | 2012 [56] | 2014 [116] | NCT02096289 | Completed | I | 2 | ||
| Sertraline | 2004 [116] | 2016 [117] | NCT02891278 | Recruiting | I | 12 | ||
| Tranylcypromine | 2012 [90] | 2014 [90] | NCT02261779 | Unknown | I/II | 2 | ||
| Metabolic | Metformin | 2010 [118] | 2015 [119] | NCT01849276 | Terminated | I | 5 | |
| Pioglitazone | 2004 [120] | 2017 [121] | NCT02942758 | Recruiting | II | 13 | ||
| Statins | Pravastatin | 1997 [104] | 2007 [122] | NA | Completed | I | 10 | |
| Lovastatin | 1997 [104] | 2001 [123] | NCT00583102 | Terminated | I/II | 4 | ||
| Atorvastatin | 2007 [124] | 2018 [124] | NCT03560882 | Recruiting | I | 11 | ||
| Cardiac | Digoxin | 2016 [125] | 2017 [126] | NCT03113071 | Terminated | I/II | 1 | |
| Compound | Year of Development | First Preclinical Study (Publication Year) | First Clinical Trial (Starting Year) | FDA Approval | Years Between Development and Preclinical Studies | Years Between Preclinical and Clinical Studies | Years Between Clinical Trial and FDA Approval | Total Drug Discovery Time |
|---|---|---|---|---|---|---|---|---|
| Midostaurin | 1986 [147] | 2002 [148] | 2005 [149] | 2017 [150] | 16 | 3 | 12 | 31 |
| Gilteritinib | 2013 * | 2014 [151] | 2013 [152] | 2018 [153] | 1 | N/A | 5 | 5 |
| Enasidenib | 2010 § [154] | 2013 [155] | 2013 [156] | 2017 [157] | 3 | 0 | 4 | 7 |
| Ivosidenib | 2012 ^ [158,159] | 2014 [160] | 2014 [161] | 2018 [162] | 2 | 0 | 4 | 6 |
| Venetoclax | 2013 [163] | 2014 [164] | 2013 [165] | 2018 [166] | 1 | N/A | 5 | 6 |
| Daurismo | 2011 [167] | 2016 [168] | 2010 [169] | 2018 [170] | 5 | N/A | 8 | 7 |
| CPX-351 | 2006 [171] | 2006 [171] | 2006 [172] | 2017 [173] | 0 | 0 | 11 | 11 |
| Median | 4 | 0.4 | 7 | 10.4 |
| Compound | First Clinical Trial in AML | Original Indication |
|---|---|---|
| Clioquinol | Oral administration | Topical application |
| OH-chloroquine | 600–1400 mg/day for 21 days | Acute malaria: 800 mg +400 mg (6h) + 400 mg (24h) + 400 mg (48h); Malaria prevention: 400 mg/week |
| Tigecycline | 50–350 mg/day (7 levels, 3-week cycles) | Initially, 100 mg, then 50 mg/day for 21 days |
| Ribavirin | 1000 mg/day | 2000 mg/day |
| Plerixafor | Escalation 0.08–0.24 mg/kg/day (= ~6–18 mg/day) | Max. 80 mg/day |
| VPA | 20 mg/kg/day for 10 days | 10–15/kg/day, max. dose 60 mg/kg/day |
| Thioridazine | 25, 50, 100 mg every 6 h/21 days (= max. 400 mg/day) | Initially, 50 × 3 mg/day, standard therapy: 200–800/day |
| Sertraline | Max. 250 mg/day | 50–200 mg/day |
| Tranylcypromine | 10–60 mg/day | 30 mg/day, max. 60 mg/day |
| Metformin | Not reported | Max. 2250 mg/day |
| Pioglitazone | 45 mg/day | 15–30 mg/day |
| Pravastatin | 40–1680 mg/day | 40–80 mg/day |
| Lovastatin | Escalation 0.5–24 mg/kg/day (~35–1680 mg/day) | Initially, 20 mg/day; maintenance: 10–80 mg/day |
| Atorvastatin | 80 mg/day 1–4 weeks | Max. 80 mg/day |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valli, D.; Gruszka, A.M.; Alcalay, M. Has Drug Repurposing Fulfilled Its Promise in Acute Myeloid Leukaemia? J. Clin. Med. 2020, 9, 1892. https://doi.org/10.3390/jcm9061892
Valli D, Gruszka AM, Alcalay M. Has Drug Repurposing Fulfilled Its Promise in Acute Myeloid Leukaemia? Journal of Clinical Medicine. 2020; 9(6):1892. https://doi.org/10.3390/jcm9061892
Chicago/Turabian StyleValli, Debora, Alicja M. Gruszka, and Myriam Alcalay. 2020. "Has Drug Repurposing Fulfilled Its Promise in Acute Myeloid Leukaemia?" Journal of Clinical Medicine 9, no. 6: 1892. https://doi.org/10.3390/jcm9061892
APA StyleValli, D., Gruszka, A. M., & Alcalay, M. (2020). Has Drug Repurposing Fulfilled Its Promise in Acute Myeloid Leukaemia? Journal of Clinical Medicine, 9(6), 1892. https://doi.org/10.3390/jcm9061892