Atherosclerosis as Pathogenetic Substrate for Sars-Cov2 Cytokine Storm

 , ,
, , {kind=link}
Abstract
:1. Introduction
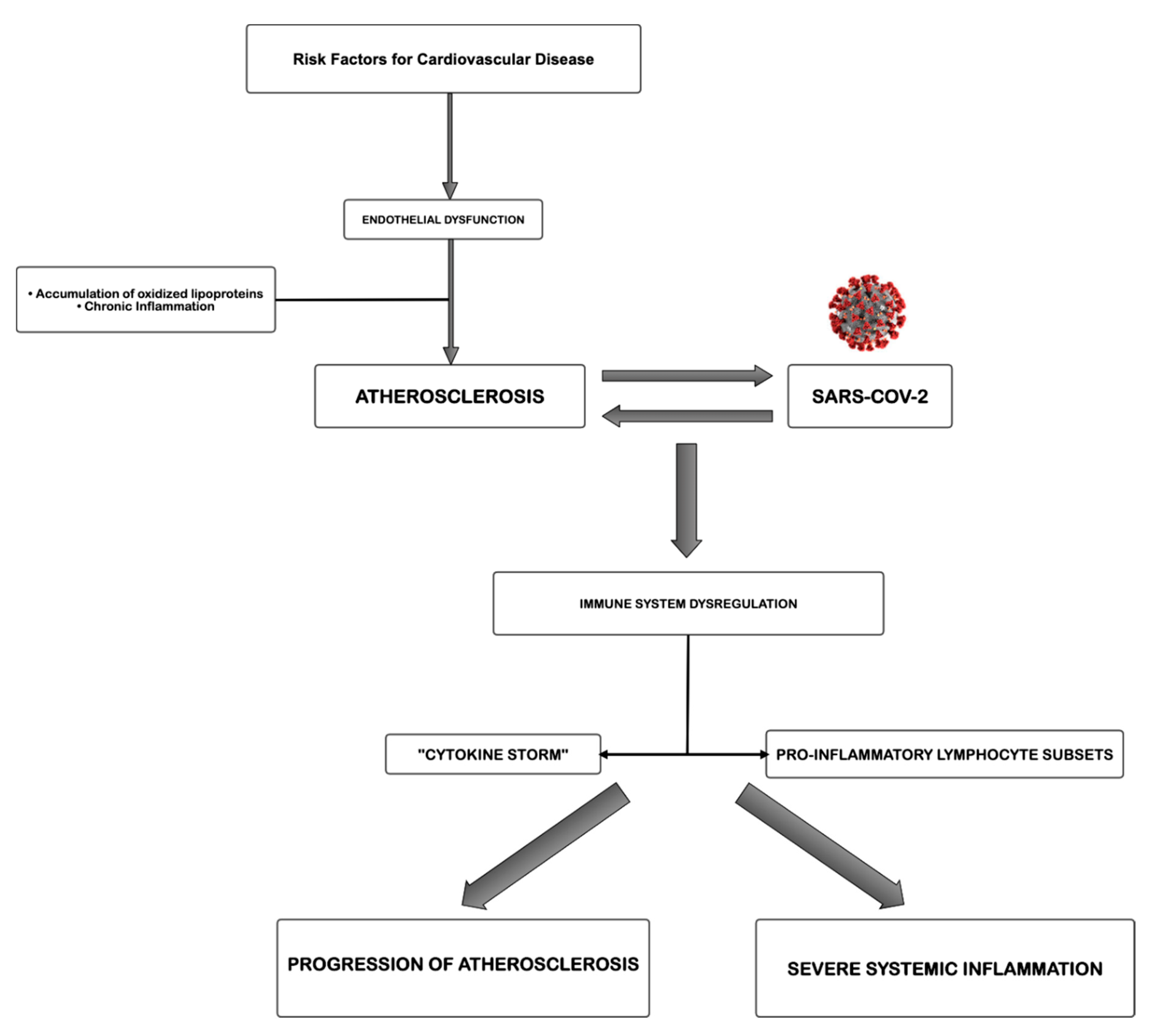
2. Pathogenesis of COVID-19
3. The Role of Atherosclerosis
4. Effects of Sars-CoV-2 on Cardiovascular System
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lauer, S.A.; Grantz, K.H.; Bi, Q.; Jones, F.K.; Zheng, Q.; Meredith, H.R.; Azman, A.S.; Reich, N.G.; Lessler, J. The incubation period of coronavirus disease 2019 (COVID-19) from publicly reported confirmed cases: estimation and application. Ann. Intern. Med. 2020, 172, 577–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Z.; Song, C.; Xu, C.; Jin, G.; Chen, Y.; Xu, X.; Ma, H.; Chen, W.; Lin, Y.; Zheng, Y.; et al. Clinical characteristics of 24 asymptomatic infections with COVID-19 screened among close contacts in Nanjing, China. Sci. China Life Sci. 2020, 63, 706–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Fan, G.; Xu, J.; Gu, X.; Cheng, Z.; Yu, T.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Yu, Y.; Xu, J.; Shu, H.; Xia, J.; Liu, H.; Wu, Y.; Zhang, L.; Yu, Z.; Fang, M.; et al. Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: a single-centered, retrospective, observational study. Lancet Resp. Med. 2020, 8, 475–481. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Mc Googan, J.M. Characteristics of and important lessons from the coronavirus disease 2019 (COVID-19) outbreak in China: Summary of a report of 72 314 cases from the Chinese Center for Disease Control and Prevention. JAMA 2020, 323, 1239–1242. [Google Scholar] [CrossRef]
- Lin, L.; Lu, L.; Cao, W.; Li, T. Hypothesis for potential pathogenesis of SARS-CoV-2 infection—A review of immune changes in patients with viral pneumonia. Emerg. Microbes Infect. 2020, 9, 727–732. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Fu, B.; Zheng, X.; Wang, D.; Zhao, C.; Qi, Y.; Sun, R.; Tian, Z.; Xu, X.; Wei, H. Pathogenic T cells and inflammatory monocytes incite inflammatory storm in severe COVID-19 patients. Natl. Sci. Rev. 2020, 7, 998–1002. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Penninger, J.M.; Li, Y.; Zhong, N.; Slutsky, A.S. Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: molecular mechanisms and potential therapeutic target. Intensive Care Med. 2020, 46, 586–590. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.; Cheng, Y.; Wu, Y. Understanding SARS-CoV-2-mediated inflammatory responses: from mechanisms to potential therapeutic tools. Virol. Sin. 2020, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Ludvigsson, J.F. Systematic review of COVID-19 in children shows milder cases and a better prognosis than adults. Acta Paediatr. 2020, 109, 1088–1095. [Google Scholar] [CrossRef]
- Zhao, X.; Zhang, B.; Li, P.; Ma, C.; Gu, J.; Hou, P.; Guo, Z.; Wu, H.; Bai, Y. Incidence, clinical characteristics and prognostic factor of patients with COVID-19: A systematic review and meta-analysis. MedRxiv 2020. [Google Scholar] [CrossRef]
- Wu, D.; Wu, T.; Liu, Q.; Yang, Z. The SARS-CoV-2 outbreak: What we know. Int. J. Infect. Dis. 2020, 94, 44–48. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Li, L. SARS-CoV-2: Virus dynamics and host response. Lancet Infect. Dis. 2020, 20, 515–516. [Google Scholar] [CrossRef] [Green Version]
- Cascella, M.; Rajnik, M.; Cuomo, A.; Dulebohn, S.C.; Di Napoli, R. Features, evaluation and treatment coronavirus (COVID-19). In StatPearls; StatPearls Publishing: St. Petersburg, Russia, 2020. [Google Scholar]
- Verdecchia, P.; Cavallini, C.; Spanevello, A.; Angeli, F. The pivotal link between ACE2 deficiency and SARS-CoV-2 infection. Eur. J. Intern. Med. 2020, 76, 14–20. [Google Scholar] [CrossRef]
- Vinciguerra, M.; Greco, E. Sars-CoV-2 and black population: ACE2 as shield or blade? Infect. Genet. Evol. 2020, 84, 104361. [Google Scholar] [CrossRef]
- Van de Veerdonk, F.L.; Netea, M.G.; van Deuren, M.; van der Meer, J.W.; de Mast, Q.; Brüggemann, R.J.; van der Hoeven, H. Kallikrein-kinin blockade in patients with COVID-19 to prevent acute respiratory distress syndrome. Elife 2020, 9, e57555. [Google Scholar] [CrossRef] [PubMed]
- Risitano, A.M.; Mastellos, D.C.; Huber-Lang, M.; Yancopoulou, D.; Garlanda, C.; Ciceri, F.; Lambris, J.D. Complement as a target in COVID-19? Nat. Rev. Immunol. 2020, 20, 343–344. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Liu, H.G.; Liu, W.; Liu, J.; Liu, K.; Shang, J.; Deng, Y.; Wei, S. Analysis of clinical features of 29 patients with 2019 novel coronavirus pneumonia. Chin. J. Tuberc. Resp. Dis. 2020, 43, E005. [Google Scholar]
- Han, H.; Yang, L.; Liu, R.; Liu, F.; Wu, K.-L.; Li, J.; Liu, X.-H.; Zhu, C. Prominent changes in blood coagulation of patients with SARS-CoV-2 infection. Clin. Chem. Lab. Med. (CCLM) 2020, 58, 1116–1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, D.; Ley, K. Immunity and Inflammation in Atherosclerosis. Circ. Res. 2019, 124, 315–327. [Google Scholar] [CrossRef]
- Deanfield, J.; Halcox, J.; Rabelink, T.J. Endothelial function and dysfunction: testing and clinical relevance. Circulation 2017, 115, 1285–1295. [Google Scholar] [CrossRef] [PubMed]
- Monteil, V.; Kwon, H.; Prado, P.; Hagelkrüys, A.; Wimmer, R.A.; Stahl, M.; Leopoldi, A.; Garreta, E.; Del Pozo, C.H.; Prosper, F.; et al. Inhibition of SARS-CoV-2 infections in engineered human tissues using clinical-grade soluble human ACE2. Cell 2020, 181, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Connors, J.M.; Levy, J.H. COVID-19 and its implications for thrombosis and anticoagulation. Blood J. Am. Soc. Hematol. 2020, 135, 2033–2040. [Google Scholar]
- Carsana, L.; Sonzogni, A.; Nasr, A.; Rossi, R.S.; Pellegrinelli, A.; Zerbi, P.; Rech, R.; Colombo, R.; Antinori, S.; Corbellino, M.; et al. Pulmonary post-mortem findings in a series of COVID-19 cases from northern Italy: A two-centre descriptive study. Lancet Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Magro, C.; Mulvey, J.J.; Berlin, D.; Nuovo, G.; Salvatore, S.; Harp, J.; Baxter-Stoltzfus, A.; Laurence, J. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: A report of five cases. Transl. Res. 2020, 220, 1–13. [Google Scholar] [CrossRef]
- Taleb, S. Inflammation in atherosclerosis: L’inflammation dans l’athérosclérose. Arch. Cardiovasc. Dis. 2016, 109, 708–715. [Google Scholar] [CrossRef]
- Abbasi, J. Cardiovascular Corner—Stable Coronary Artery Disease, An LDL “Vaccine,” and Anti-inflammatories. JAMA 2020, 323, 1233–1234. [Google Scholar] [CrossRef]
- Zhu, Y.; Xian, X.; Wang, Z.; Bi, Y.; Chen, Q.; Han, X.; Tang, D.; Chen, R. Research progress on the relationship between atherosclerosis and inflammation. Biomolecules 2018, 8, 80. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Li, W.; Li, X.; Zhou, H. Inflammation: A Novel Therapeutic Target/Direction in Atherosclerosis. Curr. Pharm. Des. 2017, 23, 1216–1227. [Google Scholar] [CrossRef]
- Miteva, K.; Madonna, R.; De Caterina, R.; Van Linthout, S. Innate and adaptive immunity in atherosclerosis. Vasc. Pharm. 2018, 107, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Wildgruber, M.; Aschenbrenner, T.; Wendorff, H.; Czubba, M.; Glinzer, A.; Haller, B.; Schiemann, M.; Zimmermann, A.; Berger, H.; Eckstein, H.-H.; et al. The “intermediate” CD14++ CD16+ monocyte subset increases in severe peripheral artery disease in humans. Sci. Rep. 2016, 6, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hovland, A.; Jonasson, L.; Garred, P.; Yndestad, A.; Aukrust, P.; Lappegard, K.T.; Espevik, T.; Mollnes, T.E. The complement system and toll-like receptors as integrated players in the pathophysiology of atherosclerosis. Atherosclerosis 2015, 241, 480–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabas, I.; Lichtman, A.H. Monocyte-Macrophages and T Cells in Atherosclerosis. Immunity 2017, 47, 621–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hedrick, C.C. Lymphocytes in atherosclerosis. Arter. Thromb. Vasc. Biol. 2015, 35, 253–257. [Google Scholar] [CrossRef] [Green Version]
- Sima, P.; Vannucci, L.; Vetvicka, V. Atherosclerosis as autoimmune disease. Ann. Transl. Med. 2018, 6, 116. [Google Scholar] [CrossRef]
- Maganto-García, E.; Tarrio, M.L.; Grabie, N.; Bu, D.-X.; Lichtman, A.H. Dynamic changes in regulatory T cells are linked to levels of diet-induced hypercholesterolemia. Circulation 2011, 124, 185–195. [Google Scholar] [CrossRef] [Green Version]
- Rial, J.G.; Tuala, M.J.C.; Calle, I.R.; Carballa, A.G.; Lopez, M.C.; Tenreiro, C.R.; Urbieta, A.D.; Velasco, C.R.; Nunez, N.R.; Pena, R.T.; et al. Increased serum levels of sCD14 and sCD163 indicate a preponderant role for monocytes in COVID-19 immunopathology. medRxiv 2020. [Google Scholar] [CrossRef]
- Wang, H.; Yuan, Z.; Pavel, M.A.; Hansen, S. Cholesterol and COVID19 lethality in elderly. bioRxiv 2020. [Google Scholar] [CrossRef]
- Cai, T.; Zhang, Y.; Ho, Y.L.; Link, N.; Sun, J.; Huang, J.; Cai, A.; Damrauer, S.; Ahuja, Y.; Honerlaw, J.; et al. Association of interleukin 6 receptor variant with cardiovascular disease effects of interleukin 6 receptor blocking therapy: A phenome-wide association study. JAMA Cardiol. 2018, 3, 849–857. [Google Scholar] [CrossRef] [Green Version]
- Ridker, P.M.; Lüscher, T.F. Anti-inflammatory therapies for cardiovascular disease. Eur. Heart J. 2014, 35, 1782–1791. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M. From C-Reactive protein to interleukin-6 to interleukin-1: Moving upstream to identify novel targets for atheroprotection. Circ. Res. 2016, 118, 145–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Abbate, A.; Van Tassell, B.W.; Biondi-Zoccai, G.G.L.; Kontos, M.C.; Grizzard, J.D.; Spillman, D.W.; Oddi, C.; Roberts, C.S.; Melchior, R.D.; Mueller, G.H.; et al. Effects of interleukin-1 blockade with anakinra on adverse cardiac remodeling and heart failure after acute myocardial infarction [from the Virginia Commonwealth University-Anakinra Remodeling Trial (2)(VCU-ART2) pilot study]. Am. J. Cardiol. 2013, 111, 1394–1400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morton, A.C.; Rothman, A.; Greenwood, J.P.; Gunn, J.; Chase, A.; Clarke, B.; Hall, I.P.; Fox, K.; Foley, C.; Banya, W.; et al. The effect of interleukin-1 receptor antagonist therapy on markers of inflammation in non-ST elevation acute coronary syndromes: The MRC-ILA Heart Study. Eur. Heart J. 2015, 36, 377–384. [Google Scholar] [CrossRef] [Green Version]
- Sarwar, N.; Butterworth, A.S.; Freitag, D.F.; Gregson, J.; Willeit, P.; Gorman, N.N.; Gao, P.; Saleheen, D.; Rendon, A.; Nelson, C.P.; et al. IL6R Genetics Consortium Emerging Risk Factors Collaboration: Interleukin-6 receptor pathways in coronary heart disease: a collaborative meta-analysis of 82 studies. Lancet 2012, 379, 1205–1213. [Google Scholar] [CrossRef] [Green Version]
- Cardillo, C.; Schinzari, F.; Melina, D.; Zoli, A.; Ferraccioli, G.; Mores, N.; Mettimano, M. Intravascular tumor necrosis factor α blockade reverses endothelial dysfunction in rheumatoid arthritis. Clin. Pharmacol. Ther. 2006, 80, 275–281. [Google Scholar] [CrossRef]
- Taguchi, H.; Nishi, K.; Suzuki, T.; Okano, Y. Anti-atherosclerotic effects of etanercept in Rheumatoid Arthritis Patients. Jpn. J. Clin. Immunol. 2012, 35, 183–187. [Google Scholar] [CrossRef] [Green Version]
- Seriolo, B.; Fasciolo, D.; Paolino, S.; Sulli, A.; Cutolo, M. Effects of anti-TNF-α treatment on lipid profile in patients with active rheumatoid arthritis. Ann. N. Y. Acad. Sci. 2006, 1069, 414–419. [Google Scholar] [CrossRef]
- Tam, L.S.; Kitas, G.D.; Gonźlez-gay, M.A. Can suppression of inflammation by anti-TNF prevent progression of subclinical atherosclerosis in inflammatory arthritis? Rheumatology 2014, 53, 1108–1119. [Google Scholar] [CrossRef] [Green Version]
- Nidorf, S.M.; Eikelboom, J.; Budgeon, C.; Thompson, P.L. Low-dose colchicine for secondary prevention of cardiovascular disease. J. Am. Coll. Cardiol. 2013, 61, 404–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavalli, G.; De Luca, G.; Campochiaro, C.; Della-Torre, E.; Ripa, M.; Canetti, D.; Oltolini, C.; Castiglioni, B.; Din, C.T.; Boffini, N.; et al. Interleukin-1 blockade with high-dose anakinra in patients with COVID-19, acute respiratory distress syndrome, and hyperinflammation: a retrospective cohort study. Lancet Rheumatol. 2020, 2, e325–e331. [Google Scholar] [CrossRef]
- Aouba, A.; Baldolli, A.; Geffray, L.; Verdon, R.; Bergot, E.; Martin-Silva, N.; Justet, A. Targeting the inflammatory cascade with anakinra in moderate to severe COVID-19 pneumonia: Case series. Ann. Rheum. Dis. 2020. [Google Scholar] [CrossRef] [PubMed]
- Bonow, R.O.; Fonarow, G.C.; O’Gara, P.T.; Yancy, C.W. Association of Coronavirus Disease 2019 (COVID-19) With Myocardial Injury and Mortality. JAMA Cardiol. 2020. [Google Scholar] [CrossRef] [Green Version]
- Smeeth, L.; Thomas, S.L.; Hall, A.; Hubbard, R.B.; Farrington, C.P.; Vallance, P. Risk of myocardial infarction and stroke after acute infection or vaccination. N. Engl. J. Med. 2004, 351, 2611–2618. [Google Scholar] [CrossRef] [Green Version]
- Albiero, R.; Seresini, G. Atherosclerotic spontaneous coronary artery dissection (A-SCAD) in a patient with COVID-19: Case report and possible mechanisms. Eur. Heart J. Case Rep. 2020. [Google Scholar] [CrossRef]
- Wu, Q.; Zhou, L.; Sun, X.; Yan, Z.; Hu, C.; Wu, J.; Xu, L.; Li, X.; Liu, H.; Yin, P.; et al. Altered lipid metabolism in recovered sars patients twelve years after infection. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef]
- Sun, J.; Yu, H.; Liu, H.; Pu, D.; Gao, J.; Jin, X.; Liu, X.; Yan, A. Correlation of pre-operative circulating inflammatory cytokines with restenosis and rapid angiographic stenotic progression risk in coronary artery disease patients underwent percutaneous coronary intervention with drug-eluting stents. J. Clin. Lab. Anal. 2019, 34, e23108. [Google Scholar] [CrossRef]
- Chen, C.; Zhou, Y.; Wang, D.W. SARS-CoV-2: A potential novel etiology of fulminant myocarditis. Herz 2020, 45, 230–232. [Google Scholar] [CrossRef] [Green Version]
- Buggey, J.; ElAmm, C.A. Myocarditis and cardiomyopathy. Curr. Opin. Cardiol. 2018, 33, 341–346. [Google Scholar] [CrossRef]
- Shi, S.; Qin, M.; Shen, B.; Cai, Y.; Liu, T.; Yang, F.; Gong, W.; Liu, X.; Liang, J.; Zhao, Q.; et al. Association of Cardiac Injury with Mortality in Hospitalized Patients with COVID-19 in Wuhan, China. JAMA Cardiol. 2020, 2020, e200950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, T.; Fan, Y.; Chen, M.; Wu, X.; Zhang, L.; He, T.; Wang, H.; Wan, J.; Wang, X.; Lu, Z. Cardiovascular Implications of Fatal Outcomes of Patients with Coronavirus Disease 2019 (COVID-19). JAMA Cardiol. 2020, 2020, e201017. [Google Scholar] [CrossRef] [PubMed] [Green Version]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vinciguerra, M.; Romiti, S.; Fattouch, K.; De Bellis, A.; Greco, E. Atherosclerosis as Pathogenetic Substrate for Sars-Cov2 Cytokine Storm. J. Clin. Med. 2020, 9, 2095. https://doi.org/10.3390/jcm9072095
Vinciguerra M, Romiti S, Fattouch K, De Bellis A, Greco E. Atherosclerosis as Pathogenetic Substrate for Sars-Cov2 Cytokine Storm. Journal of Clinical Medicine. 2020; 9(7):2095. https://doi.org/10.3390/jcm9072095
Chicago/Turabian StyleVinciguerra, Mattia, Silvia Romiti, Khalil Fattouch, Antonio De Bellis, and Ernesto Greco. 2020. "Atherosclerosis as Pathogenetic Substrate for Sars-Cov2 Cytokine Storm" Journal of Clinical Medicine 9, no. 7: 2095. https://doi.org/10.3390/jcm9072095
APA StyleVinciguerra, M., Romiti, S., Fattouch, K., De Bellis, A., & Greco, E. (2020). Atherosclerosis as Pathogenetic Substrate for Sars-Cov2 Cytokine Storm. Journal of Clinical Medicine, 9(7), 2095. https://doi.org/10.3390/jcm9072095