ACE2 as a Therapeutic Target for COVID-19; Its Role in Infectious Processes and Regulation by Modulators of the RAAS System


 ,
, 
Abstract
:1. Introduction
2. Literature Search Strategy
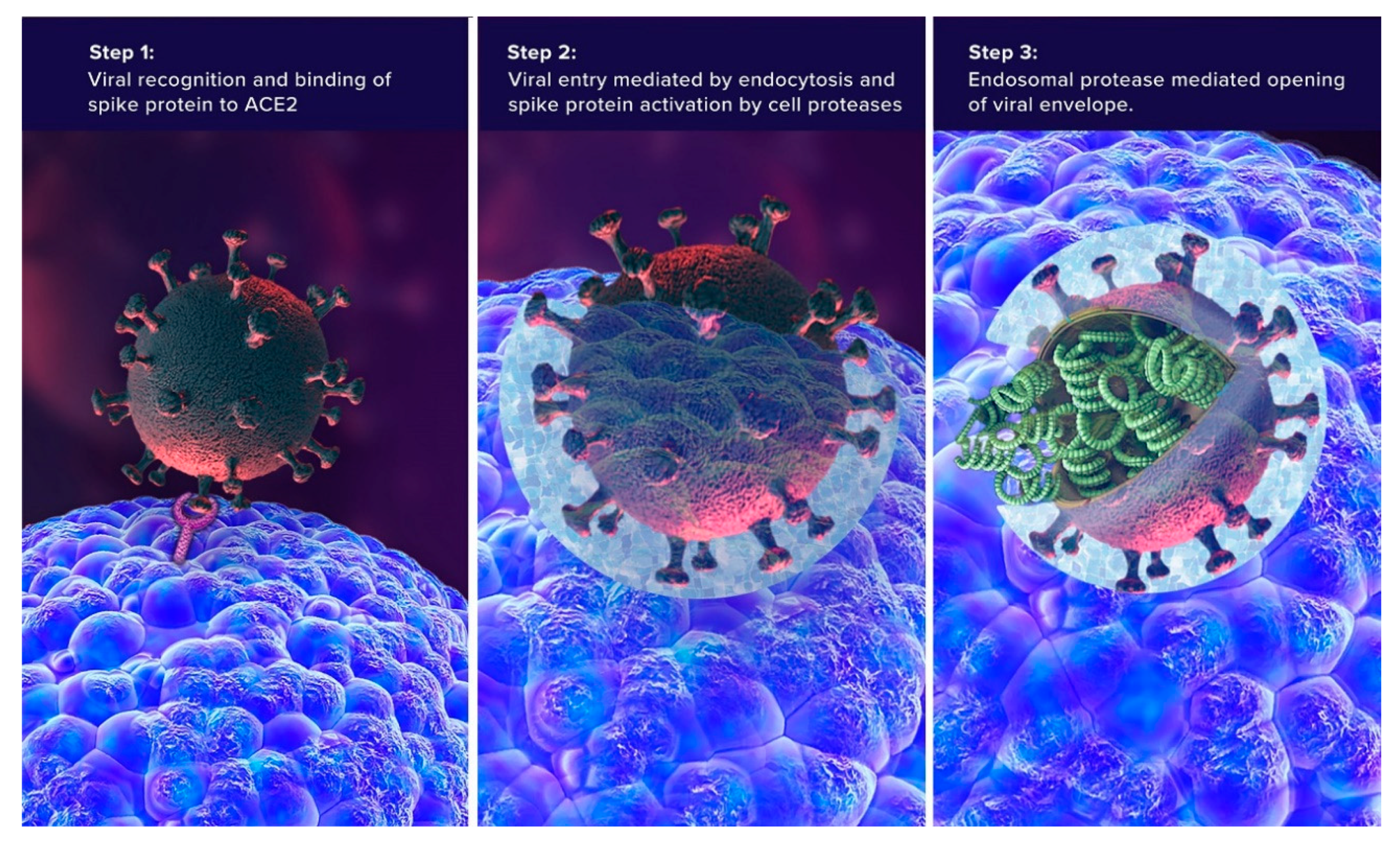
3. SARS-CoV-2
4. The Renin-Angiotensin-Aldosterone System (RAAS)
5. Role of ACE2 in the RAAS
6. ACE2 Regulation
7. Blocking or Enhancing ACE2?
8. ACE Inhibitors
9. Angiotensin Receptor Blockers (ARBs)
10. Mineralocorticoid Receptor Antagonists
11. Renin Antagonist
12. Effects of RAAS Modulators on ACE2 Expression
13. RAAS Modulators and Pneumonia Risk
14. Other Drug Classes and ACE2 Modulation: HMG-CoA Reductase Inhibitors
15. Getting Answers to the Central Question
16. Conclusions

{kind=link}
{kind=link}
| Drug Class | System Studied | Condition | Drugs | Impact on ACE2 |
|---|---|---|---|---|
| ACE-I | In vitro in CHO cells [75] | Healthy | Captopril, Enalapril, Lisinopril | No change in ACE2 activity |
| In vivo in rat renal cells [201] | Lisinopril, low sodium diet, or both | Combination lisinopril and low sodium diet decreased ACE2 mRNA | ||
| In vivo in rat cardiac (LV) cells [202] | Lisinopril | Increase in ACE2 mRNA | ||
| In vivo in rat cardiac cells [73] | MI | Ramipril | No effect on ACE2 mRNA or activity | |
| In vivo in rat cardiac (LV) and plasma cells [203] | Enalapril | Prevention of decrease in ACE2 mRNA and activity 8 weeks post-MI | ||
| In vivo in rat pulmonary tissue and in vitro in rat PMVECs [204] | ALI | Captopril | Increase in ACE2 protein level | |
| In vivo in human intestinal cells [205] | Likely HTN | Any ACE-Is | Increase in intestinal ACE2 mRNA | |
| In vivo in human urine [206] | HTN | Enalapril | No change in ACE2 protein level in urine | |
| In vivo in human heart (LV) tissue [207] | HF | Any ACE-Is | ACE and ACE2 immunoreactivity were quantitatively increased in cardiac tissue from failing hearts (n = 5) due to ischemic heart disease compared to the non-ischemic heart controls (n = 3) | |
| ARB | In vivo in rat cardiac (LV) cells [208] | Healthy | Losartan | Increase in ACE2 mRNA and activity |
| In vivo in rat cardiac (LV)/ renal cells [209] | HTN | Potentiation of renal upregulation of ACE2 mRNA | ||
| In vivo in rat aorta/carotid artery cells [210] | Olmesartan | Increase in ACE2 mRNA and activity | ||
| In vivo in rat cardiac (aorta) cells [211] | Telmisartan | Decrease in ACE2 activity | ||
| In vivo in rat cardiac cells | MI | Losartan, Olmesartan [212] | Increase in ACE2 mRNA | |
| Valsartan [213] | No effect on ACE2 mRNA or activity | |||
| HF | Eprosartan [214] | Increase in ACE2 activity | ||
| In vivo in rat BALF [215] | ARDS | Losartan | Increase in ACE2 activity | |
| In vitro in human UASMCs [211] | Healthy | Telmisartan | Decrease in ACE2 activity | |
| In vivo in human intestinal cells [205] | Likely HTN | Any ARBs | No changes in intestinal ACE2 mRNA | |
| In vivo in human urine [206] | HTN | Candesartan, Losartan Olmesartan, Telmisartan, Valsartan | ACE2 protein level was increased after treatment (>1 year) [Olmesartan only] | |
| ACE-I + ARB | In vivo in rat cardiac (LV) cells [208] | Healthy | Lisinopril + Losartan | Increase in ACE2 activity, but decrease in ACE2 mRNA |
| In vivo in rat cardiac cells [213] | MI | Ramipril + Valsartan | No effect on ACE2 mRNA or protein levels | |
| MRA | In vivo in mice peritoneal macrophages/ cardiac cells [216] | Healthy | Eplerenone | Increase in ACE2 mRNA and activity |
| In vivo in rat cardiac cells [214] | HF | Spironolactone | Trending increase in ACE2 activity (p = 0.067) | |
| In vivo in human monocyte-derived macrophages [216] | Increase in ACE2 mRNA and activity | |||
| In vivo in human plasma [217] | Any MRAs | MRAs were found to independently associate with plasma sACE2 plasma activity | ||
| RI | In vivo in rat renal cells [218] | DN | Aliskiren | Decrease in ACE2 activity |
| None | In vivo in mice pulmonary cells and in vitro in Vero E6 cells [72] | SARS-CoV LRTI | SARS-CoV Spike protein binding to ACE2 in mice lungs in vivo or in cell lines resulted in reduced surface ACE2 protein levels. | |
| In vitro in human A549 alveolar epithelial cells [72] | SARS-CoV Spike protein binding to ACE2 in human AECs resulted in reduced surface ACE2 protein level. | |||
| In vivo in mice cardiac cells [219] | Decrease in ACE2 mRNA and activity | |||
| In vivo in human cardiac cells [219] | Decrease in ACE2 protein level | |||
| In vivo in bronchoalveolar lavage fluid of rats [215] | ARDS | In bronchoalveolar lavage fluid from LPS-exposed rats, ACE activity was augmented (9-fold) while ACE2 activity was reduced (30-fold) vs controls, decreasing the ACE/ACE2 activity ratio | ||
| In vivo in rat cardiac (LV)/ plasma cells [203] | MI | ACE2 mRNA and activity were increased at week 1 post MI compared to controls, however, they were lower than controls at week 8. | ||
| In vivo in mice cardiac cells [220] | HF | Ang-(1–7) | Adverse remodeling in pressure-overloaded ACE2-deficient hearts is facilitated by a combination of pathological effects of Ang II on cardiac cells that can be successfully inhibited by Ang-(1–7). | |
| In vivo in bronchoalveolar lavage fluid of rats [215] | ARDS | In bronchoalveolar lavage fluid from LPS-exposed rats, the exposure to cAng-(1–7) increased the ACE2 activity compared to the placebo group |
| Drug Class | Treatment | Type of Study | COVID-19 Status | Setting | Severity | Phase | Country | Trial Status (Expected N) | Trial ID |
|---|---|---|---|---|---|---|---|---|---|
| ACE-I | Ramipril vs. placebo | Triple-blind, placebo-controlled; efficacy | Confirmed | HOSP or ED | Non-severe | 2 | US | Not yet recruiting (560) | NCT04366050 |
| Captopril vs. SOC | Open label; efficacy | Confirmed | HOSP | ARDS | 2 | France | Not yet recruiting (230) | NCT04355429 | |
| Captopril or enalapril vs. CQ | Open label; efficacy | Confirmed | Unspecified | Unspecified | 3 | Egypt | Not yet recruiting (60) | NCT04345406 | |
| ARB | Valsartan vs. placebo | Quadruple-blind, placebo-controlled; efficacy | Confirmed | HOSP | Mixed | 4 | Netherlands | Recruiting (651) | NCT04335786 |
| Chloroquine/Hydroxychloroquine vs. LPV/r vs. SOC vs. Rivaroxaban vs. TP vs. Candesartan vs. non-RAS AHT vs. Clazakizumab vs. Placebo | Open label; active-controlled; efficacy | Confirmed | Healthy, Outpatient, HOSP | Mixed | 2/3 | Austria | Recruiting (500) | NCT04351724 | |
| Telmisartan vs. Placebo | Triple-blind, placebo-controlled; efficacy and safety | Confirmed | Outpatient | Non-severe | 2 | US | Not yet recruiting (40) | NCT04360551 | |
| SOC vs Telmisartan + SOC | Open label; efficacy | Confirmed | Unspecified | Unspecified | 2 | Argentina | Recruiting (400) | NCT04355936 | |
| Hydroxychloroquine vs. Azithromycin vs. Telmisartan vs. SOC | Open label; efficacy | Confirmed | HOSP | Mixed | 3 | France | Not yet recruiting (1600) | NCT04359953 | |
| Vitamins vs. Hydroxychloroquine vs. Imatinib vs. Favipiravir vs. Telmisartan | Open label, multi-stage, superiority; efficacy and safety | Confirmed | Outpatient | Mild | 3 | France | Recruiting (1057) | NCT04356495 | |
| Losartan | Open label; safety | Confirmed | ICU or HOSP | ARDS | 1 | US | Recruiting (50) | NCT04335123 | |
| Losartan vs. Placebo | Quadruple-blind, placebo-controlled; efficacy | Confirmed | Outpatient | Non-severe | 2 | US | Recruiting (580) | NCT04311177 | |
| Losartan vs. Placebo | Quadruple-blind, placebo- controlled; efficacy | Confirmed or Suspected | HOSP | ARDS | 2 | US | Recruiting (200) | NCT04312009 | |
| SOC vs. Losartan + SOC | Open label; efficacy | Confirmed | HOSP | Mild/ Moderate | 4 | US | Recruiting (200) | NCT04340557 | |
| Losartan vs. Amlodipine | Open label; efficacy and safety | Confirmed | HOSP | Non-severe | 3 | Iran | Recruiting (100) | IRCT201808020 40678N4 | |
| SOC vs. ASA vs. Losartan vs. Simvastatin vs. ASA + losartan vs. ASA + Simvastatin vs. Losartan + Simvastatin vs. ASA + Losartan + Simvastatin | Open label, factorial; efficacy | Confirmed or Suspected | HOSP | Mixed | 3 | Nigeria, Pakistan | Not yet recruiting (10,000) | NCT04343001 | |
| SOC + LPV/r vs. SOC + Hydroxychloroquine vs. SOC + Losartan vs. SOC + Placebo | Quadruple-blind, placebo-controlled; efficacy | Confirmed | HOSP | Mixed | 2/3 | US | Recruiting (4000) | NCT04328012 | |
| Hydroxychloroquine + Azithromycin vs. Hydroxychloroquine + Doxycycline vs. Hydroxychloroquine + Clindamycin vs. Hydroxychloroquine + Clindamycin + Primaquine low dose vs Hydroxychloroquine + Clindamycin+ Primaquine high dose vs. Remdesivir vs. Tocilizumab vs. Methylprednisolone vs. Interferon-α2B vs. Losartan vs. Plasma | Single-blind, factorial; efficacy | Confirmed | HOSP | Unspecified | 2/3 | US | Enrolling by invitation (500) | NCT04349410 | |
| ARBs vs. SOC | Single-blinded; parallel assignment; safety and efficacy | Confirmed | HOSP | Unspecified | 4 | Australia | Recruiting (605) | NCT04394117 | |
| ARBs vs. SOC | Prospective observational; safety, efficacy, ACE2 activity | Confirmed | HOSP | COVID+ ARDS | NA | France | Recruiting (100) | NCT04337190 | |
| Chloroquine + Losartan vs. Chloroquine | Randomized, double blinded; safety and efficacy | Confirmed | HOSP | Unspecified | 2 | Mexico | Recruiting (20) | NCT04428268 | |
| ACE-I or ARB | D/C ACE-I/ARB and switch to CCB or TZD vs continue ACE-I/ARB | Open label; prevention | COVID-19 naive | Healthy | NA | 4 | Ireland | Recruiting (2414) | NCT04330300 |
| D/C vs continue ACE-I/ARB | Single-blind; efficacy | Confirmed or Suspected | HOSP | Mixed | NA | US | Enrolling by invitation (152) | NCT04338009 | |
| D/C vs continue ACE-I/ARB | Single-blind; efficacy | Confirmed | HOSP | Mild | 4 | Austria, Germany | Recruiting (208) | NCT04353596 | |
| D/C vs continue ACE-I/ARB | Single-blind; efficacy | Confirmed | HOSP | Mixed | NA | Denmark | Recruiting (215) | NCT04351581 | |
| D/C vs continue ACE-I/ARB | Open label; efficacy | Confirmed | HOSP | Mild | NA | Brazil | Recruiting (500) | NCT04364893 | |
| D/C vs continue ACE-I/ARB | Open label; efficacy and safety | Confirmed | HOSP | Mild | 3 | France | Recruiting (554) | NCT04329195 | |
| Use vs no use of ACE-I/ARB | Prospective observational; prognosis | Confirmed | HOSP | Unspecified | NA | Saudi Arabia | Recruiting (226) | NCT04357535 | |
| ACE-I/ARB/ direct renin inhibitors (DRi) | Prospective observational; prognosis and efficacy (hypertension) | Confirmed | Private practice | Unspecified | NA | Ukraine | Recruiting (10) | NCT04364984 | |
| ARB/ACE-I vs anti-malarial drugs | Prospective, observational; prognosis | Confirmed | HOSP | Unspecified | NA | France | Not yet recruiting (6,000,000) | NCT04356417 | |
| ARB/ ACE-I/ influenza vaccine vs. SOC | Prospective, observational; efficacy | Confirmed | HOSP | Unspecified | NA | Spain | Recruiting (2547) | NCT04367883 | |
| ACE-I/ARB +SOC vs. SOC | Retrospective, case-control; severity and mortality of/ from COVID | Confirmed | Unspecified | ARDS vs. non-ARDS COVID | NA | Italy | Not yet recruiting (5000) | NCT04318418 | |
| ACE-I/ARB +SOC vs. SOC | Retrospective, case-control; safety | Confirmed | HOSP | Unspecified | NA | France | Not yet recruiting (700) | NCT04374695 | |
| ACE-I/ARB + SOC vs. SOC | Open-label, case control; prognosis | Confirmed | HOSP | Any severity | NA | Italy | Recruiting (2000) | NCT04331574 | |
| MRA | Spironolactone vs. Placebo | Triple-blind, placebo-controlled; efficacy | Confirmed | ICU | ARDS | 4 | Turkey | Not yet recruiting (60) | NCT04345887 |
| Bromohexine + spironolactone vs. standard therapy | Single-blind, parallel assignment; efficacy | Confirmed | HOSP | Mild to moderate | 3 | Russia | Recruiting | NCT04424134 | |
| MAS receptor agonist | Angiotensin-(1–7) vs Placebo | Triple-blind, placebo-controlled; efficacy and safety | Confirmed or Suspected | ICU | ARDS | 2/3 | Belgium | Not yet recruiting (60) | NCT04332666 |
| Plasma derived Angiotensin-(1–7) vs. SOC | Open label; efficacy | Confirmed | HOSP | NA | NA | Turkey | Recruiting | NCT04375124 | |
| Recombinant ACE-2 | Recombinant human ACE-2 (APN01) vs. placebo | Double-blind, placebo-controlled; efficacy | Confirmed | HOSP | Mixed | 2 | Austria, Denmark, Germany | Recruiting (200) | NCT04335136 |
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- European Center for Disease Prevention and Control. Cluster of Pneumonia Cases Caused by a Novel Coronavirus, Wuhan, China. Available online: http://wjw.wuhan.gov.cn/front/web/showDetail/2019123108989 (accessed on 5 June 2020).
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Novel Coronavirus-China. Available online: https://www.who.int/csr/don/12-january-2020-novel-coronavirus-china/en/ (accessed on 5 June 2020).
- World Health Organization. Coronavirus Disease (COVID-19) Pandemic. Available online: https://www.who.int/emergencies/diseases/novel-coronavirus-2019 (accessed on 5 June 2020).
- Johns Hopkins University and Medicine. COVID-19 Dashboard by the Center for Systems Science and Engineering (CSSE) at Johns Hopkins University and Medicine. Available online: https://coronavirus.jhu.edu/map.html (accessed on 24 June 2020).
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Guan, W.-J.; Ni, Z.-Y.; Hu, Y.; Liang, W.-H.; Ou, C.-Q.; He, J.-X.; Liu, L.; Shan, H.; Lei, C.-L.; Hui, D.S.; et al. Clinical characteristics of coronavirus disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef]
- Wu, C.; Chen, X.; Cai, Y.; Xia, J.; Zhou, X.; Xu, S.; Huang, H.; Zhang, L.; Zhou, X.; Du, C.; et al. Risk factors associated with acute respiratory distress syndrome and death in patients with coronavirus disease 2019 Pneumonia in Wuhan, China. JAMA Intern. Med. 2020. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.-J.; Dong, X.; Cao, Y.-Y.; Yuan, Y.-D.; Yang, Y.-B.; Yan, Y.-Q.; Akdis, C.A.; Gao, Y.-D. Clinical characteristics of 140 patients infected with SARS-CoV-2 in Wuhan, China. Allergy 2020. [Google Scholar] [CrossRef]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Fang, L.; Karakiulakis, G.; Roth, M. Are patients with hypertension and diabetes mellitus at increased risk for COVID-19 infection? Lancet Respir. Med. 2020, 8, e21. [Google Scholar] [CrossRef]
- Esler, M.; Esler, D. Can angiotensin receptor-blocking drugs perhaps be harmful in the COVID-19 pandemic? J. Hypertens. 2020, 38, 781–782. [Google Scholar] [CrossRef]
- Watkins, J. Preventing a covid-19 pandemic. BMJ 2020, 368, m810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreutz, R.; AlGharably, E.A.E.-H.; Azizi, M.; Dobrowolski, P.; Guzik, T.; Januszewicz, A.; Persu, A.; Prejbisz, A.; Riemer, T.G.; Wang, J.-G.; et al. Hypertension, the renin–angiotensin system, and the risk of lower respiratory tract infections and lung injury: Implications for COVID-19. Cardiovasc. Res. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization. COVID-19 and the Use of Angiotensin-Converting Enzyme Inhibitors and Receptor Blockers. Available online: https://www.who.int/news-room/commentaries/detail/covid-19-and-the-use-of-angiotensin-converting-enzyme-inhibitors-and-receptor-blockers (accessed on 5 June 2020).
- Khera, R.; Clark, C.; Lu, Y.; Guo, Y.; Ren, S.; Truax, B.; Spatz, E.S.; Murugiah, K.; Lin, Z.; Omer, S.B.; et al. Association of angiotensin-converting enzyme inhibitors and angiotensin receptor blockers with the risk of hospitalization and death in hypertensive patients with coronavirus disease-19. medRxiv 2020. [Google Scholar] [CrossRef]
- Mackey, K.; King, V.J.; Gurley, S.; Kiefer, M.; Liederbauer, E.; Vela, K.; Sonnen, P.; Kansagara, D. Risks and impact of angiotensin-converting enzyme inhibitors or angiotensin-receptor blockers on sars-CoV-2 infection in adults. Ann. Int. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Dolinski, D.; Dolinska, B.; Zmaczynska-Witek, B.; Banach, M.; Kulesza, W. Unrealistic optimism in the time of coronavirus pandemic: May it help to kill, if so—Whom: Disease or the person? J. Clin. Med. 2020, 9, 1464. [Google Scholar] [CrossRef] [PubMed]
- Bingham, J.M.; Arlington, L.; Madhat, F.; Michaud, V.; Turgeon, J. Adherence takes a hit during pandemic. Pharm. Times 2020. In press. [Google Scholar]
- Lu, G.; Liu, D. SARS-like virus in the Middle East: A truly bat-related coronavirus causing human diseases. Protein Cell 2012, 3, 803–805. [Google Scholar] [CrossRef]
- Enjuanes, L.; Almazán, F.; Sola, I.; Zuñiga, S. Biochemical Aspects of Coronavirus Replication and Virus-Host Interaction. Annu. Rev. Microbiol. 2006, 60, 211–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perlman, S.; Netland, J. Coronaviruses post-SARS: Update on replication and pathogenesis. Nat. Rev. Genet. 2009, 7, 439–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, S.; Wong, G.; Shi, W.; Liu, J.; Lai, A.C.; Zhou, J.; Liu, W.; Bi, Y.; Gao, G.F. Epidemiology, genetic recombination, and pathogenesis of coronaviruses. Trends Microbiol. 2016, 24, 490–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization. Human Coronavirus Types. Available online: https://www.cdc.gov/coronavirus/types.html (accessed on 25 June 2020).
- Li, F. Structure, function, and evolution of coronavirus spike proteins. Annu. Rev. Virol. 2016, 3, 237–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wevers, B.A.; Van Der Hoek, L. Recently discovered human coronaviruses. Clin. Lab. Med. 2009, 29, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.H.; Lee, H.-J.; Kim, S.J.; Eun, B.W.; Kim, N.H.; A Lee, J.; Lee, J.H.; Song, E.K.; Kim, S.H.; Park, S.H.K.J.Y.; et al. The association of newly identified respiratory viruses with lower respiratory tract infections in Korean children, 2000–2005. Clin. Infect. Dis. 2006, 43, 585–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Li, C.; Chen, L.; Xu, B.; Zhou, Y.; Cao, L.; Shang, Y.; Fu, Z.; Chen, A.; Deng, L.; et al. A novel human coronavirus OC43 genotype detected in mainland China. Emerg. Microbes Infect. 2018, 7, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Walls, A.C.; Park, Y.-J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 2020, 181, 281–292.e6. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Li, F.; Shi, Z. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Genet. 2018, 17, 181–192. [Google Scholar] [CrossRef] [Green Version]
- Fehr, A.R.; Perlman, S. Coronaviruses: An overview of their replication and pathogenesis. Recent Results Cancer Res. 2015, 1282, 1–23. [Google Scholar] [CrossRef] [Green Version]
- Shereen, M.A.; Khan, S.; Kazmi, A.; Bashir, N.; Siddique, R. COVID-19 infection: Origin, transmission, and characteristics of human coronaviruses. J. Adv. Res. 2020, 24, 91–98. [Google Scholar] [CrossRef]
- Lu, G.; Wang, Q.; Gao, G.F. Bat-to-human: Spike features determining ‘host jump’ of coronaviruses SARS-CoV, MERS-CoV, and beyond. Trends Microbiol. 2015, 23, 468–478. [Google Scholar] [CrossRef] [Green Version]
- Belouzard, S.; Millet, J.K.; Licitra, B.N.; Whittaker, G.R. Mechanisms of coronavirus cell entry mediated by the viral spike protein. Viruses 2012, 4, 1011–1033. [Google Scholar] [CrossRef] [Green Version]
- Heald-Sargent, T.; Gallagher, T. Ready, set, fuse! The coronavirus spike protein and acquisition of fusion competence. Viruses 2012, 4, 557–580. [Google Scholar] [CrossRef] [Green Version]
- Millet, J.K.; Whittaker, G.R. Host cell proteases: Critical determinants of coronavirus tropism and pathogenesis. Virus Res. 2014, 202, 120–134. [Google Scholar] [CrossRef] [PubMed]
- Glowacka, I.; Bertram, S.; Müller, M.A.; Allen, P.; Soilleux, E.; Pfefferle, S.; Steffen, I.; Tsegaye, T.S.; He, Y.; Gnirss, K.; et al. Evidence that TMPRSS2 activates the severe acute respiratory syndrome coronavirus spike protein for membrane fusion and reduces viral control by the humoral immune response. J. Virol. 2011, 85, 4122–4134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertram, S.; Glowacka, I.; Müller, M.A.; Lavender, H.; Gnirss, K.; Nehlmeier, I.; Niemeyer, D.; He, Y.; Simmons, G.; Drosten, C.; et al. Cleavage and activation of the severe acute respiratory syndrome coronavirus spike protein by human airway trypsin-like protease. J. Virol. 2011, 85, 13363–13372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shulla, A.; Heald-Sargent, T.; Subramanya, G.; Zhao, J.; Perlman, S.; Gallagher, T. A Transmembrane serine protease is linked to the severe acute respiratory syndrome coronavirus receptor and activates virus entry. J. Virol. 2010, 85, 873–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kam, Y.-W.; Okumura, Y.; Kido, H.; Ng, L.F.P.; Bruzzone, R.; Altmeyer, R. Cleavage of the SARS coronavirus spike glycoprotein by airway proteases enhances virus entry into human bronchial epithelial cells in vitro. PLoS ONE 2009, 4, e7870. [Google Scholar] [CrossRef] [Green Version]
- Matsuyama, S.; Ujike, M.; Morikawa, S.; Tashiro, M.; Taguchi, F. Protease-mediated enhancement of severe acute respiratory syndrome coronavirus infection. Proc. Natl. Acad. Sci. USA 2005, 102, 12543–12547. [Google Scholar] [CrossRef] [Green Version]
- Li, F. Receptor Recognition Mechanisms of Coronaviruses: A Decade of Structural Studies. J. Virol. 2014, 89, 1954–1964. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Moore, M.J.; Vasilieva, N.; Sui, J.; Wong, S.K.; Berne, M.A.; Somasundaran, M.; Sullivan, J.L.; Luzuriaga, K.; Greenough, T.C.; et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003, 426, 450–454. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Yang, N.; Shen, H.-M. Targeting the Endocytic Pathway and Autophagy Process as a Novel Therapeutic Strategy in COVID-19. Int. J. Boil. Sci. 2020, 16, 1724–1731. [Google Scholar] [CrossRef]
- Wan, Y.; Shang, J.; Graham, R.; Baric, R.S.; Li, F. Receptor recognition by the novel coronavirus from Wuhan: An analysis based on decade-long structural studies of sars coronavirus. J. Virol. 2020, 94, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Zhang, C.; Sui, J.; Kuhn, J.H.; Moore, M.J.; Luo, S.; Wong, S.-K.; Huang, I.-C.; Xu, K.; Vasilieva, N.; et al. Receptor and viral determinants of SARS-coronavirus adaptation to human ACE2. EMBO J. 2005, 24, 1634–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Åkerström, S.; Mirazimi, A.; Tan, Y.-J. Inhibition of SARS-CoV replication cycle by small interference RNAs silencing specific SARS proteins, 7a/7b, 3a/3b and S. Antivir. Res. 2007, 73, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Simmons, G.; Gosalia, D.N.; Rennekamp, A.J.; Reeves, J.D.; Diamond, S.L.; Bates, P. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc. Natl. Acad. Sci. USA 2005, 102, 11876–11881. [Google Scholar] [CrossRef] [Green Version]
- Weiss, S.R.; Navas, S. Coronavirus pathogenesis and the emerging pathogen severe acute respiratory syndrome coronavirus. Microbiol. Mol. Boil. Rev. 2005, 69, 635–664. [Google Scholar] [CrossRef] [Green Version]
- Inoue, Y.; Tanaka, N.; Tanaka, Y.; Inoue, S.; Morita, K.; Zhuang, M.; Hattori, T.; Sugamura, K. Clathrin-Dependent entry of severe acute respiratory syndrome coronavirus into target cells expressing ACE2 with the cytoplasmic tail deleted. J. Virol. 2007, 81, 8722–8729. [Google Scholar] [CrossRef] [Green Version]
- Bonow, R.O.; Fonarow, G.C.; O’Gara, P.T.; Yancy, C.W. Association of Coronavirus Disease 2019 (COVID-19) with myocardial injury and mortality. JAMA Cardiol. 2020. [Google Scholar] [CrossRef] [Green Version]
- Tan, W.; Aboulhosn, J. The cardiovascular burden of coronavirus disease 2019 (COVID-19) with a focus on congenital heart disease. Int. J. Cardiol. 2020, 309, 70–77. [Google Scholar] [CrossRef]
- Bunyavanich, S.; Do, A.; Vicencio, A. Nasal gene expression of angiotensin-converting Enzyme 2 in children and adults. JAMA 2020, 323, 2427. [Google Scholar] [CrossRef]
- Wu, A.; Peng, Y.; Huang, B.; Ding, X.; Wang, X.; Niu, P.; Meng, J.; Zhu, Z.; Zhang, Z.; Wang, J.; et al. Genome composition and divergence of the novel coronavirus (2019-nCoV) originating in China. Cell Host Microbe 2020, 27, 325–328. [Google Scholar] [CrossRef] [Green Version]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef] [Green Version]
- Gheblawi, M.; Wang, K.; Viveiros, A.; Nguyen, Q.; Zhong, J.-C.; Turner, A.J.; Raizada, M.K.; Grant, M.B.; Oudit, G.Y. Angiotensin-Converting Enzyme 2: SARS-CoV-2 Receptor and regulator of the renin-angiotensin system. Circ. Res. 2020, 126, 1456–1474. [Google Scholar] [CrossRef] [PubMed]
- Skeggs, L.T.; Dorer, F.E.; Levine, M.; Lentz, K.E.; Kahn, J.R. The biochemistry of the renin-angiotensin system. Adv. Exp. Med. Biol. 1980, 130, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Johnston, C. Angiotensin converting enzyme inhibition. In The Renin-Angiotensin System; Robertson, J., Nichols, M.G., Eds.; Gower Medical Pub: London, UK, 1993; Volume 1. [Google Scholar]
- Funder, J.W. Aldosterone and mineralocorticoid receptors—Physiology and pathophysiology. Int. J. Mol. Sci. 2017, 18, 1032. [Google Scholar] [CrossRef] [Green Version]
- Campbell, D.J. Circulating and tissue angiotensin systems. J. Clin. Investig. 1987, 79, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Campbell, D.J. Tissue renin-angiotensin system. J. Cardiovasc. Pharmacol. 1987, 10 (Suppl. 7), S1–S8. [Google Scholar] [CrossRef]
- Phillips, M.I.; Speakman, E.A.; Kimura, B. Levels of angiotensin and molecular biology of the tissue renin angiotensin systems. Regul. Pept. 1993, 43, 1–20. [Google Scholar] [CrossRef]
- Romero, C.A.; Orias, M.; Weir, M.R. Novel RAAS agonists and antagonists: Clinical applications and controversies. Nat. Rev. Endocrinol. 2015, 11, 242–252. [Google Scholar] [CrossRef]
- Patel, S.; Rauf, A.; Khan, H.; Abu-Izneid, T. Renin-angiotensin-aldosterone (RAAS): The ubiquitous system for homeostasis and pathologies. Biomed. Pharmacother. 2017, 94, 317–325. [Google Scholar] [CrossRef]
- Santos, R.A.S.; Sampaio, W.O.; Alzamora, A.C.; Motta-Santos, D.; Alenina, N.; Bader, M.; Campagnole-Santos, M.J. The ACE2/Angiotensin-(1–7)/MAS Axis of the Renin-Angiotensin System: Focus on Angiotensin-(1–7). Physiol. Rev. 2018, 98, 505–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofmann, H.; Pyrc, K.; Van Der Hoek, L.; Geier, M.; Berkhout, B.; Pöhlmann, S. Human coronavirus NL63 employs the severe acute respiratory syndrome coronavirus receptor for cellular entry. Proc. Natl. Acad. Sci. 2005, 102, 7988–7993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Yang, Y.; Zhang, C.; Huang, F.; Wang, F.; Yuan, J.; Wang, Z.; Li, J.; Li, J.; Feng, C.; et al. Clinical and biochemical indexes from 2019-nCoV infected patients linked to viral loads and lung injury. Sci. China Life Sci. 2020, 63, 364–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imai, Y.; Kuba, K.; Rao, S.; Huan, Y.; Guo, F.; Guan, B.; Yang, P.; Sarao, R.; Wada, T.; Leong-Poi, H.; et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature 2005, 436, 112–116. [Google Scholar] [CrossRef]
- Kuba, K.; Imai, Y.; Rao, S.; Gao, H.; Guo, F.; Guan, B.; Huan, Y.; Yang, P.; Zhang, Y.; Deng, W.; et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus–induced lung injury. Nat. Med. 2005, 11, 875–879. [Google Scholar] [CrossRef]
- Burrell, L.M.; Johnston, C.I.; Tikellis, C.; Cooper, M.E. ACE2, a new regulator of the renin–angiotensin system. Trends Endocrinol. Metab. 2004, 15, 166–169. [Google Scholar] [CrossRef]
- Donoghue, M.; Hsieh, F.; Baronas, E.; Godbout, K.; Gosselin, M.; Stagliano, N.; Donovan, M.; Woolf, B.; Robison, K.; Jeyaseelan, R.; et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ. Res. 2000, 87, e1–e9. [Google Scholar] [CrossRef]
- Tipnis, S.R.; Hooper, N.M.; Hyde, R.; Karran, E.; Christie, G.; Turner, A.J. A Human Homolog of Angiotensin-converting Enzyme. J. Boil. Chem. 2000, 275, 33238–33243. [Google Scholar] [CrossRef] [Green Version]
- Fraga-Silva, R.A.; Costa-Fraga, F.P.; Murça, T.M.; Moraes, P.L.; Lima, A.M.; Lautner, R.Q.; Castro, C.H.; Soares, C.M.A.; Borges, C.L.; Nadu, A.P.; et al. Angiotensin-converting enzyme 2 activation improves endothelial function. Hypertension 2013, 61, 1233–1238. [Google Scholar] [CrossRef]
- Prestes, T.R.R.; Rocha, N.P.; Miranda, A.S.; Teixeira, A.L.; Simões-E-Silva, A.C.; Prestes, N.P.R.T.R.R. The Anti-Inflammatory Potential of ACE2/Angiotensin-(1-7)/Mas Receptor Axis: Evidence from Basic and Clinical Research. Curr. Drug Targets 2017, 18, 1301–1313. [Google Scholar] [CrossRef]
- The Human Protein Atlas. Angiotensin I Converting Enzyme. Available online: https://www.proteinatlas.org/ENSG00000159640-ACE (accessed on 24 April 2020).
- The Human Protein Atlas. Tissue Expression of ACE2. Available online: https://www.proteinatlas.org/ENSG00000130234-ACE2/tissue (accessed on 24 April 2020).
- Chai, X.; Hu, L.; Zhang, Y.; Han, W.; Lu, Z.; Ke, A.; Zhou, J.; Shi, G.; Fang, N.; Fan, J.; et al. Specific ACE2 Expression in Cholangiocytes May Cause Liver Damage After 2019-nCoV Infection. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Crackower, M.A.; Sarao, R.; Oudit, G.Y.; Yagil, C.; Kozieradzki, I.; Scanga, S.E.; Oliveira-Dos-Santos, A.J.; Da Costa, J.; Zhang, L.; Pei, Y.; et al. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature 2002, 417, 822–828. [Google Scholar] [CrossRef] [PubMed]
- Hamming, I.; Timens, W.; Bulthuis, M.; Lely, A.T.; Navis, G.; Van Goor, H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 2004, 203, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Tikellis, C.; Johnston, C.I.; Forbes, J.M.; Burns, W.C.; Burrell, L.M.; Risvanis, J.; Cooper, M.E. Characterization of Renal Angiotensin-Converting Enzyme 2 in Diabetic Nephropathy. Hypertension 2003, 41, 392–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, X.; Chen, K.; Zou, J.; Han, P.; Hao, J.; Han, Z.-G. Single-cell RNA-seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019-nCoV infection. Front. Med. 2020, 14, 185–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Kang, Z.; Gong, H.; Xu, D.; Wang, J.; Li, Z.; Cui, X.; Xiao, J.; Meng, T.; Zhou, W.; et al. The digestive system is a potential route of 2019-nCov infection: A bioinformatics analysis based on single-cell transcriptomes. bioRxiv 2020. [Google Scholar] [CrossRef]
- Jia, H.P.; Look, D.C.; Shi, L.; Hickey, M.; Pewe, L.; Netland, J.; Farzan, M.; Wohlford-Lenane, C.; Perlman, S.; McCray, P.B. ACE2 Receptor expression and severe acute respiratory syndrome coronavirus infection depend on differentiation of human airway epithelia. J. Virol. 2005, 79, 14614–14621. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Zhao, Z.; Wang, Y.; Zhou, Y.; Ma, Y.; Zuo, W. Single-cell RNA expression profiling of ACE2, the putative receptor of Wuhan 2019-nCov. bioRxiv 2001. [Google Scholar] [CrossRef]
- Xu, H.; Zhong, L.; Deng, J.; Peng, J.; Dan, H.; Zeng, X.; Li, T.; Chen, Q. High expression of ACE2 receptor of 2019-nCoV on the epithelial cells of oral mucosa. Int. J. Oral Sci. 2020, 12, 8. [Google Scholar] [CrossRef]
- Brann, D.; Tsukahara, T.; Weinreb, C.; Logan, D.W.; Datta, S.R. Non-neural expression of SARS-CoV-2 entry genes in the olfactory epithelium suggests mechanisms underlying anosmia in COVID-19 patients. bioRxiv 2020. [Google Scholar] [CrossRef]
- Guan, W.-j.; Ni, Z.-y.; Hu, Y.; Liang, W.-h.; Ou, C.-q.; He, J.-x.; Liu, L.; Shan, H.; Lei, C.-l.; Hui, D.S.; et al. Clinical characteristics of 2019 novel coronavirus infection in China. medRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, T.; Perlot, T.; Rehman, A.; Trichereau, J.; Ishiguro, H.; Paolino, M.; Sigl, V.; Hanada, T.; Hanada, R.; Lipinski, S.; et al. ACE2 links amino acid malnutrition to microbial ecology and intestinal inflammation. Nature 2012, 487, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Leung, W.K.; To, K.-F.; Chan, P.K.; Chan, H.L.; Wu, A.K.; Lee, N.; Yuen, K.Y.; Sung, J.J. Enteric involvement of severe acute respiratory syndrome-associated coronavirus infection. Gastroenterol. 2003, 125, 1011–1017. [Google Scholar] [CrossRef] [Green Version]
- Haga, S.; Nagata, N.; Okamura, T.; Yamamoto, N.; Sata, T.; Yamamoto, N.; Sasazuki, T.; Ishizaka, Y. TACE antagonists blocking ACE2 shedding caused by the spike protein of SARS-CoV are candidate antiviral compounds. Antivir. Res. 2010, 85, 551–555. [Google Scholar] [CrossRef]
- Heurich, A.; Hofmann-Winkler, H.; Gierer, S.; Liepold, T.; Jahn, O.; Pöhlmann, S. TMPRSS2 and ADAM17 Cleave ACE2 differentially and only proteolysis by TMPRSS2 augments entry driven by the severe acute respiratory syndrome coronavirus spike protein. J. Virol. 2014, 88, 1293–1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zunke, F.; Rose-John, S. The shedding protease ADAM17: Physiology and pathophysiology. Biochim. et Biophys. Acta (BBA)-Bioenerg. 2017, 1864, 2059–2070. [Google Scholar] [CrossRef]
- E Sama, I.; Ravera, A.; Santema, B.T.; Van Goor, H.; Ter Maaten, J.M.; Cleland, J.G.F.; Rienstra, M.; Friedrich, A.W.; Samani, N.J.; Ng, L.L.; et al. Circulating plasma concentrations of angiotensin-converting enzyme 2 in men and women with heart failure and effects of renin-angiotensin-aldosterone inhibitors. Eur. Hear. J. 2020, 41, 1810–1817. [Google Scholar] [CrossRef]
- Wang, K.; Gheblawi, M.; Oudit, G.Y. Angiotensin Converting Enzyme 2: A Double-Edged Sword. Circulation 2020. [Google Scholar] [CrossRef]
- Clarke, N.E.; Belyaev, N.D.; Lambert, D.W.; Turner, A.J. Epigenetic regulation of angiotensin-converting enzyme 2 (ACE2) by SIRT1 under conditions of cell energy stress. Clin. Sci. (Lond). 2014, 126, 507–516. [Google Scholar] [CrossRef]
- Gallagher, P.E.; Ferrario, C.M.; Tallant, E.A. Regulation of ACE2 in cardiac myocytes and fibroblasts. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H2373–H2379. [Google Scholar] [CrossRef]
- Clarke, N.E.; Turner, A.J. Angiotensin-converting enzyme 2: The first decade. Int. J. Hypertens. 2012, 2012, 307315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rice, G.I.; Jones, A.L.; Grant, P.J.; Carter, A.M.; Turner, A.J.; Hooper, N.M. Circulating activities of angiotensin-converting enzyme, its homolog, angiotensin-converting enzyme 2, and neprilysin in a family study. Hypertension 2006, 48, 914–920. [Google Scholar] [CrossRef] [PubMed]
- Varagic, J.; Ahmad, S.; Nagata, S.; Ferrario, C.M. ACE2: Angiotensin II/angiotensin-(1-7) balance in cardiac and renal injury. Curr. Hypertens. Rep. 2014, 16, 420. [Google Scholar] [CrossRef]
- Cooper, M.E.; Johnston, C.I. Optimizing treatment of hypertension in patients with diabetes. JAMA 2000, 283, 3177–3179. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.B.; Zhong, J.C.; Grant, M.B.; Oudit, G.Y. Role of the ACE2/Angiotensin 1-7 Axis of the Renin-Angiotensin System in Heart Failure. Circ. Res. 2016, 118, 1313–1326. [Google Scholar] [CrossRef] [Green Version]
- Alghamdi, I.G.; Hussain, I.I.; Almalki, S.S.; Alghamdi, M.S.; Alghamdi, M.M.; El-Sheemy, M.A. The pattern of Middle East respiratory syndrome coronavirus in Saudi Arabia: A descriptive epidemiological analysis of data from the Saudi Ministry of Health. Int. J. Gen. Med. 2014, 7, 417–423. [Google Scholar] [CrossRef] [Green Version]
- Karlberg, J.; Chong, D.S.; Lai, W.Y. Do men have a higher case fatality rate of severe acute respiratory syndrome than women do? Am. J. Epidemiol. 2004, 159, 229–231. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.Y.; Zhang, P.; Zhou, X.M.; Liu, D.; Zhong, J.C.; Zhang, C.J.; Jin, L.J.; Yu, H.M. Relationship between genetic variants of ACE2 gene and circulating levels of ACE2 and its metabolites. J. Clin. Pharm. Ther. 2018, 43, 189–195. [Google Scholar] [CrossRef]
- Fan, X.; Wang, Y.; Sun, K.; Zhang, W.; Yang, X.; Wang, S.; Zhen, Y.; Wang, J.; Li, W.; Han, Y.; et al. Polymorphisms of ACE2 gene are associated with essential hypertension and antihypertensive effects of Captopril in women. Clin. Pharmacol. Ther. 2007, 82, 187–196. [Google Scholar] [CrossRef]
- Hussain, M.; Jabeen, N.; Raza, F.; Shabbir, S.; Baig, A.A.; Amanullah, A.; Aziz, B. Structural variations in human ACE2 may influence its binding with SARS-CoV-2 spike protein. J. Med. Virol. 2020. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Li, L.; Feng, Z.; Wan, S.; Huang, P.; Sun, X.; Wen, F.; Huang, X.; Ning, G.; Wang, W. Comparative genetic analysis of the novel coronavirus (2019-nCoV/SARS-CoV-2) receptor ACE2 in different populations. Cell Discov. 2020, 6, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shenoy, V.; Grobe, J.L.; Qi, Y.; Ferreira, A.J.; Fraga-Silva, R.A.; Collamat, G.; Bruce, E.; Katovich, M.J. 17beta-Estradiol modulates local cardiac renin-angiotensin system to prevent cardiac remodeling in the DOCA-salt model of hypertension in rats. Peptides 2009, 30, 2309–2315. [Google Scholar] [CrossRef] [PubMed]
- Bukowska, A.; Spiller, L.; Wolke, C.; Lendeckel, U.; Weinert, S.; Hoffmann, J.; Bornfleth, P.; Kutschka, I.; Gardemann, A.; Isermann, B.; et al. Protective regulation of the ACE2/ACE gene expression by estrogen in human atrial tissue from elderly men. Exp. Biol. Med. (Maywood) 2017, 242, 1412–1423. [Google Scholar] [CrossRef] [PubMed]
- Channappanavar, R.; Fett, C.; Mack, M.; Ten Eyck, P.P.; Meyerholz, D.K.; Perlman, S. Sex-based differences in susceptibility to severe acute respiratory syndrome coronavirus infection. J. Immunol. 2017, 198, 4046–4053. [Google Scholar] [CrossRef] [PubMed]
- Verdecchia, P.; Cavallini, C.; Spanevello, A.; Angeli, F. The pivotal link between ACE2 deficiency and SARS-CoV-2 infection. Eur. J. Intern. Med. 2020. [Google Scholar] [CrossRef]
- Zou, Z.; Yan, Y.; Shu, Y.; Gao, R.; Sun, Y.; Li, X.; Ju, X.; Liang, Z.; Liu, Q.; Zhao, Y.; et al. Angiotensin-converting enzyme 2 protects from lethal avian influenza A H5N1 infections. Nat. Commun. 2014, 5, 3594. [Google Scholar] [CrossRef]
- Bodor, C.; Nagy, J.; Végh, B.; Nemeth, A.; Jenei, A.; Mirzahosseini, S.; Sebe, A.; Rosivall, L. Angiotensin II increases the permeability and PV-1 expression of endothelial cells. Am. J. Physiol. Cell Physiol. 2012, 302, C267–C276. [Google Scholar] [CrossRef] [Green Version]
- Elferink, J.G.; de Koster, B.M. The stimulation of human neutrophil migration by angiotensin IL: Its dependence on Ca2+ and the involvement of cyclic GMP. Br. J. Pharmacol. 1997, 121, 643–648. [Google Scholar] [CrossRef] [Green Version]
- Guzik, T.J.; Hoch, N.E.; Brown, K.A.; McCann, L.A.; Rahman, A.; Dikalov, S.; Goronzy, J.; Weyand, C.; Harrison, D.G. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J. Exp. Med. 2007, 204, 2449–2460. [Google Scholar] [CrossRef]
- Jurewicz, M.; McDermott, D.H.; Sechler, J.M.; Tinckam, K.; Takakura, A.; Carpenter, C.B.; Milford, E.; Abdi, R. Human T and natural killer cells possess a functional renin-angiotensin system: Further mechanisms of angiotensin II-induced inflammation. J. Am. Soc. Nephrol. 2007, 18, 1093–1102. [Google Scholar] [CrossRef]
- Lijnen, P.; Fagard, R.; Petrov, V. Cytosolic calcium changes induced by angiotensin II in human peripheral blood mononuclear cells are mediated via angiotensin II subtype 1 receptors. J. Hypertens. 1997, 15, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Nahmod, K.A.; Vermeulen, M.E.; Raiden, S.; Salamone, G.; Gamberale, R.; Fernández-Calotti, P.; Alvarez, A.; Nahmod, V.; Giordano, M.; Geffner, J.R. Control of dendritic cell differentiation by angiotensin II. FASEB J. 2003, 17, 491–493. [Google Scholar] [CrossRef] [PubMed]
- Nataraj, C.; Oliverio, M.I.; Mannon, R.B.; Mannon, P.J.; Audoly, L.P.; Amuchastegui, C.S.; Ruiz, P.; Smithies, O.; Coffman, T.M. Angiotensin II regulates cellular immune responses through a calcineurin-dependent pathway. J. Clin. Investig. 1999, 104, 1693–1701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, M.I.; Kagiyama, S. Angiotensin II as a pro-inflammatory mediator. Curr. Opin. Investig. Drugs 2002, 3, 569–577. [Google Scholar] [PubMed]
- Platten, M.; Youssef, S.; Hur, E.M.; Ho, P.P.; Han, M.H.; Lanz, T.V.; Phillips, L.K.; Goldstein, M.J.; Bhat, R.; Raine, C.S.; et al. Blocking angiotensin-converting enzyme induces potent regulatory T cells and modulates TH1- and TH17-mediated autoimmunity. Proc. Natl. Acad. Sci. USA 2009, 106, 14948–14953. [Google Scholar] [CrossRef] [Green Version]
- Sadoshima, J. Cytokine actions of angiotensin II. Circ. Res. 2000, 86, 1187–1189. [Google Scholar] [CrossRef] [Green Version]
- Swirski, F.K.; Nahrendorf, M.; Etzrodt, M.; Wildgruber, M.; Cortez-Retamozo, V.; Panizzi, P.; Figueiredo, J.L.; Kohler, R.H.; Chudnovskiy, A.; Waterman, P.; et al. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science 2009, 325, 612–616. [Google Scholar] [CrossRef] [Green Version]
- Lau, Y.L.; Peiris, J.S. Pathogenesis of severe acute respiratory syndrome. Curr. Opin. Immunol. 2005, 17, 404–410. [Google Scholar] [CrossRef]
- Perlman, S.; Dandekar, A.A. Immunopathogenesis of coronavirus infections: Implications for SARS. Nat. Rev. Immunol. 2005, 5, 917–927. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Penninger, J.M.; Li, Y.; Zhong, N.; Slutsky, A.S. Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: Molecular mechanisms and potential therapeutic target. Intensiv. Care Med. 2020, 46, 586–590. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.-Y.; Ma, Y.-T.; Zhang, J.-Y.; Xie, X. COVID-19 and the cardiovascular system. Nat. Rev. Cardiol. 2020, 17, 259–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byrnes, J.J.; Gross, S.; Ellard, C.; Connolly, K.; Donahue, S.; Picarella, D. Effects of the ACE2 inhibitor GL1001 on acute dextran sodium sulfate-induced colitis in mice. Inflamm. Res. 2009, 58, 819–827. [Google Scholar] [CrossRef] [PubMed]
- Towler, P.; Staker, B.; Prasad, S.G.; Menon, S.; Tang, J.; Parsons, T.; Ryan, D.; Fisher, M.; Williams, D.; Dales, N.A.; et al. ACE2 X-ray structures reveal a large hinge-bending motion important for inhibitor binding and catalysis. J. Biol. Chem. 2004, 279, 17996–18007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, N.E.; Hooper, N.M.; Turner, A.J. Angiotensin-converting enzyme-2. Handb. Proteolytic Enzym. 2013, 499. [Google Scholar]
- Hernández Prada, J.A.; Ferreira, A.J.; Katovich, M.J.; Shenoy, V.; Qi, Y.; Santos, R.A.; Castellano, R.K.; Lampkins, A.J.; Gubala, V.; Ostrov, D.A.; et al. Structure-based identification of small-molecule angiotensin-converting enzyme 2 activators as novel antihypertensive agents. Hypertension 2008, 51, 1312–1317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, Y.; Haga, S.; Ishizaka, Y.; Mimori, A. Autoantibodies to angiotensin-converting enzyme 2 in patients with connective tissue diseases. Arthritis. Res. Ther. 2010, 12, R85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bryan, J. From snake venom to ACE inhibitor-The discovery and rise of captopril. Pharm. J. 2009, 282, 455. [Google Scholar]
- Heran, B.S.; Wong, M.M.; Heran, I.K.; Wright, J.M. Blood pressure lowering efficacy of angiotensin converting enzyme (ACE) inhibitors for primary hypertension. Cochrane Database Syst. Rev. 2008, 2008, Cd003823. [Google Scholar] [CrossRef]
- Campbell, D.J.; Lawrence, A.C.; Towrie, A.; Kladis, A.; Valentijn, A.J. Differential regulation of angiotensin peptide levels in plasma and kidney of the rat. Hypertension 1991, 18, 763–773. [Google Scholar] [CrossRef] [Green Version]
- Ohkubo, H.; Nakayama, K.; Tanaka, T.; Nakanishi, S. Tissue distribution of rat angiotensinogen mRNA and structural analysis of its heterogeneity. J. Biol. Chem. 1986, 261, 319–323. [Google Scholar] [PubMed]
- Bader, M.; Peters, J.; Baltatu, O.; Müller, D.N.; Luft, F.C.; Ganten, D. Tissue renin-angiotensin systems: New insights from experimental animal models in hypertension research. J. Mol. Med. (Berl. Germany) 2001, 79, 76–102. [Google Scholar] [CrossRef] [PubMed]
- Engeli, S.; Negrel, R.; Sharma, A.M. Physiology and pathophysiology of the adipose tissue renin-angiotensin system. Hypertension 2000, 35, 1270–1277. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, S.; Sigmund, C.D. Angiotensin mutant mice: A focus on the brain renin-angiotensin system. Neuropeptides 2002, 36, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Sernia, C. A critical appraisal of the intrinsic pancreatic angiotensin-generating system. JOP 2001, 2, 50–55. [Google Scholar] [PubMed]
- Dzau, V.J.; Bernstein, K.; Celermajer, D.; Cohen, J.; Dahlöf, B.; Deanfield, J.; Diez, J.; Drexler, H.; Ferrari, R.; van Gilst, W.; et al. The relevance of tissue angiotensin-converting enzyme: Manifestations in mechanistic and endpoint data. Am. J. Cardiol. 2001, 88, 1l–20l. [Google Scholar] [CrossRef] [Green Version]
- Ranadive, S.A.; Chen, A.X.; Serajuddin, A.T. Relative lipophilicities and structural-pharmacological considerations of various angiotensin-converting enzyme (ACE) inhibitors. Pharm. Res. 1992, 9, 1480–1486. [Google Scholar] [CrossRef]
- Singh, K.D.; Karnik, S.S. Angiotensin Receptors: Structure, function, signaling and clinical applications. J. Cell Signal. 2016, 1, 111. [Google Scholar] [CrossRef]
- Guimond, M.-O.; Hallberg, M.; Gallo-Payet, N.; Wallinder, C. Saralasin and Sarile Are AT2 receptor agonists. ACS Med. Chem. Lett. 2014, 5, 1129–1132. [Google Scholar] [CrossRef] [Green Version]
- Moore, A.F.; Fulton, R.W. Angiotensin II antagonists—saralasin. Drug Dev. Res. 1984, 4, 331–349. [Google Scholar] [CrossRef]
- Yang, R.; Luo, Z.; Liu, Y.; Sun, M.; Zheng, L.; Chen, Y.; Li, Y.; Wang, H.; Chen, L.; Wu, M.; et al. Drug interactions with angiotensin receptor blockers: role of human cytochromes P450. Curr. Drug Metab. 2016, 17, 681–691. [Google Scholar] [CrossRef]
- Siragy, H.M. Angiotensin receptor blockers: How important is selectivity? Am. J. Hypertens. 2002, 15, 1006–1014. [Google Scholar] [CrossRef] [Green Version]
- Suwannakul, S.; Ieiri, I.; Kimura, M.; Kawabata, K.; Kusuhara, H.; Hirota, T.; Irie, S.; Sugiyama, Y.; Higuchi, S. Pharmacokinetic interaction between pravastatin and olmesartan in relation to SLCO1B1 polymorphism. J. Hum. Genet. 2008, 53, 899–904. [Google Scholar] [CrossRef] [Green Version]
- De Kloet, E.R.; Vreugdenhil, E.; Oitzl, M.S.; Joëls, M. Brain corticosteroid receptor balance in health and disease. Endocr. Rev. 1998, 19, 269–301. [Google Scholar] [CrossRef]
- Funder, J.W. Mineralocorticoid receptors: Distribution and activation. Heart Fail. Rev. 2005, 10, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Sanchez, C.E.; Warden, M.; Gomez-Sanchez, M.T.; Hou, X.; Gomez-Sanchez, E.P. Diverse immunostaining patterns of mineralocorticoid receptor monoclonal antibodies. Steroids 2011, 76, 1541–1545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sica, D.A. Pharmacokinetics and pharmacodynamics of mineralocorticoid blocking agents and their effects on potassium homeostasis. Heart Fail. Rev. 2005, 10, 23–29. [Google Scholar] [CrossRef]
- Levy, D.G.; Rocha, R.; Funder, J.W. Distinguishing the antihypertensive and electrolyte effects of eplerenone. J. Clin. Endocrinol. Metab. 2004, 89, 2736–2740. [Google Scholar] [CrossRef] [Green Version]
- Tolbert, D.; Reid, S.; Roniker, B. Pharmacokinetics of eplerenone coadministered with other medications. Pharmacotherapy 2020, 22, 1331. [Google Scholar]
- Pitt, B.; Remme, W.; Zannad, F.; Neaton, J.; Martinez, F.; Roniker, B.; Bittman, R.; Hurley, S.; Kleiman, J.; Gatlin, M. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N. Engl. J. Med. 2003, 348, 1309–1321. [Google Scholar] [CrossRef]
- Pitt, B.; Zannad, F.; Remme, W.J.; Cody, R.; Castaigne, A.; Perez, A.; Palensky, J.; Wittes, J. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N. Engl. J. Med. 1999, 341, 709–717. [Google Scholar] [CrossRef] [Green Version]
- Vaidyanathan, S.; Jarugula, V.; Dieterich, H.A.; Howard, D.; Dole, W.P. Clinical pharmacokinetics and pharmacodynamics of aliskiren. Clin. Pharmacokinet. 2008, 47, 515–531. [Google Scholar] [CrossRef] [PubMed]
- Azizi, M.; Webb, R.; Nussberger, J.; Hollenberg, N.K. Renin inhibition with aliskiren: Where are we now, and where are we going? J. Hypertens. 2006, 24, 243–256. [Google Scholar] [CrossRef] [PubMed]
- Lijnen, P.; Fagard, R.; Staessen, J.; Amery, A. Effect of chronic diuretic treatment on the plasma renin-angiotensin-aldosterone system in essential hypertension. Br. J. Clin. Pharmacol. 1981, 12, 387–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Z.; McGoogan, J.M. Characteristics of and important lessons from the coronavirus disease 2019 (COVID-19) outbreak in China: Summary of a report of 72 314 cases from the Chinese center for disease control and prevention. JAMA 2020, 13, 1239–1242. [Google Scholar] [CrossRef]
- Arai, T.; Yasuda, Y.; Takaya, T.; Toshima, S.; Kashiki, Y.; Shibayama, M.; Yoshimi, N.; Fujiwara, H. Angiotensin-converting enzyme inhibitors, angiotensin-II receptor antagonists, and pneumonia in elderly hypertensive patients with stroke. Chest 2001, 119, 660–661. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, R.; Watabe, N.; Konno, T.; Mishina, N.; Sekizawa, K.; Sasaki, H. High incidence of silent aspiration in elderly patients with community-acquired pneumonia. Am. J. Respir. Crit. Care Med. 1994, 150, 251–253. [Google Scholar] [CrossRef] [PubMed]
- Sekizawa, K.; Ujiie, Y.; Itabashi, S.; Sasaki, H.; Takishima, T. Lack of cough reflex in aspiration pneumonia. Lancet 1990, 335, 1228–1229. [Google Scholar] [CrossRef]
- Arai, T.; Yasuda, Y.; Takaya, T.; Toshima, S.; Kashiki, Y.; Yoshimi, N.; Fujiwara, H. ACE inhibitors and symptomless dysphagia. Lancet 1998, 352, 115–116. [Google Scholar] [CrossRef]
- Arai, T.; Yoshimi, N.; Fujiwara, H.; Sekizawa, K. Serum substance P concentrations and silent aspiration in elderly patients with stroke. Neurology 2003, 61, 1625–1626. [Google Scholar] [CrossRef]
- El Bekay, R.; Alvarez, M.; Monteseirín, J.; Alba, G.; Chacón, P.; Vega, A.; Martin-Nieto, J.; Jiménez, J.; Pintado, E.; Bedoya, F.J.; et al. Oxidative stress is a critical mediator of the angiotensin II signal in human neutrophils: Involvement of mitogen-activated protein kinase, calcineurin, and the transcription factor NF-kappaB. Blood 2003, 102, 662–671. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Castelo, S.; Arzt, E.S.; Pesce, A.; Criscuolo, M.E.; Diaz, A.; Finkielman, S.; Nahmod, V.E. Angiotensin II regulates interferon-gamma production. J. Interferon. Res. 1987, 7, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Fukuzawa, M.; Satoh, J.; Sagara, M.; Muto, G.; Muto, Y.; Nishimura, S.; Miyaguchi, S.; Qiang, X.L.; Sakata, Y.; Nakazawa, T.; et al. Angiotensin converting enzyme inhibitors suppress production of tumor necrosis factor-alpha in vitro and in vivo. Immunopharmacology 1997, 36, 49–55. [Google Scholar] [CrossRef]
- Hernández-Presa, M.; Bustos, C.; Ortego, M.; Tuñon, J.; Renedo, G.; Ruiz-Ortega, M.; Egido, J. Angiotensin-converting enzyme inhibition prevents arterial nuclear factor-kappa B activation, monocyte chemoattractant protein-1 expression, and macrophage infiltration in a rabbit model of early accelerated atherosclerosis. Circulation 1997, 95, 1532–1541. [Google Scholar] [CrossRef] [PubMed]
- Kranzhöfer, R.; Browatzki, M.; Schmidt, J.; Kübler, W. Angiotensin II activates the proinflammatory transcription factor nuclear factor-kappaB in human monocytes. Biochem. Biophys. Res. Commun. 1999, 257, 826–828. [Google Scholar] [CrossRef] [PubMed]
- Nahmod, K.A.; Nahmod, V.E.; Szvalb, A.D. Potential mechanisms of AT1 receptor blockers on reducing pneumonia-related mortality. Clin. Infect. Dis. 2013, 56, 1193–1194. [Google Scholar] [CrossRef] [Green Version]
- Arai, T.; Sekizawa, K.; Ohrui, T.; Fujiwara, H.; Yoshimi, N.; Matsuoka, H.; Sasaki, H. ACE inhibitors and protection against pneumonia in elderly patients with stroke. Neurology 2005, 64, 573–574. [Google Scholar] [CrossRef]
- Ohkubo, T.; Chapman, N.; Neal, B.; Woodward, M.; Omae, T.; Chalmers, J. Effects of an angiotensin-converting enzyme inhibitor-based regimen on pneumonia risk. Am. J. Respir. Crit. Care Med. 2004, 169, 1041–1045. [Google Scholar] [CrossRef]
- Okaishi, K.; Morimoto, S.; Fukuo, K.; Niinobu, T.; Hata, S.; Onishi, T.; Ogihara, T. Reduction of risk of pneumonia associated with use of angiotensin I converting enzyme inhibitors in elderly inpatients. Am. J. Hypertens. 1999, 12, 778–783. [Google Scholar] [CrossRef] [Green Version]
- Etminan, M.; Zhang, B.; Fitzgerald, M.; Brophy, J.M. Do angiotensin-converting enzyme inhibitors or angiotensin II receptor blockers decrease the risk of hospitalization secondary to community-acquired pneumonia? A nested case-control study. Pharmacotherapy 2006, 26, 479–482. [Google Scholar] [CrossRef]
- Mortensen, E.M.; Restrepo, M.I.; Anzueto, A.; Pugh, J. The impact of prior outpatient ACE inhibitor use on 30-day mortality for patients hospitalized with community-acquired pneumonia. BMC Pulm. Med. 2005, 5, 12. [Google Scholar] [CrossRef] [Green Version]
- van de Garde, E.M.; Souverein, P.C.; Hak, E.; Deneer, V.H.; van den Bosch, J.M.; Leufkens, H.G. Angiotensin-converting enzyme inhibitor use and protection against pneumonia in patients with diabetes. J. Hypertens. 2007, 25, 235–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liappis, A.P.; Kan, V.L.; Rochester, C.G.; Simon, G.L. The effect of statins on mortality in patients with bacteremia. Clin. Infect. Dis. 2001, 33, 1352–1357. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, E.M.; Restrepo, M.I.; Copeland, L.A.; Pugh, J.A.; Anzueto, A. Association of hydrophilic versus lipophilic angiotensin-converting enzyme inhibitor use on pneumonia-related mortality. Am. J. Med. Sci. 2008, 336, 462–466. [Google Scholar] [CrossRef]
- Mortensen, E.M.; Nakashima, B.; Cornell, J.; Copeland, L.A.; Pugh, M.J.; Anzueto, A.; Good, C.; Restrepo, M.I.; Downs, J.R.; Frei, C.R.; et al. Population-based study of statins, angiotensin II receptor blockers, and angiotensin-converting enzyme inhibitors on pneumonia-related outcomes. Clin. Infect. Dis. 2012, 55, 1466–1473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.-L.; Shau, W.-Y.; Chang, C.-H.; Wu, C.-S.; Lai, M.-S. Pneumonia risk and use of angiotensin-converting enzyme inhibitors and angiotensin II receptor blockers. J. Epidemiol. 2013, 23, 344–350. [Google Scholar] [CrossRef] [Green Version]
- Soto, M.; Bang, S.I.; McCombs, J.; Rodgers, K.E. Renin Angiotensin system-modifying therapies are associated with improved pulmonary health. Clin. Diabetes Endocrinol. 2017, 3, 6. [Google Scholar] [CrossRef] [Green Version]
- Caldeira, D.; Alarcão, J.; Vaz-Carneiro, A.; Costa, J. Risk of pneumonia associated with use of angiotensin converting enzyme inhibitors and angiotensin receptor blockers: Systematic review and meta-analysis. BMJ 2012, 345, e4260. [Google Scholar] [CrossRef] [Green Version]
- Fedson, D.S. Treating the host response to emerging virus diseases: Lessons learned from sepsis, pneumonia, influenza and Ebola. Ann. Transl. Med. 2016, 4, 421. [Google Scholar] [CrossRef] [Green Version]
- Reiner, Ž.; Hatamipour, M.; Banach, M.; Pirro, M.; Al-Rasadi, K.; Jamialahmadi, T.; Radenkovic, D.; Montecucco, F.; Sahebkar, A. Statins and the COVID-19 main protease: In silico evidence on direct interaction. Arch. Med. Sci. 2020, 16, 490–496. [Google Scholar] [CrossRef]
- Rodrigues Díez, R.; Rodrigues-Díez, R.; Lavoz, C.; Rayego-Mateos, S.; Civantos, E.; Rodríguez-Vita, J.; Mezzano, S.; Ortiz, A.; Egido, J.; Ruiz-Ortega, M. Statins inhibit angiotensin II/Smad pathway and related vascular fibrosis, by a TGF-β-independent process. PLoS ONE 2010, 5, e14145. [Google Scholar] [CrossRef] [Green Version]
- Aguilar, C.; Ventura, F.; Rodríguez-Delfín, L. [Atorvastatin induced increase in homologous angiotensin I converting enzyme (ACE2) mRNA is associated to decreased fibrosis and decreased left ventricular hypertrophy in a rat model of diabetic cardiomyopathy]. Rev. Peru. Med. Exp. Salud Publica 2011, 28, 264–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.H.; Wang, Q.X.; Zhou, J.W.; Chu, X.M.; Man, Y.L.; Liu, P.; Ren, B.B.; Sun, T.R.; An, Y. Effects of rosuvastatin on expression of angiotensin-converting enzyme 2 after vascular balloon injury in rats. J. Geriatr. Cardiol. 2013, 10, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.H.; Min, J.J.; Lee, J.H.; Kim, E.H.; Kim, G.E.; Kim, M.H.; Lee, J.J.; Ahn, H.J. The effect of fluvastatin on cardiac fibrosis and angiotensin-converting enzyme-2 expression in glucose-controlled diabetic rat hearts. Heart Vessels 2017, 32, 618–627. [Google Scholar] [CrossRef] [PubMed]
- Sasidhar, M.V.; Chevooru, S.K.; Eickelberg, O.; Hartung, H.P.; Neuhaus, O. Downregulation of monocytic differentiation via modulation of CD147 by 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors. PLoS ONE 2017, 12, e0189701. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Gao, X.; Zhao, Z.; Pan, S. [Expression of cyclophilin A/CD147 in carotid atherosclerotic plaque and the intervention of atorvastatin]. Zhong Nan Da Xue Xue Bao Yi Xue Ban 2016, 41, 482–488. [Google Scholar] [CrossRef]
- Wang, K.; Chen, W.; Zhou, Y.-S.; Lian, J.-Q.; Zhang, Z.; Du, P.; Gong, L.; Zhang, Y.; Cui, H.-Y.; Geng, J.-J.; et al. SARS-CoV-2 invades host cells via a novel route: CD147-spike protein. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Mi, L.; Xu, J.; Yu, J.; Wang, X.; Jiang, J.; Xing, J.; Shang, P.; Qian, A.; Li, Y. Function of HAb18G/CD147 in invasion of host cells by severe acute respiratory syndrome coronavirus. J. Infect. Dis. 2005, 191, 755–760. [Google Scholar] [CrossRef] [Green Version]
- Katsiki, N.; Banach, M.; Mikhailidis, D.P. Lipid-lowering therapy and renin-angiotensin-aldosterone system inhibitors in the era of the COVID-19 pandemic. Arch. Med. Sci. 2020, 16, 485–489. [Google Scholar] [CrossRef]
- Guo, J.; Huang, Z.; Lin, L.; Lv, J. Coronavirus Disease 2019 (COVID-19) and Cardiovascular Disease: A viewpoint on the potential influence of angiotensin-converting enzyme inhibitors/angiotensin receptor blockers on onset and severity of severe acute respiratory syndrome coronavirus 2 infection. J. Am. Heart Assoc. 2020, 9, e016219. [Google Scholar] [CrossRef]
- Sommerstein, R.; Kochen, M.M.; Messerli, F.H.; Gräni, C. Coronavirus Disease 2019 (COVID-19): Do Angiotensin-converting enzyme inhibitors/angiotensin receptor blockers have a biphasic effect? J. Am. Heart Assoc. 2020, 9, e016509. [Google Scholar] [CrossRef]
- Hanff, T.C.; Harhay, M.O.; Brown, T.S.; Cohen, J.B.; Mohareb, A.M. Is there an association between COVID-19 mortality and the renin-angiotensin system-a call for epidemiologic investigations. Clin. Infect. Dis. 2020. [Google Scholar] [CrossRef] [Green Version]
- Hamming, I.; van Goor, H.; Turner, A.J.; Rushworth, C.A.; Michaud, A.A.; Corvol, P.; Navis, G. Differential regulation of renal angiotensin-converting enzyme (ACE) and ACE2 during ACE inhibition and dietary sodium restriction in healthy rats. Exp. Physiol. 2008, 93, 631–638. [Google Scholar] [CrossRef] [PubMed]
- Ferrario, C.M.; Jessup, J.; Chappell, M.C.; Averill, D.B.; Brosnihan, K.B.; Tallant, E.A.; Diz, D.I.; Gallagher, P.E. Effect of angiotensin-converting enzyme inhibition and angiotensin II receptor blockers on cardiac angiotensin-converting Enzyme 2. Circulation 2005, 111, 2605–2610. [Google Scholar] [CrossRef] [Green Version]
- Ocaranza, M.P.; Godoy, I.; Jalil, J.E.; Varas, M.; Collantes, P.; Pinto, M.; Roman, M.; Ramirez, C.; Copaja, M.; Diaz-Araya, G.; et al. Enalapril attenuates downregulation of Angiotensin-converting enzyme 2 in the late phase of ventricular dysfunction in myocardial infarcted rat. Hypertension 2006, 48, 572–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Zeng, Z.; Cao, Y.; Liu, Y.; Ping, F.; Liang, M.; Xue, Y.; Xi, C.; Zhou, M.; Jiang, W. Angiotensin-converting enzyme 2 prevents lipopolysaccharide-induced rat acute lung injury via suppressing the ERK1/2 and NF-κB signaling pathways. Sci. Rep. 2016, 6, 27911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vuille-dit-Bille, R.N.; Camargo, S.M.; Emmenegger, L.; Sasse, T.; Kummer, E.; Jando, J.; Hamie, Q.M.; Meier, C.F.; Hunziker, S.; Forras-Kaufmann, Z.; et al. Human intestine luminal ACE2 and amino acid transporter expression increased by ACE-inhibitors. Amino Acids 2015, 47, 693–705. [Google Scholar] [CrossRef] [Green Version]
- Furuhashi, M.; Moniwa, N.; Mita, T.; Fuseya, T.; Ishimura, S.; Ohno, K.; Shibata, S.; Tanaka, M.; Watanabe, Y.; Akasaka, H.; et al. Urinary angiotensin-converting enzyme 2 in hypertensive patients may be increased by olmesartan, an angiotensin II receptor blocker. Am. J. Hypertens. 2015, 28, 15–21. [Google Scholar] [CrossRef] [Green Version]
- Burrell, L.M.; Risvanis, J.; Kubota, E.; Dean, R.G.; MacDonald, P.S.; Lu, S.; Tikellis, C.; Grant, S.L.; Lew, R.A.; Smith, A.I.; et al. Myocardial infarction increases ACE2 expression in rat and humans. Eur. Heart J. 2005, 26, 369–375, discussion 322–364. [Google Scholar] [CrossRef] [Green Version]
- Ferrario, C.M.; Jessup, J.; Gallagher, P.E.; Averill, D.B.; Brosnihan, K.B.; Ann Tallant, E.; Smith, R.D.; Chappell, M.C. Effects of renin-angiotensin system blockade on renal angiotensin-(1-7) forming enzymes and receptors. Kidney Int. 2005, 68, 2189–2196. [Google Scholar] [CrossRef] [Green Version]
- Klimas, J.; Olvedy, M.; Ochodnicka-Mackovicova, K.; Kruzliak, P.; Cacanyiova, S.; Kristek, F.; Krenek, P.; Ochodnicky, P. Perinatally administered losartan augments renal ACE2 expression but not cardiac or renal Mas receptor in spontaneously hypertensive rats. J. Cell Mol. Med. 2015, 19, 1965–1974. [Google Scholar] [CrossRef] [PubMed]
- Igase, M.; Strawn, W.B.; Gallagher, P.E.; Geary, R.L.; Ferrario, C.M. Angiotensin II AT1 receptors regulate ACE2 and angiotensin-(1-7) expression in the aorta of spontaneously hypertensive rats. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H1013–H1019. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.C.; Ye, J.Y.; Jin, H.Y.; Yu, X.; Yu, H.M.; Zhu, D.L.; Gao, P.J.; Huang, D.Y.; Shuster, M.; Loibner, H.; et al. Telmisartan attenuates aortic hypertrophy in hypertensive rats by the modulation of ACE2 and profilin-1 expression. Regul. Pept. 2011, 166, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Ishiyama, Y.; Gallagher, P.E.; Averill, D.B.; Tallant, E.A.; Brosnihan, K.B.; Ferrario, C.M. Upregulation of angiotensin-converting enzyme 2 after myocardial infarction by blockade of angiotensin II receptors. Hypertension 2004, 43, 970–976. [Google Scholar] [CrossRef] [Green Version]
- Burchill, L.J.; Velkoska, E.; Dean, R.G.; Griggs, K.; Patel, S.K.; Burrell, L.M. Combination renin-angiotensin system blockade and angiotensin-converting enzyme 2 in experimental myocardial infarction: Implications for future therapeutic directions. Clin. Sci. (Lond.) 2012, 123, 649–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karam, C.N.; Nuwayri-Salti, N.; Usta, J.A.; Zwainy, D.S.; Abrahamian, R.E.; Al Jaroudi, W.A.; Baasisri, M.J.; Abdallah, S.M.; Bitar, K.M.; Bikhazi, A.B. Effect of systemic insulin and angiotensin II receptor subtype-1 antagonist on endothelin-1 receptor subtype(s) regulation and binding in diabetic rat heart. Endothelium 2005, 12, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Wösten-van Asperen, R.M.; Lutter, R.; Specht, P.A.; Moll, G.N.; van Woensel, J.B.; van der Loos, C.M.; van Goor, H.; Kamilic, J.; Florquin, S.; Bos, A.P. Acute respiratory distress syndrome leads to reduced ratio of ACE/ACE2 activities and is prevented by angiotensin-(1-7) or an angiotensin II receptor antagonist. J. Pathol. 2011, 225, 618–627. [Google Scholar] [CrossRef]
- Keidar, S.; Gamliel-Lazarovich, A.; Kaplan, M.; Pavlotzky, E.; Hamoud, S.; Hayek, T.; Karry, R.; Abassi, Z. Mineralocorticoid receptor blocker increases angiotensin-converting enzyme 2 activity in congestive heart failure patients. Circ. Res. 2005, 97, 946–953. [Google Scholar] [CrossRef] [Green Version]
- Epelman, S.; Tang, W.H.; Chen, S.Y.; Van Lente, F.; Francis, G.S.; Sen, S. Detection of soluble angiotensin-converting enzyme 2 in heart failure: Insights into the endogenous counter-regulatory pathway of the renin-angiotensin-aldosterone system. J. Am. Coll Cardiol. 2008, 52, 750–754. [Google Scholar] [CrossRef] [Green Version]
- Ding, W.; Li, X.; Wu, W.; He, H.; Li, Y.; Gao, L.; Gan, L.; Wang, M.; Ou, S.; Liu, J. [Aliskiren inhibits angiotensin II/angiotensin 1–7(Ang II/Ang1-7) signal pathway in rats with diabetic nephropathy]. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi 2018, 34, 891–895. [Google Scholar]
- Oudit, G.Y.; Kassiri, Z.; Jiang, C.; Liu, P.P.; Poutanen, S.M.; Penninger, J.M.; Butany, J. SARS-coronavirus modulation of myocardial ACE2 expression and inflammation in patients with SARS. Eur. J. Clin. Investig. 2009, 39, 618–625. [Google Scholar] [CrossRef]
- Patel, V.B.; Bodiga, S.; Fan, D.; Das, S.K.; Wang, Z.; Wang, W.; Basu, R.; Zhong, J.; Kassiri, Z.; Oudit, G.Y. Cardioprotective effects mediated by angiotensin II type 1 receptor blockade and enhancing angiotensin 1-7 in experimental heart failure in angiotensin-converting enzyme 2-null mice. Hypertension 2012, 59, 1195–1203. [Google Scholar] [CrossRef] [PubMed] [Green Version]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Michaud, V.; Deodhar, M.; Arwood, M.; Al Rihani, S.B.; Dow, P.; Turgeon, J. ACE2 as a Therapeutic Target for COVID-19; Its Role in Infectious Processes and Regulation by Modulators of the RAAS System. J. Clin. Med. 2020, 9, 2096. https://doi.org/10.3390/jcm9072096
Michaud V, Deodhar M, Arwood M, Al Rihani SB, Dow P, Turgeon J. ACE2 as a Therapeutic Target for COVID-19; Its Role in Infectious Processes and Regulation by Modulators of the RAAS System. Journal of Clinical Medicine. 2020; 9(7):2096. https://doi.org/10.3390/jcm9072096
Chicago/Turabian StyleMichaud, Veronique, Malavika Deodhar, Meghan Arwood, Sweilem B Al Rihani, Pamela Dow, and Jacques Turgeon. 2020. "ACE2 as a Therapeutic Target for COVID-19; Its Role in Infectious Processes and Regulation by Modulators of the RAAS System" Journal of Clinical Medicine 9, no. 7: 2096. https://doi.org/10.3390/jcm9072096
APA StyleMichaud, V., Deodhar, M., Arwood, M., Al Rihani, S. B., Dow, P., & Turgeon, J. (2020). ACE2 as a Therapeutic Target for COVID-19; Its Role in Infectious Processes and Regulation by Modulators of the RAAS System. Journal of Clinical Medicine, 9(7), 2096. https://doi.org/10.3390/jcm9072096