Familial Dysalbuminemic Hyperthyroxinemia: An Underdiagnosed Entity

 , , , and
, , , and
Abstract
:1. Introduction
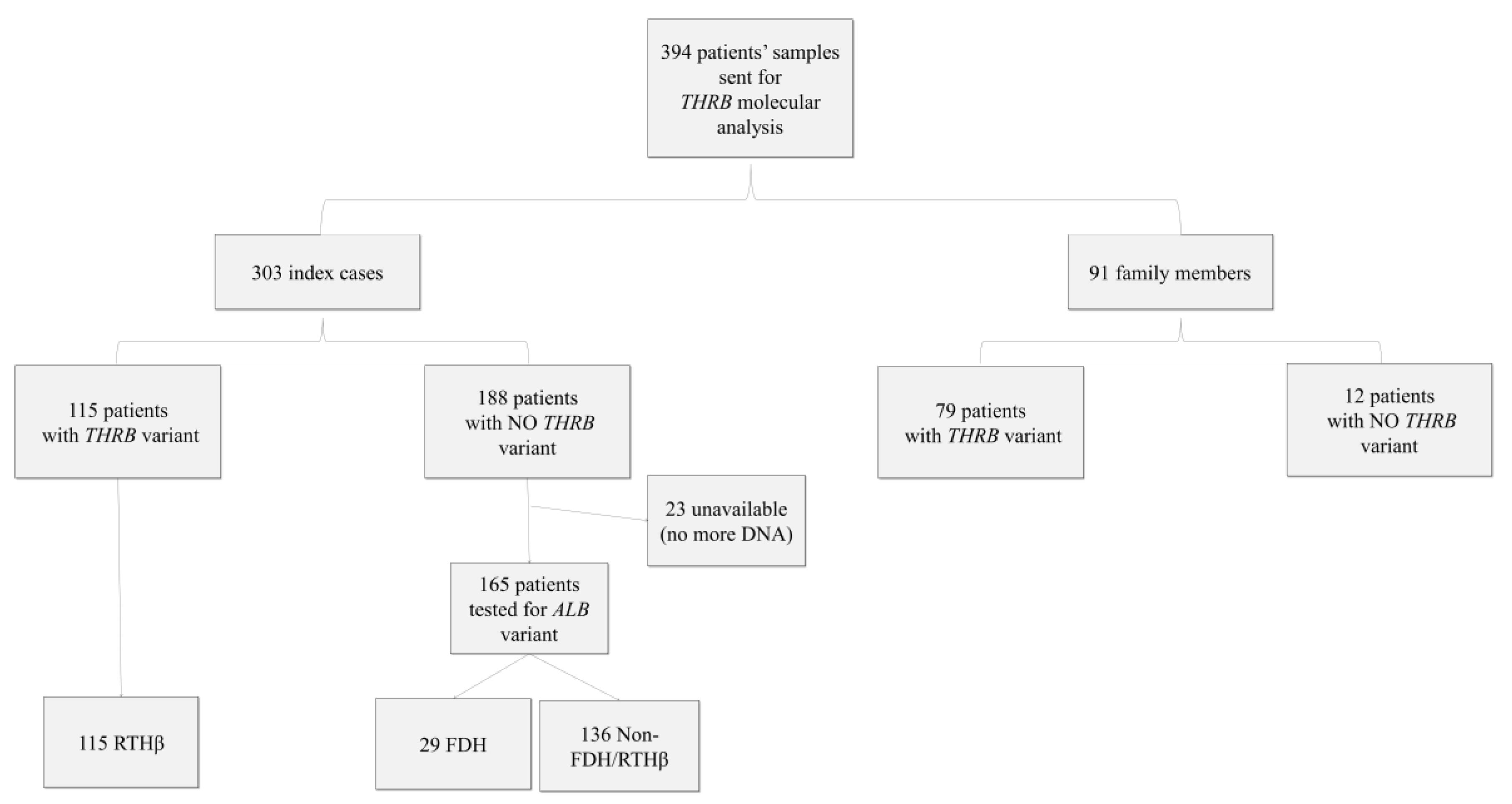
2. Materials and Methods
2.1. Patients
2.2. THRB and ALB Gene Mutation
2.3. Hormonal Assays and Clinical Information
2.4. Statistical Analysis
2.4.1. Univariate Analysis
2.4.2. Multivariate Analysis
3. Results
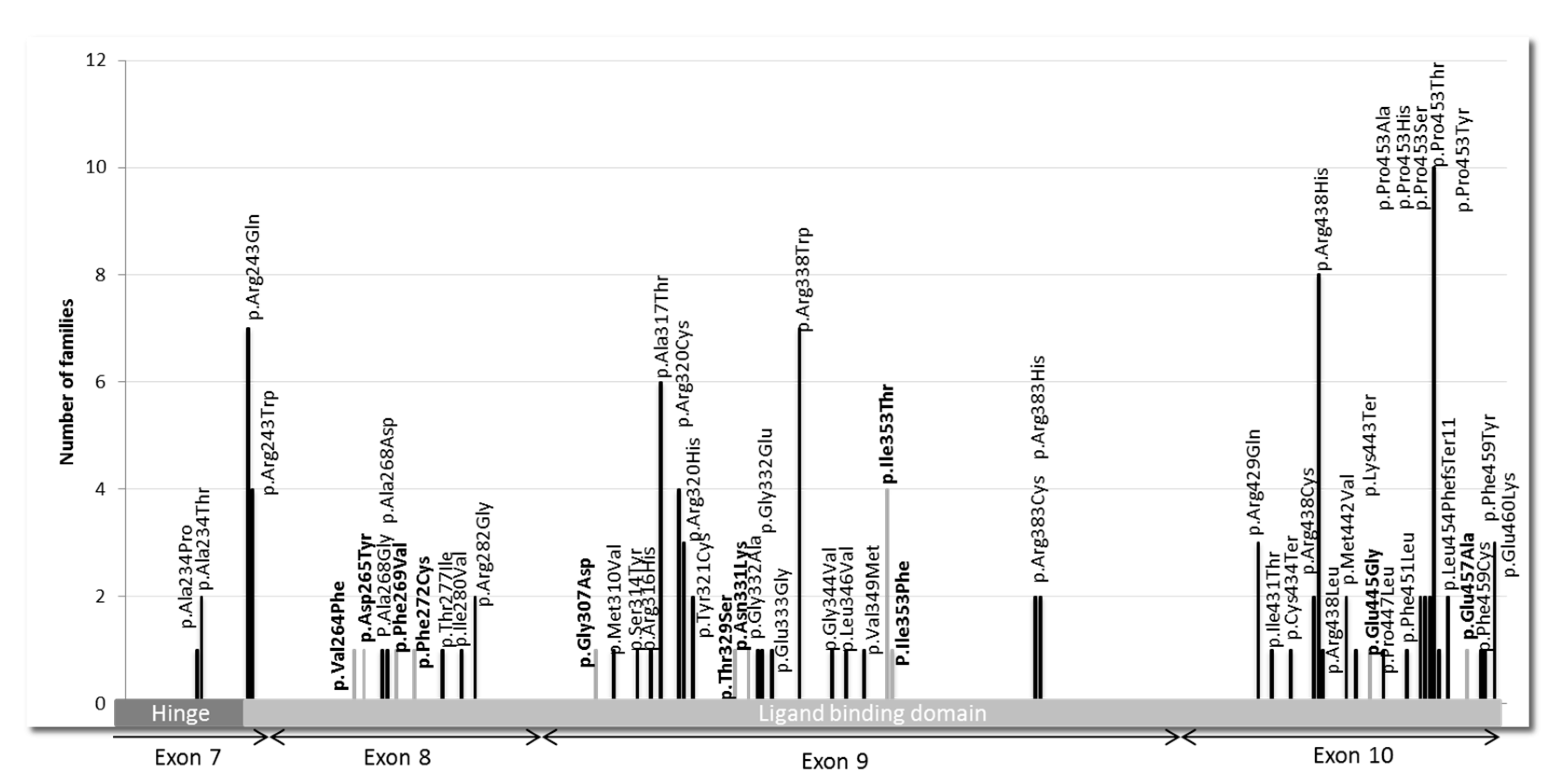
3.1. Pathogenic Variants and VUS in THRB Gene
3.2. Patients with FDH
3.3. Patients without FDH or RTHβ (Non FDH/RTHβ)
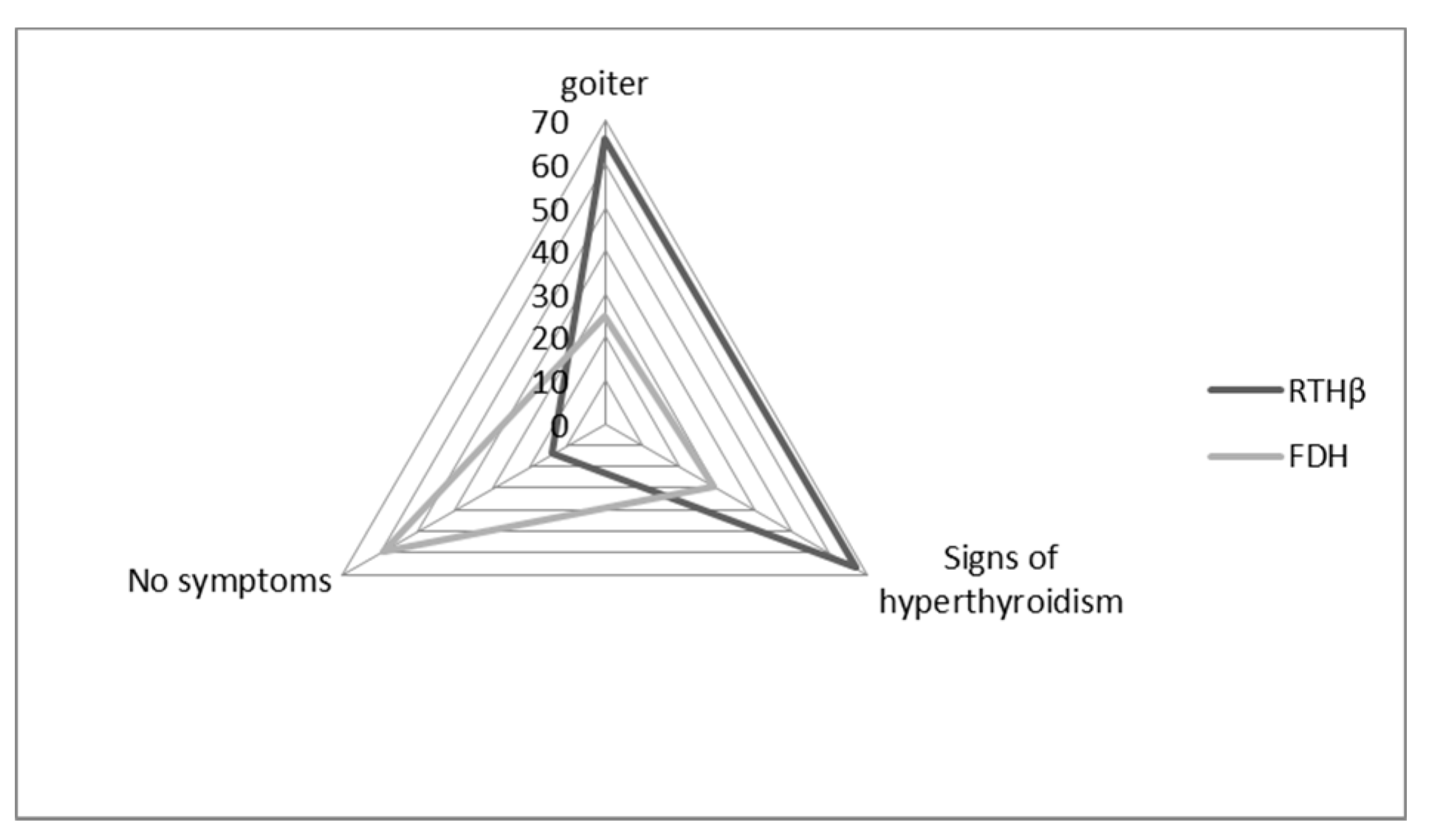
3.4. Clinical Information
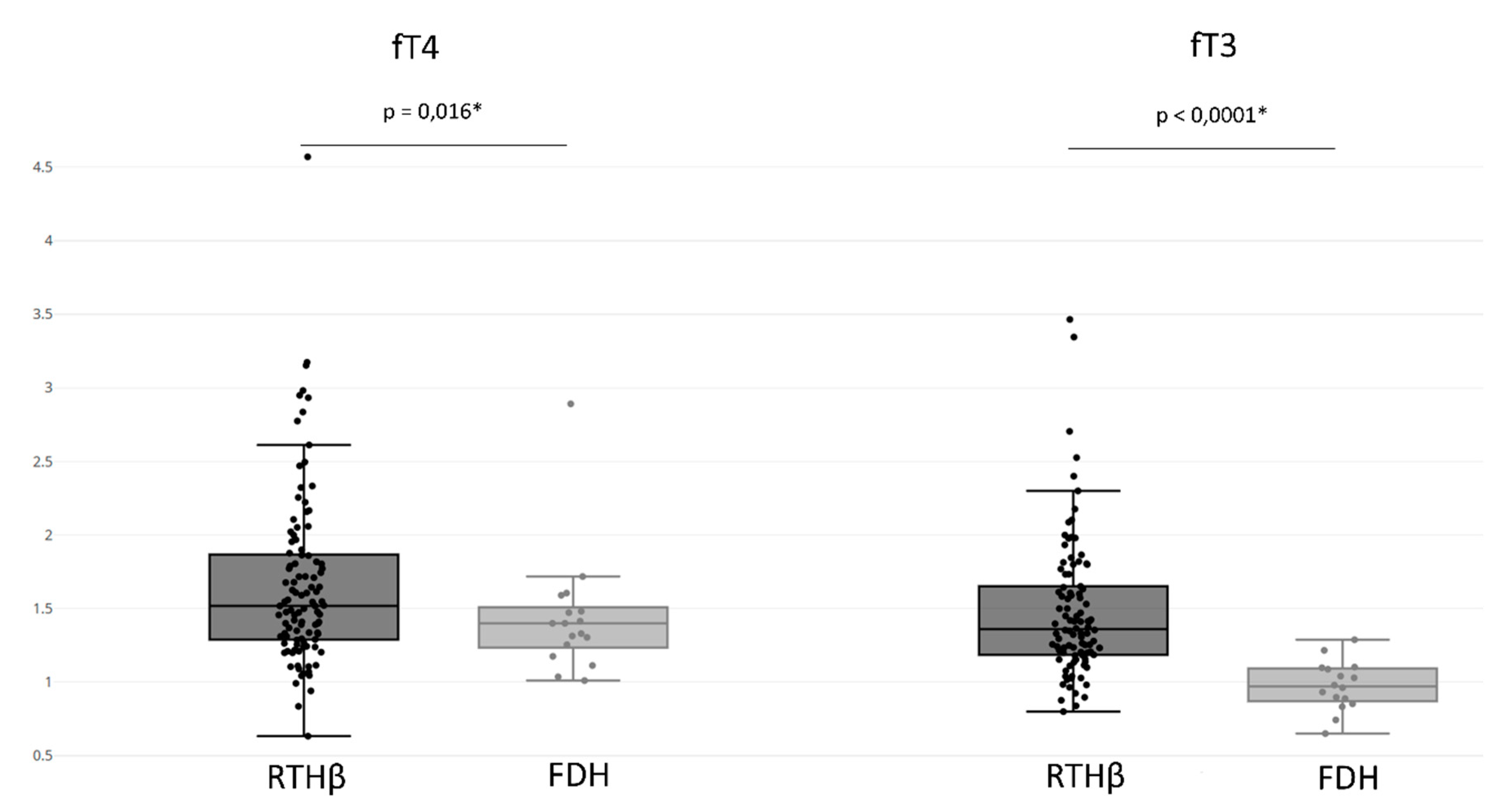
3.5. Hormone Assays
3.6. Multivariate Model to Differentiate between THRB and ALB Variants
- •
- If a patient’s fT3 is above 1.11, then he/she has a THRB variant.
- •
- For all the other cases, the patient has FDH.
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Refetoff, S.; DeWind, L.T.; DeGroot, L.J. Familial syndrome combining deaf-mutism, stuppled epiphyses, goiter and abnormally high PBI: Possible target organ refractoriness to thyroid hormone. J. Clin. Endocrinol. Metab. 1967, 27, 279–294. [Google Scholar] [CrossRef] [PubMed]
- Dumitrescu, A.M.; Refetoff, S. The syndromes of reduced sensitivity to thyroid hormone. Biochim. Biophys. Acta 2013, 1830, 3987–4003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakurai, A.; Takeda, K.; Ain, K.; Ceccarelli, P.; Nakai, A.; Seino, S.; Bell, G.I.; Refetoff, S.; DeGroot, L.J. Generalized resistance to thyroid hormone associated with a mutation in the ligand-binding domain of the human thyroid hormone receptor beta. Proc. Natl. Acad. Sci. U.S.A. 1989, 86, 8977–8981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrara, A.M.; Onigata, K.; Ercan, O.; Woodhead, H.; Weiss, R.E.; Refetoff, S. Homozygous thyroid hormone receptor β-gene mutations in resistance to thyroid hormone: Three new cases and review of the literature. J. Clin. Endocrinol. Metab. 2012, 97, 1328–1336. [Google Scholar] [CrossRef] [Green Version]
- Ortiga-Carvalho, T.M.; Sidhaye, A.R.; Wondisford, F.E. Thyroid hormone receptors and resistance to thyroid hormone disorders. Nat. Rev. Endocrinol. 2014, 10, 582–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vella, K.R.; Hollenberg, A.N. The actions of thyroid hormone signaling in the nucleus. Mol. Cell. Endocrinol. 2017, 458, 127–135. [Google Scholar] [CrossRef]
- Weiss, R.E.; Hayashi, Y.; Nagaya, T.; Petty, K.J.; Murata, Y.; Tunca, H.; Seo, H.; Refetoff, S. Dominant inheritance of resistance to thyroid hormone not linked to defects in the thyroid hormone receptor alpha or beta genes may be due to a defective cofactor. J. Clin. Endocrinol. Metab. 1996, 81, 4196–4203. [Google Scholar] [CrossRef] [Green Version]
- Pohlenz, J.; Weiss, R.E.; Macchia, P.E.; Pannain, S.; Lau, I.T.; Ho, H.; Refetoff, S. Five new families with resistance to thyroid hormone not caused by mutations in the thyroid hormone receptor β Gene 1. J. Clin. Endocrinol. Metab. 1999, 84, 3919–3928. [Google Scholar] [CrossRef]
- Mamanasiri, S.; Yesil, S.; Dumitrescu, A.M.; Liao, X.-H.; Demir, T.; Weiss, R.E.; Refetoff, S. Mosaicism of a thyroid hormone receptor-β gene mutation in resistance to thyroid hormone. J. Clin. Endocrinol. Metab. 2006, 91, 3471–3477. [Google Scholar] [CrossRef] [Green Version]
- Reutrakul, S.; Sadow, P.M.; Pannain, S.; Pohlenz, J.; Carvalho, G.A.; Macchia, P.E.; Weiss, R.E.; Refetoff, S. Search for abnormalities of nuclear corepressors, coactivators, and a coregulator in families with resistance to thyroid hormone without mutations in thyroid hormone receptor β or α Genes 1. J. Clin. Endocrinol. Metab. 2000, 85, 3609–3617. [Google Scholar] [CrossRef] [Green Version]
- Brent, G.A. Mechanisms of thyroid hormone action. J. Clin. Invest. 2012, 122, 3035–3043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koulouri, O.; Moran, C.; Halsall, D.; Chatterjee, K.; Gurnell, M. Pitfalls in the measurement and interpretation of thyroid function tests. Best Pract. Res. Clin. Endocrinol. Metab. 2013, 27, 745–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piketty, M.-L.; Polak, M.; Flechtner, I.; Gonzales-Briceño, L.; Souberbielle, J.-C. False biochemical diagnosis of hyperthyroidism in streptavidin-biotin-based immunoassays: The problem of biotin intake and related interferences. Clin. Chem. Lab. Med. 2017, 55, 780–788. [Google Scholar] [CrossRef] [PubMed]
- Ohba, K.; Noh, J.Y.; Unno, T.; Satoh, T.; Iwahara, K.; Matsushita, A.; Sasaki, S.; Oki, Y.; Nakamura, H. Falsely elevated thyroid hormone levels caused by anti-ruthenium interference in the Elecsys assay resembling the syndrome of inappropriate secretion of thyrotropin. Endocr. J. 2012, 59, 663–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Favresse, J.; Burlacu, M.-C.; Maiter, D.; Gruson, D. Interferences with thyroid function immunoassays: Clinical implications and detection algorithm. Endocr. Rev. 2018, 39, 830–850. [Google Scholar] [CrossRef] [PubMed]
- Holm, S.S.; Hansen, S.H.; Faber, J.; Staun-Olsen, P. Reference methods for the measurement of free thyroid hormones in blood: Evaluation of potential reference methods for free thyroxine. Clin. Biochem. 2004, 37, 85–93. [Google Scholar] [CrossRef]
- Pappa, T.; Ferrara, A.M.; Refetoff, S. Inherited defects of thyroxine-binding proteins. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 735–747. [Google Scholar] [CrossRef] [Green Version]
- Kragh-Hansen, U.; Galliano, M.; Minchiotti, L. Clinical, genetic, and protein structural aspects of familial dysalbuminemic hyperthyroxinemia and hypertriiodothyroninemia. Front. Endocrinol 2017, 8, 297. [Google Scholar] [CrossRef] [Green Version]
- Janssen, S.T.; Janssen, O.E. Directional thyroid hormone distribution via the blood stream to target sites. Mol. Cell. Endocrinol. 2017, 458, 16–21. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Exome Aggregation Consortium; Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [Green Version]
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Albarca Aguilera, M.; Meyer, R.; Massouras, A. VarSome: The human genomic variant search engine. Bioinformatics 2019, 35, 1978–1980. [Google Scholar] [CrossRef] [PubMed]
- Cartwright, D.; O’Shea, P.; Rajanayagam, O.; Agostini, M.; Barker, P.; Moran, C.; Macchia, E.; Pinchera, A.; John, R.; Agha, A.; et al. Familial dysalbuminemic hyperthyroxinemia: A persistent diagnostic challenge. Clin. Chem. 2009, 55, 1044–1046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, H.A.; de Rijke, Y.B.; Sweep, F.C.G.J. Spuriously high free thyroxine values in familial dysalbuminemic hyperthyroxinemia. Clin. Chem. 2011, 57, 524–525. [Google Scholar] [CrossRef] [PubMed]
- Sturgeon, C.M.; Viljoen, A. Analytical error and interference in immunoassay: Minimizing risk. Ann. Clin. Biochem. 2011, 48, 418–432. [Google Scholar] [CrossRef]
- Zouwail, S.A.; O’Toole, A.M.; Clark, P.M.S.; Begley, J.P. Influence of thyroid hormone autoantibodies on 7 thyroid hormone assays. Clin. Chem. 2008, 54, 927–928. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ALL | RTHβ | FDH | Non-FDH/RTHβ | Not Available | |
|---|---|---|---|---|---|
| Age (years) | 32.3 ± 20.9 | 29.4 ± 21.2 | 28.5 ± 21.5 | 37.9 ± 19.2 | 37.8 ± 12.6. |
| Males (%) | 39 | 43 | 48 | 33 | 22 |
| Index cases | 303 | 115 | 29 | 136 | 23 |
| Next of kin | 91 | 79 | 4 | 8 | 0 |
| EXON | HGVSc1 NM_000461.4 | HGVSp2 NP_000452.2 | RS ID | gnomAD | Families | Patients | In Silico Predictions Algorithms | Classification Following ACMG |
|---|---|---|---|---|---|---|---|---|
| 8 | c.790G > T | p.(Val264Phe) | rs1559415493 | 0 | 1 | 1 | 7 pathogenic vs. 1 benign * | Likely pathogenic (PM1, PM2, PP2, PP3) |
| 8 | c.793G > T | p.(Asp265Tyr) | none | 0 | 1 | 1 | 7 pathogenic vs. 1 benign * | Likely pathogenic (PM1, PM2, PP2, PP3) |
| 8 | c.805T > G | p.(Phe269Val) | none | 0 | 1 | 2 | 8 pathogenic ** | Likely pathogenic (PM1, PM2, PP2, PP3) |
| 8 | c.815T > G | p.(Phe272Cys) | none | 0 | 1 | 4 | 7 pathogenic vs. 1 benign * | Likely pathogenic (PM1, PM2, PP2, PP3) |
| 9 | c.920G > A | p.(Gly307Asp) | rs1553611101 | 0 | 1 | 1 | 8 pathogenic ** | Likely pathogenic (PM1, PM2, PP2, PP3) |
| 9 | c.985A > T | p(.Thr329Ser) | none | 0 | 1 | 1 | 7 pathogenic vs. 1 benign *** | Likely pathogenic (PM1, PM2, PP2, PP3) |
| 9 | c.993T > A | p.(Asn331Lys) | none | 0 | 1 | 1 | 6 pathogenic vs. 2 benign **** | Likely pathogenic (PM1, PM2, PP2, PP3, PM5) |
| 9 | c.1058T > C | p.(Ile353Thr) | none | 0 | 4 | 7 | 8 pathogenic ** | Likely pathogenic (PM1, PM2, PP2, PP3) |
| 9 | c.1057A > T | p.(Ile353Phe) | none | 0 | 1 | 1 | 8 pathogenic ** | Likely pathogenic (PM1, PM2, PP2, PP3) |
| 10 | c.1334A > G | p.(Glu445Gly) | none | 0 | 1 | 2 | 8 pathogenic ** | Likely pathogenic (PM1, PM2, PP2, PP3) |
| 10 | c.1370A > C ***** | p.(Glu457Ala) ***** | none | 0 | 1 | 1 | 8 pathogenic ** | Likely pathogenic (PM1, PM2, PP2, PP3) |
| fT4 ↗ fT3 ↗ | fT4 ↗ fT3 Nl | fT4 Nl fT3 ↗ | |
|---|---|---|---|
| RTHβ | 89% | 8% | 3% |
| FDH | 44% | 56% | 0% |
| Genetic Variant | |||
|---|---|---|---|
| THRB | ALB | ||
| Model prediction about the presence of a THRB variant | positive | 22 | 1 |
| negative | 5 | 4 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dieu, X.; Bouzamondo, N.; Briet, C.; Illouz, F.; Moal, V.; Boux de Casson, F.; Bouhours-Nouet, N.; Reynier, P.; Coutant, R.; Rodien, P.; et al. Familial Dysalbuminemic Hyperthyroxinemia: An Underdiagnosed Entity. J. Clin. Med. 2020, 9, 2105. https://doi.org/10.3390/jcm9072105
Dieu X, Bouzamondo N, Briet C, Illouz F, Moal V, Boux de Casson F, Bouhours-Nouet N, Reynier P, Coutant R, Rodien P, et al. Familial Dysalbuminemic Hyperthyroxinemia: An Underdiagnosed Entity. Journal of Clinical Medicine. 2020; 9(7):2105. https://doi.org/10.3390/jcm9072105
Chicago/Turabian StyleDieu, Xavier, Nathalie Bouzamondo, Claire Briet, Frédéric Illouz, Valérie Moal, Florence Boux de Casson, Natacha Bouhours-Nouet, Pascal Reynier, Régis Coutant, Patrice Rodien, and et al. 2020. "Familial Dysalbuminemic Hyperthyroxinemia: An Underdiagnosed Entity" Journal of Clinical Medicine 9, no. 7: 2105. https://doi.org/10.3390/jcm9072105
APA StyleDieu, X., Bouzamondo, N., Briet, C., Illouz, F., Moal, V., Boux de Casson, F., Bouhours-Nouet, N., Reynier, P., Coutant, R., Rodien, P., & Mirebeau-Prunier, D. (2020). Familial Dysalbuminemic Hyperthyroxinemia: An Underdiagnosed Entity. Journal of Clinical Medicine, 9(7), 2105. https://doi.org/10.3390/jcm9072105