Pathophysiology and Diagnosis of Drug-Induced Immune Thrombocytopenia

 , and
, and
Abstract
:1. Introduction
2. Drugs and Mechanisms Involved in Drug-Induced Immune Thrombocytopenia
2.1. Thrombocytopenia Induced by Hapten-Dependent Antibodies
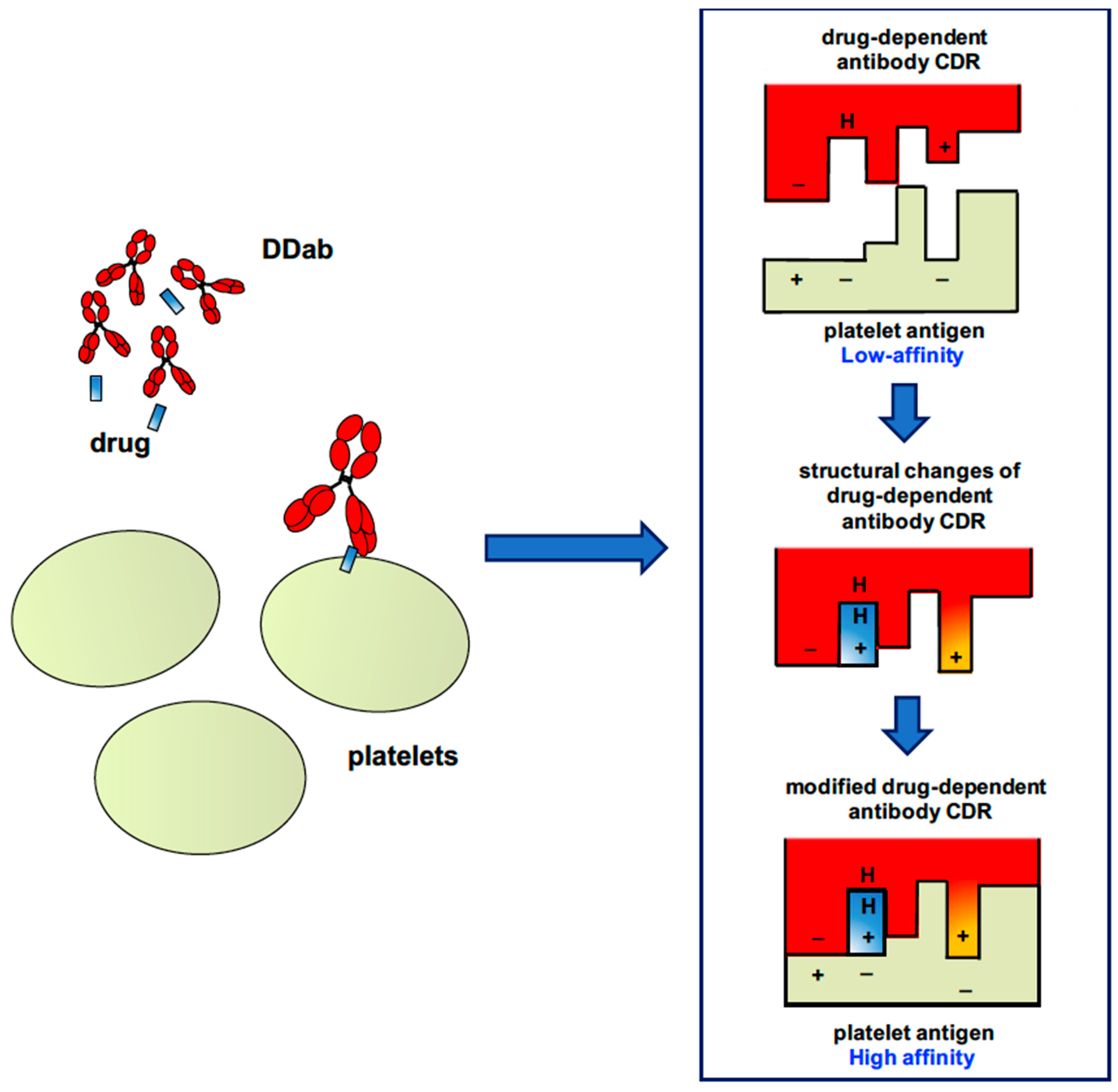
2.2. “Quinine-Type” Drug-Induced Immune Thrombocytopenia
2.3. Immune Thrombocytopenia Induced by GPIIb/IIIa Inhibitors
2.3.1. Thrombocytopenia Induced by Abciximab
2.3.2. Immune Thrombocytopenia Induced by Ligand-Mimetic Fibrinogen-Receptor Antagonists
2.4. Drug-Induced Immune Thrombocytopenia from Other Causes
2.4.1. Thrombocytopenia Induced by Platelet-Specific Auto-Antibodies
2.4.2. Thrombocytopenia Induced by Immune Complexes: Heparin-Induced Thrombocytopenia
2.5. Unresolved Questions on the Pathogenesis of Drug-Induced Immune Thrombocytopenia
2.5.1. Role of Complement in Drug-Induced Immune Thrombocytopenia
2.5.2. Role of Fcγ Receptors in Drug-Induced Immune Thrombocytopenia
2.5.3. Effect of Drug-Dependent Antibodies on Platelet Production
3. How to Diagnose Drug-Induced Immune Thrombocytopenia
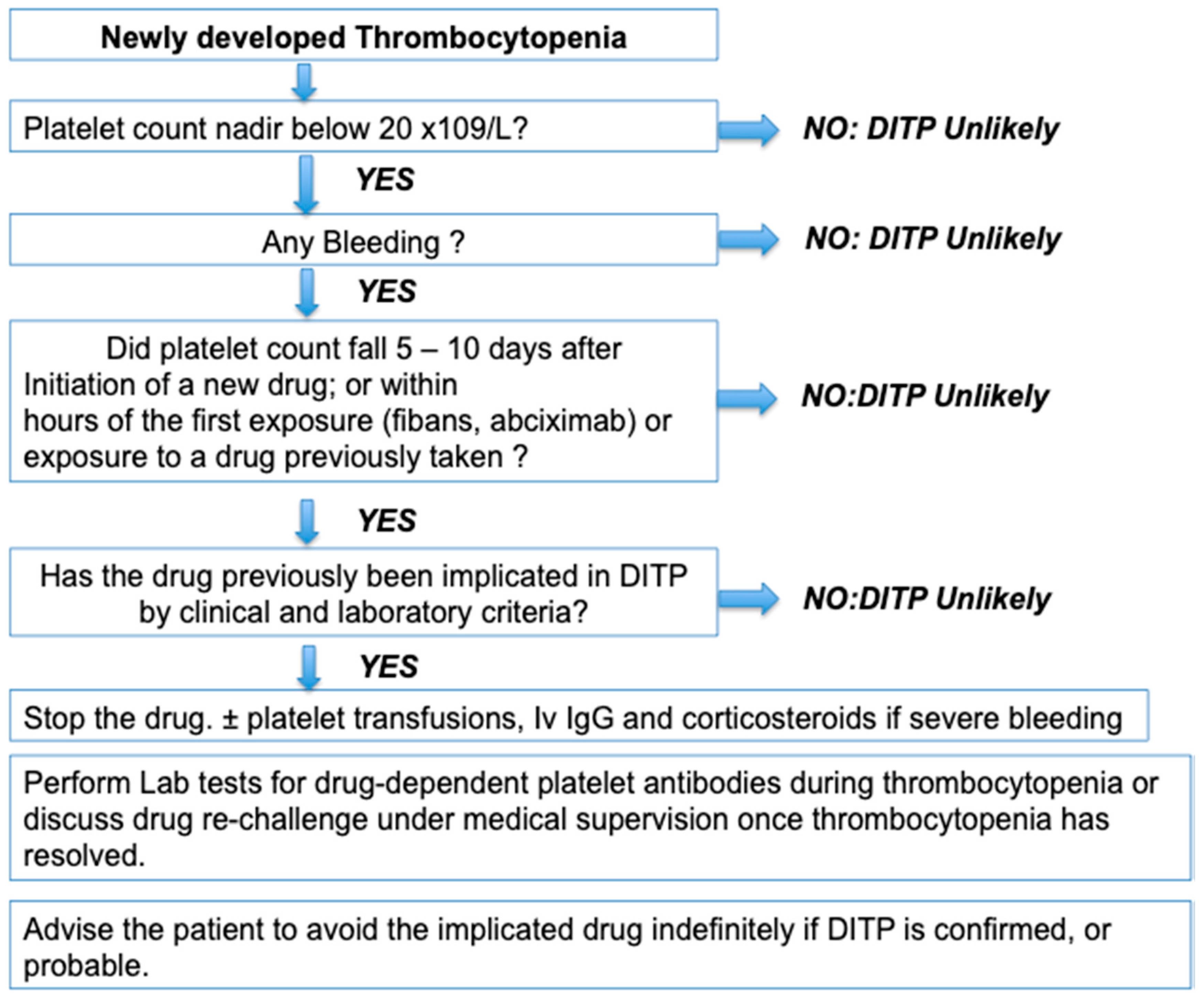
3.1. Clinical Features
3.2. Laboratory Assays for the Diagnosis of Drug Immune Thrombocytopenia Are Poorly Standardized
3.3. The Diagnosis of Heparin-Induced Thrombocytopenia Is Easier to Confirm
Author Contributions
Funding
Conflicts of Interest
References
- Aster, R.H. Drug-induced Thrombocytopenia. In Platelets, 4th ed.; Michelson, A.D., Cattaneo, M., Frelinger, A., Newman, P.J., Eds.; Academic Press: Cambridge, MA, USA, 2019; pp. 725–739. [Google Scholar]
- Curtis, B.R. Drug-induced immune thrombocytopenia: Incidence, clinical features, laboratory testing, and pathogenic mechanisms. Immunohematology 2014, 30, 55–65. [Google Scholar] [PubMed]
- Aster, R.H.; Curtis, B.R.; McFarland, J.G.; Bougie, D.W. Drug-induced immune thrombocytopenia: Pathogenesis, diagnosis, and management. J. Thromb. Haemost. 2009, 7, 911–918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aster, R.H.; Bougie, D.W. Drug-induced immune thrombocytopenia. N. Engl. J. Med. 2007, 357, 580–587. [Google Scholar] [CrossRef] [Green Version]
- Arnold, D.M.; Nazi, I.; Warkentin, T.E.; Smith, J.W.; Toltl, L.J.; George, J.N.; Kelton, J.G. Approach to the diagnosis and management of drug-induced immune thrombocytopenia. Transfus. Med. Rev. 2013, 27, 137–145. [Google Scholar] [CrossRef] [Green Version]
- Reese, J.A.; Li, X.; Hauben, M.; Aster, R.H.; Bougie, D.W.; Curtis, B.R.; George, J.N.; Vesely, S.K. Identifying drugs that cause acute thrombocytopenia: An analysis using 3 distinct methods. Blood 2010, 116, 2127–2133. [Google Scholar] [CrossRef] [Green Version]
- Hackett, T.; Kelton, J.G.; Powers, P. Drug-induced platelet destruction. Semin. Thromb. Hemost. 1982, 8, 116–137. [Google Scholar] [CrossRef] [PubMed]
- George, J.N.; Raskob, G.E.; Shah, S.R.; Rizvi, M.A.; Hamilton, S.A.; Osborne, S.; Vondracek, T. Drug-induced thrombocytopenia: A systematic review of published case reports. Ann. Intern. Med. 1998, 129, 886–890. [Google Scholar] [CrossRef] [PubMed]
- Arnold, D.M.; Kukaswadia, S.; Nazi, I.; Esmail, A.; Dewar, L.; Smith, J.W.; Warkentin, T.E.; Kelton, J.G. A systematic evaluation of laboratory testing for drug-induced immune thrombocytopenia. J. Thromb. Haemost. 2013, 11, 169–176. [Google Scholar] [CrossRef] [Green Version]
- Kaufman, D.W.; Kelly, J.P.; Johannes, C.B.; Sandler, A.; Harmon, D.; Stolley, P.D.; Shapiro, S. Acute thrombocytopenic purpura in relation to the use of drugs. Blood 1993, 82, 2714–2718. [Google Scholar] [CrossRef] [Green Version]
- Gruel, Y.; De Maistre, E.; Pouplard, C.; Mullier, F.; Susen, S.; Roullet, S.; Blais, N.; Le Gal, G.; Vincentelli, A.; Lasne, D.; et al. Diagnosis and management of heparin-induced thrombocytopenia. Anaesth. Crit. Care Pain Med. 2020. [Google Scholar] [CrossRef]
- Greinacher, A. Clinical Practice. Heparin-Induced Thrombocytopenia. N. Engl. J. Med. 2015, 373, 252–261. [Google Scholar] [CrossRef]
- Salamon, D.J.; Nusbacher, J.; Stroupe, T.; Wilson, J.H.; Hanrahan, J.B. Red cell and platelet-bound IgG penicillin antibodies in a patient with thrombocytopenia. Transfusion 1984, 24, 395–398. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.F.; Riordan, T.; Minchinton, R.M.; Chapman, J.F.; Amess, J.A.; Shaw, E.J.; Waters, A.H. Demonstration of an immune-mediated mechanism of penicillin-induced neutropenia and thrombocytopenia. Br. J. Haematol. 1983, 55, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Lang, R.; Lishner, M.; Ravid, M. Adverse reactions to prolonged treatment with high doses of carbenicillin and ureidopenicillins. Rev. Infect. Dis. 1991, 13, 68–72. [Google Scholar] [CrossRef] [PubMed]
- Grossjohann, B.; Eichler, P.; Greinacher, A.; Santoso, S.; Kroll, H. Ceftriaxone causes drug-induced immune thrombocytopenia and hemolytic anemia: Characterization of targets on platelets and red blood cells. Transfusion 2004, 44, 1033–1040. [Google Scholar] [CrossRef] [PubMed]
- Kam, T.; Alexander, M. Drug-induced immune thrombocytopenia. J. Pharm. Pract. 2014, 27, 430–439. [Google Scholar] [CrossRef]
- Loo, A.S.; Gerzenshtein, L.; Ison, M.G. Antimicrobial drug-induced thrombocytopenia: A review of the literature. Semin. Thromb. Hemost. 2012, 38, 818–829. [Google Scholar] [CrossRef]
- Berkowitz, S.D.; Sane, D.C.; Sigmon, K.N.; Shavender, J.H.; Harrington, R.A.; Tcheng, J.E.; Topol, E.J.; Califf, R.M. Occurrence and clinical significance of thrombocytopenia in a population undergoing high-risk percutaneous coronary revascularization. Evaluation of c7E3 for the Prevention of Ischemic Complications (EPIC) Study Group. J. Am. Coll. Cardiol. 1998, 32, 311–319. [Google Scholar] [CrossRef] [Green Version]
- Dery, J.P.; Braden, G.A.; Lincoff, A.M.; Kereiakes, D.J.; Browne, K.; Little, T.; George, B.S.; Sane, D.C.; Cines, D.B.; Effron, M.B.; et al. Final results of the ReoPro readministration registry. Am. J. Cardiol. 2004, 93, 979–984. [Google Scholar] [CrossRef]
- Curtis, B.R.; Divgi, A.; Garritty, M.; Aster, R.H. Delayed thrombocytopenia after treatment with abciximab: A distinct clinical entity associated with the immune response to the drug. J. Thromb. Haemost. 2004, 2, 985–992. [Google Scholar] [CrossRef] [Green Version]
- Topol, E.J.; Byzova, T.V.; Plow, E.F. Platelet GPIIb-IIIa blockers. Lancet (London, UK) 1999, 353, 227–231. [Google Scholar] [CrossRef]
- Adachi, J.D.; Bensen, W.G.; Kassam, Y.; Powers, P.J.; Bianchi, F.A.; Cividino, A.; Kean, W.F.; Rooney, P.J.; Craig, G.L.; Buchanan, W.W.; et al. Gold induced thrombocytopenia: 12 cases and a review of the literature. Semin. Arthritis Rheum. 1987, 16, 287–293. [Google Scholar] [CrossRef]
- Von dem Borne, A.E.; Pegels, J.G.; Van der Stadt, R.J.; Van der Plas-van Dalen, C.M.; Helmerhorst, F.M. Thrombocytopenia associated with gold therapy: A drug-induced autoimmune disease? Br. J. Haematol. 1986, 63, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Landrum, E.M.; Siegert, E.A.; Hanlon, J.T.; Currie, M.S. Prolonged thrombocytopenia associated with procainamide in an elderly patient. Ann. Pharmacother. 1994, 28, 1172–1176. [Google Scholar] [CrossRef] [PubMed]
- Giner, V.; Rueda, D.; Salvador, A.; Hernandez, J.C.; Esteban, M.J.; Redon, J. Thrombocytopenia associated with levodopa treatment. Arch. Intern. Med. 2003, 163, 735–736. [Google Scholar] [CrossRef] [PubMed]
- Kumar, J.; Bhargava, M.; Aggarwal, S. Bevacizumab-induced reversible thrombocytopenia in a patient with adenocarcinoma of colon: Rare adverse effect of bevacizumab. Case Rep. Oncol. Med. 2012, 2012, 695430. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, S.; Mashima, H. Infliximab-induced thrombocytopenia in a patient with ulcerative colitis. Int. J. Colorectal Dis. 2016, 31, 921–922. [Google Scholar] [CrossRef]
- Mocciaro, F.; Russo, G.; Di Mitri, R.; Marino, A. Infliximab-induced thrombocytopenia in an elderly patient with ileocolonic Crohn’s disease. Inflamm. Bowel Dis. 2013, 19, E52–E53. [Google Scholar] [CrossRef]
- Yi, J.H.; Kim, S.J.; Ahn, H.K.; Lee, S.J.; Chang, M.H.; Kim, W.S. Rituximab-induced acute thrombocytopenia: A case report and review of the literature. Med. Oncol. (Northwood, London, UK) 2009, 26, 45–48. [Google Scholar] [CrossRef]
- Omura, Y.; Shimazu, H.; Takahashi, T. Rituximab-induced Acute Thrombocytopenia in a Patient with Follicular Lymphoma: A Case Report and Review of the Literature. Intern. Med. (Tokyo, Japan) 2018, 57, 1151–1154. [Google Scholar] [CrossRef] [Green Version]
- Cachia, D.; Izzy, S.; Berriosmorales, I.; Ionete, C. Drug-induced thrombocytopenia secondary to natalizumab treatment. BMJ Case Rep. 2014, 2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiuan, E.; Beckermann, K.E.; Ozgun, A.; Kelly, C.; McKean, M.; McQuade, J.; Thompson, M.A.; Puzanov, I.; Greer, J.P.; Rapisuwon, S.; et al. Thrombocytopenia in patients with melanoma receiving immune checkpoint inhibitor therapy. J. Immunother. Cancer 2017, 5, 8. [Google Scholar] [CrossRef] [Green Version]
- Bakchoul, T.; Zollner, H.; Amiral, J.; Panzer, S.; Selleng, S.; Kohlmann, T.; Brandt, S.; Delcea, M.; Warkentin, T.E.; Sachs, U.J.; et al. Anti-protamine-heparin antibodies: Incidence, clinical relevance, and pathogenesis. Blood 2013, 121, 2821–2827. [Google Scholar] [CrossRef] [Green Version]
- Lee, G.M.; Welsby, I.J.; Phillips-Bute, B.; Ortel, T.L.; Arepally, G.M. High incidence of antibodies to protamine and protamine/heparin complexes in patients undergoing cardiopulmonary bypass. Blood 2013, 121, 2828–2835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pouplard, C.; Leroux, D.; Rollin, J.; Amiral, J.; May, M.A.; Gruel, Y. Incidence of antibodies to protamine sulfate/heparin complexes incardiac surgery patients and impact on platelet activation and clinical outcome. Thromb. Haemost. 2013, 109, 1141–1147. [Google Scholar]
- Leger, R.M.; Arndt, P.A.; Garratty, G. Serological studies of piperacillin antibodies. Transfusion 2008, 48, 2429–2434. [Google Scholar] [CrossRef]
- Mitta, A.; Curtis, B.R.; Reese, J.A.; George, J.N. Drug-induced thrombocytopenia: 2019 Update of clinical and laboratory data. Am. J. Hematol. 2019, 94, 76–78. [Google Scholar]
- Burgess, J.K.; Lopez, J.A.; Berndt, M.C.; Dawes, I.; Chesterman, C.N.; Chong, B.H. Quinine-dependent antibodies bind a restricted set of epitopes on the glycoprotein Ib-IX complex: Characterization of the epitopes. Blood 1998, 92, 2366–2373. [Google Scholar] [CrossRef] [Green Version]
- Peterson, J.A.; Nelson, T.N.; Kanack, A.J.; Aster, R.H. Fine specificity of drug-dependent antibodies reactive with a restricted domain of platelet GPIIIA. Blood 2008, 111, 1234–1239. [Google Scholar] [CrossRef] [Green Version]
- Von Drygalski, A.; Curtis, B.R.; Bougie, D.W.; McFarland, J.G.; Ahl, S.; Limbu, I.; Baker, K.R.; Aster, R.H. Vancomycin-induced immune thrombocytopenia. N. Engl. J. Med. 2007, 356, 904–910. [Google Scholar] [CrossRef] [Green Version]
- Cheah, C.Y.; De Keulenaer, B.; Leahy, M.F. Fluoroquinolone-induced immune thrombocytopenia: A report and review. Intern. Med. J. 2009, 39, 619–623. [Google Scholar] [CrossRef] [PubMed]
- Curtis, B.R.; McFarland, J.G.; Wu, G.G.; Visentin, G.P.; Aster, R.H. Antibodies in sulfonamide-induced immune thrombocytopenia recognize calcium-dependent epitopes on the glycoprotein IIb/IIIa complex. Blood 1994, 84, 176–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garner, S.F.; Campbell, K.; Smith, G.; Hurd, C.; Davidson, S.J.; Treacy, M.; Burman, J.F.; Kroll, H.; Ouwehand, W.H. Teicoplanin-dependent antibodies: Detection and characterization. Br. J. Haematol. 2005, 129, 279–281. [Google Scholar] [CrossRef] [PubMed]
- Pereira, J.; Hidalgo, P.; Ocqueteau, M.; Blacutt, M.; Marchesse, M.; Nien, Y.; Letelier, L.; Mezzano, D. Glycoprotein Ib/IX complex is the target in rifampicin-induced immune thrombocytopenia. Br. J. Haematol. 2000, 110, 907–910. [Google Scholar] [CrossRef]
- Asvadi, P.; Ahmadi, Z.; Chong, B.H. Drug-induced thrombocytopenia: Localization of the binding site of GPIX-specific quinine-dependent antibodies. Blood 2003, 102, 1670–1677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmadi, Z.; Perdomo, J.; Wong, R.; Chong, B.H. Drug-induced immune thrombocytopenia: Mapping of the drug binding site to the membrane-proximal region of platelet GPIX. Platelets 2019, 30, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Zhu, J.; Bougie, D.W.; Aster, R.H.; Springer, T.A. Structural basis for quinine-dependent antibody binding to platelet integrin alphaIIbbeta3. Blood 2015, 126, 2138–2145. [Google Scholar] [CrossRef]
- Bougie, D.W.; Peterson, J.; Rasmussen, M.; Aster, R.H. Mechanism of quinine-dependent monoclonal antibody binding to platelet glycoprotein IIb/IIIa. Blood 2015, 126, 2146–2152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perdomo, J.; Yan, F.; Ahmadi, Z.; Jiang, X.M.; Stocker, R.; Chong, B.H. Quinine-induced thrombocytopenia: Drug-dependent GPIb/IX antibodies inhibit megakaryocyte and proplatelet production in vitro. Blood 2011, 117, 5975–5986. [Google Scholar] [CrossRef] [Green Version]
- Coller, B.S. Platelet GPIIb/IIIa antagonists: The first anti-integrin receptor therapeutics. J. Clin. Invest. 1997, 100, 57–60. [Google Scholar] [CrossRef]
- Lajus, S.; Clofent-Sanchez, G.; Jais, C.; Coste, P.; Nurden, P.; Nurden, A. Thrombocytopenia after abciximab use results from different mechanisms. Thromb. Haemost. 2010, 103, 651–661. [Google Scholar] [PubMed]
- Lown, J.A.; Hughes, A.S.; Cannell, P. Prolonged profound abciximab associated immune thrombocytopenia complicated by transient multispecific platelet antibodies. Heart (British Cardiac Society) 2004, 90, e55. [Google Scholar] [CrossRef]
- Mascelli, M.A.; Lance, E.T.; Damaraju, L.; Wagner, C.L.; Weisman, H.F.; Jordan, R.E. Pharmacodynamic profile of short-term abciximab treatment demonstrates prolonged platelet inhibition with gradual recovery from GP IIb/IIIa receptor blockade. Circulation 1998, 97, 1680–1688. [Google Scholar] [CrossRef] [PubMed]
- Bougie, D.W.; Rasmussen, M.; Zhu, J.; Aster, R.H. Antibodies causing thrombocytopenia in patients treated with RGD-mimetic platelet inhibitors recognize ligand-specific conformers of alphaIIb/beta3 integrin. Blood 2012, 119, 6317–6325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Billheimer, J.T.; Dicker, I.B.; Wynn, R.; Bradley, J.D.; Cromley, D.A.; Godonis, H.E.; Grimminger, L.C.; He, B.; Kieras, C.J.; Pedicord, D.L.; et al. Evidence that thrombocytopenia observed in humans treated with orally bioavailable glycoprotein IIb/IIIa antagonists is immune mediated. Blood 2002, 99, 3540–3546. [Google Scholar] [CrossRef] [Green Version]
- Bougie, D.W.; Wilker, P.R.; Wuitschick, E.D.; Curtis, B.R.; Malik, M.; Levine, S.; Lind, R.N.; Pereira, J.; Aster, R.H. Acute thrombocytopenia after treatment with tirofiban or eptifibatide is associated with antibodies specific for ligand-occupied GPIIb/IIIa. Blood 2002, 100, 2071–2076. [Google Scholar] [CrossRef] [Green Version]
- Greinacher, A.; Fuerll, B.; Zinke, H.; Mullejans, B.; Kruger, W.; Michetti, N.; Motz, W.; Schwertz, H. Megakaryocyte impairment by eptifibatide-induced antibodies causes prolonged thrombocytopenia. Blood 2009, 114, 1250–1253. [Google Scholar] [CrossRef]
- Dezsi, D.A.; Bokori, G.; Falukozy, J.; Bujaky, C.; Fogarassy, G.; Veress, G.; Aradi, D. Eptifibatide-induced thrombocytopenia leading to acute stent thrombosis. J. Thromb. Thrombolysis 2016, 41, 522–524. [Google Scholar] [CrossRef]
- Dunkley, S.; Evans, S.; Gaudry, L.; Jepson, N. Two distinct subgroups of tirofiban-induced thrombocytopenia exist due to drug dependent antibodies that cause platelet activation and increased ischaemic events. Platelets 2005, 16, 462–468. [Google Scholar] [CrossRef]
- Gao, C.; Boylan, B.; Bougie, D.; Gill, J.C.; Birenbaum, J.; Newman, D.K.; Aster, R.H.; Newman, P.J. Eptifibatide-induced thrombocytopenia and thrombosis in humans require FcgammaRIIa and the integrin beta3 cytoplasmic domain. J. Clin. Invest. 2009, 119, 504–511. [Google Scholar] [CrossRef] [Green Version]
- Garner, S.F.; Campbell, K.; Metcalfe, P.; Keidan, J.; Huiskes, E.; Dong, J.F.; Lopez, J.A.; Ouwehand, W.H. Glycoprotein V: The predominant target antigen in gold-induced autoimmune thrombocytopenia. Blood 2002, 100, 344–346. [Google Scholar] [CrossRef]
- Kiorpelidou, D.; Tsiouri, G.; Gaitanis, G.; Akritidis, N.; Bassukas, I.D. Efalizumab-induced thrombocytopenia: Report of relapse after re-administration. Clin. Exp. Dermatol. 2009, 34, e914–e916. [Google Scholar] [CrossRef] [PubMed]
- Warkentin, T.E.; Kwon, P. Immune thrombocytopenia associated with efalizumab therapy for psoriasis. Ann. Intern. Med. 2005, 143, 761–763. [Google Scholar] [CrossRef] [Green Version]
- Casanova, M.J.; Chaparro, M.; Martinez, S.; Vicuna, I.; Gisbert, J.P. Severe adalimumab-induced thrombocytopenia in a patient with Crohn’s disease. J. Crohns Colitis 2012, 6, 1034–1037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dior, M.; Coriat, R.; Mir, O.; Brezault, C.; Perkins, G.; Dhooge, M.; Goldwasser, F.; Chaussade, S. A rare hematological adverse event induced by bevacizumab: Severe thrombocytopenia. Am. J. Med. 2012, 125, 828–830. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Lewis, M.; Corrie, P.; Iddawela, M. Ipilimumab-induced thrombocytopenia in a patient with metastatic melanoma. J. Oncol. Pharm. Pract. 2012, 18, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Brandt, S.; Krauel, K.; Gottschalk, K.E.; Renné, T.; Helm, C.A.; Greinacher, A.; Block, S. Characterisation of the conformational changes in platelet factor 4 induced by polyanions: Towards in vitro prediction of antigenicity. Thromb. Haemost. 2014, 112, 53–64. [Google Scholar] [CrossRef]
- Greinacher, A.; Gopinadhan, M.; Gunther, J.U.; Omer-Adam, M.A.; Strobel, U.; Warkentin, T.E.; Papastavrou, G.; Weitschies, W.; Helm, C.A. Close approximation of two platelet factor 4 tetramers by charge neutralization forms the antigens recognized by HIT antibodies. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2386–2393. [Google Scholar] [CrossRef] [Green Version]
- Greinacher, A.; Alban, S.; Dummel, V.; Franz, G.; Mueller-Eckhardt, C. Characterization of the structural requirements for a carbohydrate-based anticoagulant with a reduced risk of inducing the immunological type of heparin-associated thrombocytopenia. Thromb. Haemost. 1995, 74, 886–892. [Google Scholar] [CrossRef]
- Selleng, K.; Schutt, A.; Selleng, S.; Warkentin, T.E.; Greinacher, A. Studies of the anti-platelet factor 4/heparin immune response: Adapting the enzyme-linked immunosorbent spot assay for detection of memory B cells against complex antigens. Transfusion 2010, 50, 32–39. [Google Scholar] [CrossRef]
- Greinacher, A.; Kohlmann, T.; Strobel, U.; Sheppard, J.A.; Warkentin, T.E. The temporal profile of the anti-PF4/heparin immune response. Blood 2009, 113, 4970–4976. [Google Scholar] [CrossRef] [PubMed]
- Krauel, K.; Potschke, C.; Weber, C.; Kessler, W.; Furll, B.; Ittermann, T.; Maier, S.; Hammerschmidt, S.; Broker, B.M.; Greinacher, A. Platelet factor 4 binds to bacteria, [corrected] inducing antibodies cross-reacting with the major antigen in heparin-induced thrombocytopenia. Blood 2011, 117, 1370–1378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nazy, I.; Clare, R.; Staibano, P.; Warkentin, T.E.; Larche, M.; Moore, J.C.; Smith, J.W.; Whitlock, R.P.; Kelton, J.G.; Arnold, D.M. Cellular immune responses to platelet factor 4 and heparin complexes in patients with heparin-induced thrombocytopenia. J. Thromb. Haemost. 2018, 16, 1402–1412. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Yu, M.; Podd, A.; Yuan, L.; Newman, D.K.; Wen, R.; Arepally, G.; Wang, D. Critical role for mouse marginal zone B cells in PF4/heparin antibody production. Blood 2013, 121, 3484–3492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.; Yu, M.; Padmanabhan, A.; Aster, R.H.; Yuan, L.; Wen, R.; Wang, D. Critical role of CD4 T cells in PF4/heparin antibody production in mice. Blood 2015, 125, 1826–1829. [Google Scholar] [CrossRef] [Green Version]
- Khandelwal, S.; Ravi, J.; Rauova, L.; Johnson, A.; Lee, G.M.; Gilner, J.B.; Gunti, S.; Notkins, A.L.; Kuchibhatla, M.; Frank, M.; et al. Polyreactive IgM initiates complement activation by PF4/heparin complexes through the classical pathway. Blood 2018, 132, 2431–2440. [Google Scholar] [CrossRef] [Green Version]
- Greinacher, A.; Alban, S.; Omer-Adam, M.A.; Weitschies, W.; Warkentin, T.E. Heparin-induced thrombocytopenia: A stoichiometry-based model to explain the differing immunogenicities of unfractionated heparin, low-molecular-weight heparin, and fondaparinux in different clinical settings. Thromb. Res. 2008, 122, 211–220. [Google Scholar] [CrossRef]
- Visentin, G.; Ford, S.; Scott, J.; Aster, R. Antibodies from patients with heparin-induced thrombocytopenia/thrombosis are specific for platelet factor 4 complexed with heparin or bound to endothelial cells. J. Clin. Invest. 1994, 93, 81–88. [Google Scholar] [CrossRef]
- Pouplard, C.; Amiral, J.; Borg, J.Y.; Vissac, A.M.; Delahousse, B.; Gruel, Y. Differences in specificity of heparin-dependent antibodies developed in heparin-induced thrombocytopenia and consequences on cross-reactivity with danaparoid sodium. Br. J. Haematol. 1997, 99, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.H.; Medvedev, N.; Delcea, M.; Greinacher, A. Anti-platelet factor 4/polyanion antibodies mediate a new mechanism of autoimmunity. Nat. Commun. 2017, 8, 14945. [Google Scholar] [CrossRef] [PubMed]
- Greinacher, A.; Selleng, K.; Warkentin, T.E. Autoimmune heparin-induced thrombocytopenia. J. Thromb. Haemost. 2017, 15, 2099–2114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arepally, G.M. Heparin-induced thrombocytopenia. Blood 2017, 129, 2864–2872. [Google Scholar] [CrossRef] [PubMed]
- Rollin, J.; Pouplard, C.; Gruel, Y. Risk factors for heparin-induced thrombocytopenia: Focus on Fcgamma receptors. Thromb. Haemost. 2016, 116, 799–805. [Google Scholar] [CrossRef]
- Warkentin, T.; Hayward, C.; Boshkov, L.; Santos, A.; Sheppard, J.; Bode, A.; Kelton, J. Sera from patients with heparin-induced thrombocytopenia generate platelet-derived microparticles with procoagulant activity: An explanation for the thrombotic complications of heparin-induced thrombocytopenia. Blood 1994, 84, 3691–3699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, M.; Hayward, C.P.; Warkentin, T.E.; Horsewood, P.; Chorneyko, K.A.; Kelton, J.G. Morphological analysis of microparticle generation in heparin-induced thrombocytopenia. Blood 2000, 96, 188–194. [Google Scholar] [CrossRef]
- Pouplard, C.; Iochmann, S.; Renard, B.; Herault, O.; Colombat, P.; Amiral, J.; Gruel, Y. Induction of monocyte tissue factor expression by antibodies to heparin- platelet factor 4 complexes developed in heparin-induced thrombocytopenia. Blood 2001, 97, 3300–3302. [Google Scholar] [CrossRef] [Green Version]
- Tutwiler, V.; Madeeva, D.; Ahn, H.S.; Andrianova, I.; Hayes, V.; Zheng, X.L.; Cines, D.B.; McKenzie, S.E.; Poncz, M.; Rauova, L. Platelet transactivation by monocytes promotes thrombosis in heparin-induced thrombocytopenia. Blood 2016, 127, 464–472. [Google Scholar] [CrossRef] [Green Version]
- Gollomp, K.; Kim, M.; Johnston, I.; Hayes, V.; Welsh, J.; Arepally, G.M.; Kahn, M.; Lambert, M.P.; Cuker, A.; Cines, D.B.; et al. Neutrophil accumulation and NET release contribute to thrombosis in HIT. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Perdomo, J.; Leung, H.H.L.; Ahmadi, Z.; Yan, F.; Chong, J.J.H.; Passam, F.H.; Chong, B.H. Neutrophil activation and NETosis are the major drivers of thrombosis in heparin-induced thrombocytopenia. Nat. Commun. 2019, 10, 1322. [Google Scholar] [CrossRef] [Green Version]
- Johnston, I.; Sarkar, A.; Hayes, V.; Koma, G.T.; Arepally, G.M.; Chen, J.; Chung, D.W.; López, J.A.; Cines, D.B.; Rauova, L.; et al. Recognition of PF4-VWF complexes by heparin-induced thrombocytopenia antibodies contributes to thrombus propagation. Blood 2020, 135, 1270–1280. [Google Scholar] [CrossRef]
- Bakchoul, T.; Jouni, R.; Warkentin, T.E. Protamine (heparin)-induced thrombocytopenia: A review of the serological and clinical features associated with anti-protamine/heparin antibodies. J. Thromb. Haemost. 2016, 14, 1685–1695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shulman, N.R. Immunoreactions involving platelets. IV. Studies on the pathogenesis of thrombocytopenia in drug purpura using test doses of quinidine in sensitized individuals; their implications in idiopathic thrombocytopenic purpura. J. Exp. Med. 1958, 107, 711–729. [Google Scholar] [CrossRef] [PubMed]
- Kiefel, V.; Salama, A.; Mueller-Eckhardt, C. In vitro fixation of C3d and C5b-9 on platelets by human platelet reactive antibodies. Blut 1989, 58, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Kenney, B.; Tormey, C.A. Acute vancomycin-dependent immune thrombocytopenia as an anamnestic response. Platelets 2008, 19, 379–383. [Google Scholar] [CrossRef] [PubMed]
- Gales, B.J.; Sulak, L.B. Severe thrombocytopenia associated with alatrofloxacin. Ann. Pharmacother. 2000, 34, 330–334. [Google Scholar] [CrossRef] [PubMed]
- Bruhns, P. Properties of mouse and human IgG receptors and their contribution to disease models. Blood 2012, 119, 5640–5649. [Google Scholar] [CrossRef] [PubMed]
- Rollin, J.; Pouplard, C.; Sung, H.C.; Leroux, D.; Saada, A.; Gouilleux-Gruart, V.; Thibault, G.; Gruel, Y. Increased risk of thrombosis in FcgammaRIIA 131RR patients with HIT due to defective control of platelet activation by plasma IgG2. Blood 2015, 125, 2397–2404. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, L.E.; Santoso, S.; Baurichter, G.; Kroll, H.; Papenberg, S.; Eichler, P.; Westerdaal, N.A.; Kiefel, V.; Van de Winkel, J.G.; Greinacher, A. Heparin-induced thrombocytopenia: New insights into the impact of the FcgammaRIIa-R-H131 polymorphism. Blood 1998, 92, 1526–1531. [Google Scholar] [CrossRef]
- Gruel, Y.; Vayne, C.; Rollin, J.; Weber, P.; Faille, D.; Bauters, A.; Macchi, L.; Ahlenc-Gelas, M.; Lebreton, A.; de Maistre, E.; et al. Comparative analysis of a French prospective series of 144 patients with heparin-induced thrombocytopenia (FRIGTIH) and the literature. Thromb. Haemost. 2020, in press. [Google Scholar] [CrossRef]
- McKenzie, S.E.; Taylor, S.M.; Malladi, P.; Yuhan, H.; Cassel, D.L.; Chien, P.; Schwartz, E.; Schreiber, A.D.; Surrey, S.; Reilly, M.P. The role of the human Fc receptor Fc gamma RIIA in the immune clearance of platelets: A transgenic mouse model. J. Immunol. 1999, 162, 4311–4318. [Google Scholar]
- Arepally, G.; McKenzie, S.E.; Jiang, X.M.; Poncz, M.; Cines, D.B. Fc gamma RIIA H/R(131) polymorphism, subclass-specific IgG anti-heparin/platelet factor 4 antibodies and clinical course in patients with heparin-induced thrombocytopenia and thrombosis. Blood 1997, 89, 370–375. [Google Scholar] [CrossRef] [PubMed]
- Gruel, Y.; Pouplard, C.; Lasne, D.; Magdelaine-Beuzelin, C.; Charroing, C.; Watier, H. The homozygous FcgammaRIIIa-158V genotype is a risk factor for heparin-induced thrombocytopenia in patients with antibodies to heparin-platelet factor 4 complexes. Blood 2004, 104, 2791–2793. [Google Scholar] [CrossRef] [PubMed]
- Nagelkerke, S.Q.; Kuijpers, T.W. Immunomodulation by IVIg and the Role of Fc-Gamma Receptors: Classic Mechanisms of Action after all? Front. Immunol. 2014, 5, 674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, S.X.; Pinkevych, M.; Khachigian, L.M.; Parish, C.R.; Davenport, M.P.; Chong, B.H. Drug-induced thrombocytopenia: Development of a novel NOD/SCID mouse model to evaluate clearance of circulating platelets by drug-dependent antibodies and the efficacy of IVIG. Blood 2010, 116, 1958–1960. [Google Scholar] [CrossRef] [Green Version]
- Bose, S.; Wurm, E.; Popovich, M.J.; Silver, B.J. Drug-induced immune-mediated thrombocytopenia in the intensive care unit. J. Clin. Anesth. 2015, 27, 602–605. [Google Scholar] [CrossRef]
- Warkentin, T.E. High-dose intravenous immunoglobulin for the treatment and prevention of heparin-induced thrombocytopenia: A review. Expert Rev. Hematol. 2019, 12, 685–698. [Google Scholar] [CrossRef]
- Andres, E.; Dali-Youcef, N.; Serraj, K.; Zimmer, J. Recognition and management of drug-induced cytopenias: The example of idiosyncratic drug-induced thrombocytopenia. Expert Opin. Drug Saf. 2009, 8, 183–190. [Google Scholar] [CrossRef]
- Chang, M.; Nakagawa, P.A.; Williams, S.A.; Schwartz, M.R.; Imfeld, K.L.; Buzby, J.S.; Nugent, D.J. Immune thrombocytopenic purpura (ITP) plasma and purified ITP monoclonal autoantibodies inhibit megakaryocytopoiesis in vitro. Blood 2003, 102, 887–895. [Google Scholar] [CrossRef]
- McMillan, R.; Wang, L.; Tomer, A.; Nichol, J.; Pistillo, J. Suppression of in vitro megakaryocyte production by antiplatelet autoantibodies from adult patients with chronic ITP. Blood 2004, 103, 1364–1369. [Google Scholar] [CrossRef] [Green Version]
- Perdomo, J.; Yan, F.; Chong, B.H. A megakaryocyte with no platelets: Anti-platelet antibodies, apoptosis, and platelet production. Platelets 2013, 24, 98–106. [Google Scholar] [CrossRef]
- Bakchoul, T.; Marini, I. Drug-associated thrombocytopenia. Hematol. Am. Soc. Hematol. Educ. Progr. 2018, 2018, 576–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samaranayake, C.B.; Yap, E. Fatal quinine-induced thrombocytopenia from pulmonary haemorrhage. Intern. Med. J. 2014, 44, 423–425. [Google Scholar] [CrossRef] [PubMed]
- Reddy, J.C.; Shuman, M.A.; Aster, R.H. Quinine/quinidine-induced thrombocytopenia: A great imitator. Arch. Intern. Med. 2004, 164, 218–220. [Google Scholar] [CrossRef] [PubMed]
- Arnold, D.M.; Curtis, B.R.; Bakchoul, T. Recommendations for standardization of laboratory testing for drug-induced immune thrombocytopenia: Communication from the SSC of the ISTH. J. Thromb. Haemost. 2015, 13, 676–678. [Google Scholar] [CrossRef] [Green Version]
- Tardy, B.; Lecompte, T.; Mullier, F.; Vayne, C.; Pouplard, C. Detection of Platelet-Activating Antibodies Associated with Heparin-Induced Thrombocytopenia. J. Clin. Med. 2020, 9, 1226. [Google Scholar] [CrossRef]
- Cuker, A.; Arepally, G.M.; Chong, B.H.; Cines, D.B.; Greinacher, A.; Gruel, Y.; Linkins, L.A.; Rodner, S.B.; Selleng, S.; Warkentin, T.E.; et al. American Society of Hematology 2018. Guidelines for management of venous thromboembolism: Heparin-Induced Thrombocytopenia. Blood Adv. 2018, 2, 3360–3392. [Google Scholar] [CrossRef] [Green Version]
- Nagler, M.; Bachmann, L.M.; ten Cate, H.; ten Cate-Hoek, A. Diagnostic value of immunoassays for heparin-induced thrombocytopenia: A systematic review and meta-analysis. Blood 2016, 127, 546–557. [Google Scholar] [CrossRef] [Green Version]
- Warkentin, T.E.; Greinacher, A.; Gruel, Y.; Aster, R.H.; Chong, B.H. Laboratory testing for heparin-induced thrombocytopenia: A conceptual framework and implications for diagnosis. J. Thromb. Haemost. 2011, 9, 2498–2500. [Google Scholar] [CrossRef]
- Minet, V.; Dogne, J.M.; Mullier, F. Functional Assays in the Diagnosis of Heparin-Induced Thrombocytopenia: A Review. Molecules 2017, 22, 617. [Google Scholar] [CrossRef] [Green Version]
- Vayne, C.; Guery, E.A.; Charuel, N.; Besombes, J.; Lambert, W.C.; Rollin, J.; Gruel, Y.; Pouplard, C. Evaluation of functional assays for the diagnosis of heparin induced thrombocytopenia using 5B9, a monoclonal IgG that mimics human antibodies. J. Thromb. Haemost. 2020, 18, 968–975. [Google Scholar] [CrossRef]
- Gruel, Y.; Pouplard, C. Post-operative platelet count profile: The most reliable tool for identifying patients with true heparin-induced thrombocytopenia after cardiac surgery. J. Thromb. Haemost. 2010, 8, 27–29. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Type | Mechanism | Examples | References |
|---|---|---|---|
| Hapten-induced antibody | Drug binds to platelet membrane and promotes antibody response | Penicillin and derivatives, cephalosporins | [13,14,15,16] |
| “Quinine-type” antibody | Drug binds to antibody Fab and/or membrane glycoprotein (GP), thereby enhancing antibody affinity and binding to platelet GP | Quinidine, quinine, antibiotics (vancomycin, rifampicin, sulfamethoxazol), anticonvulsants | [3,4,5,17,18] |
| Drug-specific antibody | Antibody recognizes the monoclonal antibody bound to its target | abciximab | [19,20,21] |
| Fibrinogen receptor antagonist-dependent antibody | Drug binds to GPIIb/IIIa inducing conformational changes, then recognized by antibody | tirofiban, eptifibatide | [22] |
| Autoantibody induction | Drug induces formation of autoantibody that binds alone to platelet GP | procainamide, gold salts, L-dopa, and likely several therapeutic monoclonal antibodies | [23,24,25,26,27,28,29,30,31,32,33] |
| Immune complexes | Drug binds to PF4 inducing antibodies that activate platelets via FcγRIIa receptors | heparin, protamine | [11,12,34,35,36] |
| HIT | Other DITPs | |
|---|---|---|
| frequency | frequent | rare |
| main mechanism of thrombocytopenia | activation | consumption/destruction |
| contribution of other cell types | yes: leukocytes, endothelial cells | no |
| time to occurrence after drug initiation | mostly: 5–10 days | few hours to few days |
| depth of thrombocytopenia | moderate: nadir close to 50 × 109/L in most cases | severe: nadir < 10–20 × 109/L in most cases |
| clinical manifestations | thrombosis in 30-50% of cases; bleeding in < 10% of patients, in case of DIC | bleeding |
| diagnosis | affordable: well-established diagnostic approach, first-line tests: immunoassays, confirmation tests: functional assays | difficult: few assays available (immunoassays or flow cytometry-based assays) of unknown sensitivity, and restricted to specialized laboratories |
| recurrence on re-exposure to the drug | not systematically | very likely |
| I. Sample Collection |
|---|
| Timing: - preferentially during the acute episode of thrombocytopenia - at least on a sample collected up to 3 weeks after the acute event Anticoagulant: - clotted serum or citrated plasma; avoid EDTA |
| II. Preparation of Test Platelets |
| Use fresh platelets from healthy donors or stored platelets (0.1% sodium azide). - collect blood in citrate-containing tubes using a 21-gauge needle (avoid vacuum suction), from donors with blood group O and known to express the HPA-1a antigen - centrifuge whole blood for obtaining PRP (200× g, 10 min) - wash platelets twice with phosphate-buffered saline containing BSA 0.1%. |
| III. Test Methods |
| Drug preparation: Dissolve each drug to be tested in adequate solution, according to its solubility. The suspected drug should be tested at therapeutic concentration (i.e., 0.3 mg/mL for vancomycin) Flow cytometry and Enzyme immunoassays (EIAs) can be used for detecting DDabs. In both assays, healthy donor platelets are incubated with patient serum or plasma in the presence and absence of drug. When required, the drug must be present in all buffers and during all steps, including washings. After washings, platelet-associated DDabs will be detected using fluorescent-labeled anti-human IgG and IgM (flow cytometry), or using an enzyme-labeled goat anti-Human IgG and IgM (EIA). |
| IV. Patient Samples and Controls |
| Patient samples and negative/positive controls (usually serum) must always be tested. Positive control: serum or plasma sample from one previous patient with DITP, or with anti-HPA1 antibody (WHO standard 106/05 or patient sample), but test HPA1 positive platelets. Negative control: serum or plasma sample from patient treated with the drug and normal platelet count, or from healthy control. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vayne, C.; Guéry, E.-A.; Rollin, J.; Baglo, T.; Petermann, R.; Gruel, Y. Pathophysiology and Diagnosis of Drug-Induced Immune Thrombocytopenia. J. Clin. Med. 2020, 9, 2212. https://doi.org/10.3390/jcm9072212
Vayne C, Guéry E-A, Rollin J, Baglo T, Petermann R, Gruel Y. Pathophysiology and Diagnosis of Drug-Induced Immune Thrombocytopenia. Journal of Clinical Medicine. 2020; 9(7):2212. https://doi.org/10.3390/jcm9072212
Chicago/Turabian StyleVayne, Caroline, Eve-Anne Guéry, Jérôme Rollin, Tatiana Baglo, Rachel Petermann, and Yves Gruel. 2020. "Pathophysiology and Diagnosis of Drug-Induced Immune Thrombocytopenia" Journal of Clinical Medicine 9, no. 7: 2212. https://doi.org/10.3390/jcm9072212
APA StyleVayne, C., Guéry, E. -A., Rollin, J., Baglo, T., Petermann, R., & Gruel, Y. (2020). Pathophysiology and Diagnosis of Drug-Induced Immune Thrombocytopenia. Journal of Clinical Medicine, 9(7), 2212. https://doi.org/10.3390/jcm9072212