Current State of Knowledge on Primary Sjögren’s Syndrome, an Autoimmune Exocrinopathy



Abstract
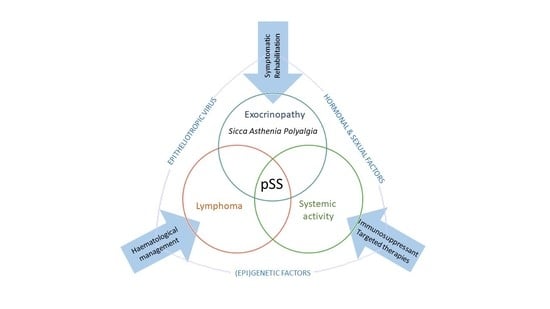
:
1. Introduction
2. Epidemiology
2.1. Prevalence
2.2. Incidence
3. Physiopathology of Sjögren’s Syndrome
3.1. Trigger Phase
3.1.1. Environmental Factors
3.1.2. Genetic Predisposition
3.1.3. Epigenetic Factors
3.1.4. Sex Hormones Deregulation and X-Chromosome Linked Factors
3.2. SGEC Deregulation
3.2.1. Upregulation of Adhesion Molecules
3.2.2. Antigen-Presenting Cell Properties
3.2.3. Chemokines Production
3.2.4. Apoptosis and Expression of Self-Antigens
3.2.5. Alteration of Proteins Involved in Saliva Secretion
3.3. Chronic Inflammation
3.3.1. T-Cell Infiltration
3.3.2. Breakdown of B Cells Tolerance
3.3.3. Formation of Germinal-Like Structures
3.3.4. Local Production of Autoantibodies
3.3.5. Damage of Salivary Acini Architecture
4. Clinical Manifestations
4.1. General Manifestations
4.2. Ocular Manifestations
4.3. Stomatologic Manifestations
4.4. Musculoskeletal Manifestations
4.5. Neurological Manifestations
4.6. Pulmonary Manifestations
4.7. Dermatological Manifestations
4.8. Cardiovascular Manifestations
4.9. Oeso-Gastrointestinal Manifestations
4.10. Pancreatic and Hepatobiliary Manifestations
4.11. Uronephrologic Manifestations
4.12. Haematological Manifestations
4.13. Ear–Nose–Throat (ENT) Manifestations
4.14. Gynaecological and Obstetrical Manifestations
5. Diagnosis Workup
5.1. Diagnosis Versus Classification Criteria
5.2. Sicca Syndrome and Glandular Assessment
5.3. Labial Minor SG Biopsy
5.4. Antinuclear Antibodies (ANA) Profile
5.5. Blood Workup
5.6. Sjögren’s Syndrome Differential Diagnosis
5.7. Primary versus Secondary Sjögren’s Syndrome
6. Prognosis
6.1. Death
6.2. Disease Activity
6.3. Damage Accrual
6.4. Discomfort and Disability
7. Therapeutic
7.1. Sicca Syndrome and Non-Visceral Manifestations
7.2. Systemic Manifestations
7.3. pSS-Associated Lymphoma
7.4. Obstetrical Considerations
7.5. Targeted Therapies: Revolution or Disillusion?
8. Conclusions
- SS is characterized by lymphoplasmacytic infiltration of exocrine glands. The cause of SS is complex and influenced by a combination of genetic, epigenetic, hormonal and environmental factors.
- The pathogenic mechanisms remain unclear. However, the immune system-mediated loss of glands function, specifically of salivary and lacrimal glands, certainly explains the common symptoms of dry mouth and dry eyes. In this inflammatory environment, T-cells mediate a direct destruction of glandular tissue and B-cell activation, leading to the production of autoantibodies. More than 20 autoantibodies could be involved in SS, but the most commonly used for SS diagnosis are anti-Ro/SSA and anti-La/SSB.
- Although often reduced to its sicca syndrome due to its tropism for glandular tissue, pSS remains a systemic disease that can affect virtually all organs. These clinical manifestations can be due to various mechanisms: dryness secondary to exocrinopathy, autoimmune epithelitis with periepithelial lymphocytic infiltration of target organs, autoimmunity and clonal lymphocytic expansion.
- Due to its protean and willingly insidious presentation, pSS is sometimes difficult to recognize and may delay diagnosis by more than 10 years. Classification criteria are used to create cohorts for study purposes and should not be used blindly as diagnostic criteria but as a guide in clinical practice. For these various reasons, the gold standard for individual diagnosis of pSS remains the opinion of an expert clinician.
- From a serohistological point of view, so-called “secondary Sjögren’s syndrome” in SLE and SScl patients does not differ from pSS. It is therefore preferable to forget this historical dichotomy. In this way, the clinician avoids three pitfalls: (1) minimizing the SS-related symptoms, which decrease the quality of life of the patients; (2) forget that overlap may change the clinical phenotype and (3) forget the risk of lymphoma.
- Although overall pSS mortality is low and similar to the general population, a subgroup of patients will have a poorer vital prognosis linked to cardiovascular events, solid-organ and lymphoid malignancies and infections. Biomarkers associated with the development of MALT lymphoma are mainly signs associated with exuberant B cell proliferation and immune-complex production.
- The impact of pSS can be assessed according to three clinical dimensions: “sicca asthenia polyalgia” complex, inflammatory disease activity and structural damage. They are assessed by the ESSPRI, ESSDAI and SSD(D)I scores, respectively. Even in the absence of florid systemic manifestations, pSS can be disabling and associated with significant functional status impairment related to oral and/or ocular dryness, systemic activity, pain, fatigue and daytime somnolence, anxiety and depression symptoms.
- The treatment of manifestations linked to the “sicca asthenia polyalgia” complex mainly involves symptomatic measures and rehabilitation. To date, no immunosuppressant has demonstrated a favourable risk–benefit balance in this indication.
- The treatment of manifestations related to inflammatory disease activity is currently based on scarce evidence. Therapeutic regimen must be tailored to organ specific involvement and severity of the disorder. Mild manifestations will be treated with hydroxychloroquine or local corticosteroids while moderate to severe systemic involvement will require the use of systemic corticosteroid therapy, combined or not with a broad-spectrum immunosuppressant. Rituximab will only be used as a third line, except in cases of cryoglobulinemia where it is the treatment of choice.
- Despite targeted therapies having revolutionized rheumatology in recent years and the impressive number of molecules tested so far in pSS, a revolution like the one known in the field of RA has not yet occurred.
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACA | Anti-centromere antibodies |
| ACPA | Anti-citrullinated protein antibodies |
| ACR | American College of Rheumatology |
| AECG | American European Consensus Group |
| AH | Autoimmune Hepatitis |
| ANA | Antinuclear antibodies |
| anti-M3R | Anti-muscarinic receptor 3 |
| APRIL | A proliferation-inducing ligand |
| ASAP | “Abatacept Sjögren Active Patients” study |
| AZA | Azathioprine |
| BAFF | B cell Activating Factor |
| BCR | B cell receptor |
| BUT | Break-up Time |
| CCP | Cyclic Citrullinated Peptide |
| circRNA | Circular RNA |
| ciRNAs | Intronic circRNAs |
| ClinESSDAI | Clinical ESSDAI variant |
| CPK | Creatine phosphokinase |
| CRISP-3 | Cysteine-Rich Secretory Protein 3 () |
| CT-scan | Computerized tomography |
| CyA | Ciclosporin A |
| DAMPS | Danger-associated molecular patterns |
| DAP-kinase | Pro-apoptotic death associated protein kinase |
| DHEA | Dehydroepiandrosterone |
| DHT | Dihydrotestosterone |
| DLBCL | Diffuse large B cell lymphoma |
| DMARD | Disease Modifying Anti-Rheumatic Drug |
| DNMTs | DNA methyltransferases |
| DREAM | “Dry Eye Assessment and Management” study |
| EBV | Epstein-Barr virus |
| ecircRNAs | Exonic circRNAs |
| EIciRNAs | Exon-intron circRNAs |
| ELISA | Enzyme-linked immunosorbent assay |
| ENT | Ear-Nose-Throat |
| ESSDAI | EULAR Sjögren’s syndrome disease activity index |
| ESSPRI | EULAR Sjögren’s Syndrome Patient Reported Index |
| EULAR | European League Against Rheumatism |
| FASl | Fas ligand |
| FDC | Follicular dendritic cells |
| GCs | Germinal centres |
| HCQ | Hydroxychloroquine |
| HCV | Hepatitis C virus |
| HLH | Hemophagocytic lymphohistiocytosis |
| HTLV1 | Human T-lymphotropic virus type I |
| ICAM-1 | InterCellular Adhesion Molecule 1 |
| IF | Immunofluorescence |
| IFN | Interferon |
| IgG,A,M | Immunoglobulin G, A and M |
| IL- | Interleukin |
| ILD | Interstitial lung disease(s) |
| IRF | Interferon Regulatory Factor |
| IV | Intravenous therapy |
| IVIG | Intravenous Immunoglobulin |
| KCS | Keratoconjunctivitis sicca |
| LEMA | Myoepithelial sialadenitis |
| LESA | lymphoepithelial sialadenitis |
| LIP | Lymphocytic interstitial pneumonitis |
| LMP1 | Latent membrane protein 1 |
| lncRNA | Long non-coding RNAs |
| LPR | Laryngopharyngeal reflux |
| LSG | Labial SG |
| MALT | mucosa-associated lymphoid tissue |
| MHC | Major histocompatibility genes |
| MMF | Mycophenolate mofetil |
| MMP | Matrix metalloproteinases |
| MPGN | Mesangioproliferative glomerulonephritis |
| MRI | Magnetic Resonance Imaging |
| MS | Multiple Sclerosis |
| MSGB | Minor salivary gland biopsy |
| MTX | Methotrexate |
| NAC | N-acetylcystein |
| NFkB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NHL | Non-Hodgkin’s lymphoma |
| NICE | National Institute for Health and Care Excellence |
| NMOSD | Neuromyelitis optica spectrum disorder |
| NOD | Non-obese diabetic |
| NSAID | Nonsteroidal anti-inflammatory drugs |
| NSIP | Nonspecific interstitial pneumonia |
| OMERACT | Outcome Measures in Rheumatology group |
| OSDI | Ocular Surface Disease Index |
| OSS | Ocular Staining Score |
| PAMPs | Pathogen-associated molecular patterns |
| PBC | Primary Biliary Cirrhosis |
| PDC | Plasmacytoid dendritic cells |
| PDL1 | Programmed death ligand 1 |
| PET scan | Positron emission tomography |
| PGA | Patient Global Assessment |
| PIP | Prolactin inducible protein |
| PO | per os |
| PSP | Parotid secretory protein |
| pSS | Primary Sjögren’s Syndrome |
| pSS-ILD | pSS-related interstitial lung disease |
| q6h, q8h | Every 6 h, every 8 h |
| RA | Rheumatoid Arthritis |
| RCT | Randomized controlled trial |
| RF | Rheumatoid Factor |
| RTA | Renal tubular acidosis |
| RTX | Rituximab |
| RX1 | Runt-related transcription factor |
| SAM | Methyl donor S-adenosylmethionine |
| SAP | Sicca Asthenia Polyalgia |
| SF-36 | Short Form 36 health survey |
| SG | Salivary Gland |
| SGS | Salivary glands scintigraphy |
| SGUS | Salivary glands ultrasound |
| SICCA | Sjögren’s International Collaborative Clinical Alliance |
| SLE | Systemic lupus erythematosus |
| SNP | Single nucleotide polymorphism |
| SP-1 | Salivary protein 1 |
| SSDDI | Sjögren’s Syndrome Disease Damage Index |
| SSDI | Sjögren’s Syndrome Damage Index |
| sSS | Secondary Sjögren’s Syndrome |
| SWSF | Stimulated Whole Salivary Flow rate |
| TACI | Transmembrane Activator and CAML Interactor |
| TEARS | “Tolerance and efficacy of rituximab in primary Sjögren syndrome” trial |
| Tfh | Follicular helper T cells |
| TLRs | Toll Like Receptors |
| TNF-α | Tumour necrosis factor-α |
| TPHA | Treponema Pallidum Hemagglutinations Assay |
| TRACTISS | “TRial of Anti-B-Cell Therapy In patients with primary Sjögren’s Syndrome” trial |
| TSH | Thyroid-stimulating hormone |
| TTP | Thrombotic Thrombocytopenic Purpura |
| UCLH | University College London Hospitals |
| UIP | Usual interstitial pneumonia |
| UWSF | Unstimulated Whole Saliva Flow rate |
| VAS | Visual analogue scales |
| VDRL | Venereal Disease Research Laboratory |
References
- Fox, R.I. Sjögren’s syndrome. Lancet 2005, 366, 321–331. [Google Scholar] [CrossRef]
- Gerli, R.; Bartoloni, E.; Alunno, A. (Eds.) Sjögren’s Syndrome: Novel Insights in Pathogenic, Clinical, and Therapeutic Aspects; Elsevier: Amsterdam, The Netherlands; Academic Press: Cambridge, MA, USA, 2016; ISBN 978-0-12-803604-4. [Google Scholar]
- Ghafoor, M. Sjögren’s Before Sjögren: Did Henrik Sjögren (1899–1986) Really Discover Sjögren’s Disease? J. Maxillofac. Oral Surg. 2012, 11, 373–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murube, J. Henrik Sjögren, 1899–1986. Ocul. Surf. 2010, 8, 2–7. [Google Scholar] [CrossRef]
- Wollheim, F.A. Henrik Sjögren and Sjögren’s syndrome. Scand. J. Rheumatol. Suppl. 1986, 61, 11–16. [Google Scholar]
- Binard, A.; Devauchelle-Pensec, V.; Fautrel, B.; Jousse, S.; Youinou, P.; Saraux, A. Epidemiology of Sjögren’s syndrome: Where are we now? Clin. Exp. Rheumatol. 2007, 25, 1–4. [Google Scholar]
- Mavragani, C.P.; Moutsopoulos, H.M. The geoepidemiology of Sjögren’s syndrome. Autoimmun. Rev. 2010, 9, A305–A310. [Google Scholar] [CrossRef]
- Qin, B.; Wang, J.; Yang, Z.; Yang, M.; Ma, N.; Huang, F.; Zhong, R. Epidemiology of primary Sjögren’s syndrome: A systematic review and meta-analysis. Ann. Rheum. Dis. 2015, 74, 1983–1989. [Google Scholar] [CrossRef]
- Delaleu, N.; Jonsson, M.V.; Appel, S.; Jonsson, R. New concepts in the pathogenesis of Sjögren’s syndrome. Rheum. Dis. Clin. N. Am. 2008, 34, 833–845. [Google Scholar] [CrossRef]
- Konttinen, Y.T.; Käsnä-Ronkainen, L. Sjögren’s syndrome: Viewpoint on pathogenesis. One of the reasons I was never asked to write a textbook chapter on it. Scand. J. Rheumatol. Suppl. 2002, 116, 15–22. [Google Scholar] [CrossRef]
- Mitsias, D.I.; Kapsogeorgou, E.K.; Moutsopoulos, H.M. Sjögren’s syndrome: Why autoimmune epithelitis? Oral Dis. 2006, 12, 523–532. [Google Scholar] [CrossRef]
- Igoe, A.; Scofield, R.H. Autoimmunity and infection in Sjögren’s syndrome. Curr. Opin. Rheumatol. 2013, 25, 480–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Björk, A.; Mofors, J.; Wahren-Herlenius, M. Environmental factors in the pathogenesis of primary Sjögren’s syndrome. J. Intern. Med. 2020, 287, 475–492. [Google Scholar] [CrossRef] [PubMed]
- Ascherio, A.; Munger, K.L. Epstein-barr virus infection and multiple sclerosis: A review. J. Neuroimmune Pharmacol. 2010, 5, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Toussirot, E.; Roudier, J. Epstein-Barr virus in autoimmune diseases. Best Pract. Res. Clin. Rheumatol. 2008, 22, 883–896. [Google Scholar] [CrossRef] [PubMed]
- Saito, I.; Servenius, B.; Compton, T.; Fox, R.I. Detection of Epstein-Barr virus DNA by polymerase chain reaction in blood and tissue biopsies from patients with Sjogren’s syndrome. J. Exp. Med. 1989, 169, 2191–2198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mariette, X.; Gozlan, J.; Clerc, D.; Bisson, M.; Morinet, F. Detection of Epstein-Barr virus DNA by in situ hybridization and polymerase chain reaction in salivary gland biopsy specimens from patients with Sjögren’s syndrome. Am. J. Med. 1991, 90, 286–294. [Google Scholar] [CrossRef]
- Dimitriou, I.; Xanthou, G.; Kapsogeorgou, E.; Abu-Helu, R.; Moutsopoulos, H.; Manoussakis, M. High spontaneous CD40 expression by salivary gland epithelial cells in Sjogren’s syndrome: Possible evidence for intrinsic activation of epithelial cells. Arthritis Res. 2001, 3, P018. [Google Scholar] [CrossRef] [Green Version]
- Kivity, S.; Arango, M.T.; Ehrenfeld, M.; Tehori, O.; Shoenfeld, Y.; Anaya, J.-M.; Agmon-Levin, N. Infection and autoimmunity in Sjogren’s syndrome: A clinical study and comprehensive review. J. Autoimmun. 2014, 51, 17–22. [Google Scholar] [CrossRef]
- Iwakiri, D.; Zhou, L.; Samanta, M.; Matsumoto, M.; Ebihara, T.; Seya, T.; Imai, S.; Fujieda, M.; Kawa, K.; Takada, K. Epstein-Barr virus (EBV)-encoded small RNA is released from EBV-infected cells and activates signaling from Toll-like receptor 3. J. Exp. Med. 2009, 206, 2091–2099. [Google Scholar] [CrossRef] [Green Version]
- Murray, R.J.; Wang, D.; Young, L.S.; Wang, F.; Rowe, M.; Kieff, E.; Rickinson, A.B. Epstein-Barr virus-specific cytotoxic T-cell recognition of transfectants expressing the virus-coded latent membrane protein LMP. J. Virol. 1988, 62, 3747–3755. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, H.; Takahashi, Y.; Yamamoto-Fukuda, T.; Horai, Y.; Nakashima, Y.; Arima, K.; Nakamura, T.; Koji, T.; Kawakami, A. Direct Infection of Primary Salivary Gland Epithelial Cells by Human T Lymphotropic Virus Type I in Patients With Sjögren’s Syndrome. Arthritis Rheumatol. 2015, 67, 1096–1106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terada, K.; Katamine, S.; Eguchi, K.; Moriuchi, R.; Kita, M.; Shimada, H.; Yamashita, I.; Iwata, K.; Tsuji, Y.; Nagataki, S. Prevalence of serum and salivary antibodies to HTLV-1 in Sjögren’s syndrome. Lancet 1994, 344, 1116–1119. [Google Scholar] [CrossRef]
- Stathopoulou, E.A.; Routsias, J.G.; Stea, E.A.; Moutsopoulos, H.M.; Tzioufas, A.G. Cross-reaction between antibodies to the major epitope of Ro60 kD autoantigen and a homologous peptide of Coxsackie virus 2B protein. Clin. Exp. Immunol. 2005, 141, 148–154. [Google Scholar] [CrossRef]
- Gottenberg, J.-E.; Pallier, C.; Ittah, M.; Lavie, F.; Miceli-Richard, C.; Sellam, J.; Nordmann, P.; Cagnard, N.; Sibilia, J.; Mariette, X. Failure to confirm coxsackievirus infection in primary Sjögren’s syndrome. Arthritis Rheum. 2006, 54, 2026–2028. [Google Scholar] [CrossRef] [PubMed]
- Flores-Chávez, A.; Carrion, J.A.; Forns, X.; Ramos-Casals, M. Extrahepatic manifestations associated with Chronic Hepatitis C Virus Infection. Rev. Espanola Sanid. Penit. 2017, 19, 87–97. [Google Scholar] [CrossRef]
- Kang, H.I.; Fei, H.M.; Saito, I.; Sawada, S.; Chen, S.L.; Yi, D.; Chan, E.; Peebles, C.; Bugawan, T.L.; Erlich, H.A. Comparison of HLA class II genes in Caucasoid, Chinese, and Japanese patients with primary Sjögren’s syndrome. J. Immunol. 1950 1993, 150, 3615–3623. [Google Scholar]
- Kerttula, T.O.; Collin, P.; Polvi, A.; Korpela, M.; Partanen, J.; Mäki, M. Distinct immunologic features of Finnish Sjögren’s syndrome patients with HLA alleles DRB1*0301, DQA1*0501, and DQB1*0201. Alterations in circulating T cell receptor gamma/delta subsets. Arthritis Rheum. 1996, 39, 1733–1739. [Google Scholar] [CrossRef]
- Mountz, J.D.; Zhou, T.; Su, X.; Wu, J.; Cheng, J. The role of programmed cell death as an emerging new concept for the pathogenesis of autoimmune diseases. Clin. Immunol. Immunopathol. 1996, 80, S2–S14. [Google Scholar] [CrossRef]
- Adachi, M.; Watanabe-Fukunaga, R.; Nagata, S. Aberrant transcription caused by the insertion of an early transposable element in an intron of the Fas antigen gene of lpr mice. Proc. Natl. Acad. Sci. USA 1993, 90, 1756–1760. [Google Scholar] [CrossRef] [Green Version]
- Bolstad, A.I.; Wargelius, A.; Nakken, B.; Haga, H.J.; Jonsson, R. Fas and Fas ligand gene polymorphisms in primary Sjögren’s syndrome. J. Rheumatol. 2000, 27, 2397–2405. [Google Scholar]
- Nakken, B.; Jonsson, R.; Bolstad, A.I. Polymorphisms of the Ro52 gene associated with anti-Ro 52-kd autoantibodies in patients with primary Sjögren’s syndrome. Arthritis Rheum. 2001, 44, 638–646. [Google Scholar] [CrossRef]
- Hulkkonen, J.; Pertovaara, M.; Antonen, J.; Lahdenpohja, N.; Pasternack, A.; Hurme, M. Genetic association between interleukin-10 promoter region polymorphisms and primary Sjögren’s syndrome. Arthritis Rheum. 2001, 44, 176–179. [Google Scholar] [CrossRef]
- Qin, B.; Wang, J.; Liang, Y.; Yang, Z.; Zhong, R. The association between TNF-α, IL-10 gene polymorphisms and primary Sjögren’s syndrome: A meta-analysis and systemic review. PLoS ONE 2013, 8, e63401. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Casals, M.; Font, J.; Brito-Zeron, P.; Trejo, O.; García-Carrasco, M.; Lozano, F. Interleukin-4 receptor alpha polymorphisms in primary Sjögren’s syndrome. Clin. Exp. Rheumatol. 2004, 22, 374. [Google Scholar]
- Imgenberg-Kreuz, J.; Rasmussen, A.; Sivils, K.; Nordmark, G. Genetics and epigenetics in primary Sjögren’s syndrome. Rheumatology 2019. [Google Scholar] [CrossRef] [Green Version]
- Traianos, E.Y.; Locke, J.; Lendrem, D.; Bowman, S.; Hargreaves, B.; Macrae, V. Serum CXCL13 levels are associated with lymphoma risk and lymphoma occurrence in primary Sjögren’s syndrome. Rheumatol. Int. 2020, 40, 541–548. [Google Scholar] [CrossRef] [Green Version]
- Ben-Eli, H.; Gomel, N.; Aframian, D.J.; Abu-Seir, R.; Perlman, R.; Ben-Chetrit, E.; Mevorach, D.; Kleinstern, G.; Paltiel, O.; Solomon, A. SNP variations in IL10, TNFα and TNFAIP3 genes in patients with dry eye syndrome and Sjogren’s syndrome. J. Inflamm. 2019, 16, 6. [Google Scholar] [CrossRef] [Green Version]
- Nocturne, G.; Tarn, J.; Boudaoud, S.; Locke, J.; Miceli-Richard, C.; Hachulla, E.; Dubost, J.J.; Bowman, S.; Gottenberg, J.E.; Criswell, L.A.; et al. Germline variation of TNFAIP3 in primary Sjögren’s syndrome-associated lymphoma. Ann. Rheum. Dis. 2016, 75, 780–783. [Google Scholar] [CrossRef]
- Nezos, A.; Gkioka, E.; Koutsilieris, M.; Voulgarelis, M.; Tzioufas, A.G.; Mavragani, C.P. TNFAIP3 F127C Coding Variation in Greek Primary Sjogren’s Syndrome Patients. J. Immunol. Res. 2018, 2018, 6923213. [Google Scholar] [CrossRef] [Green Version]
- Fragkioudaki, S.; Nezos, A.; Souliotis, V.L.; Chatziandreou, I.; Saetta, A.A.; Drakoulis, N.; Tzioufas, A.G.; Voulgarelis, M.; Sfikakis, P.P.; Koutsilieris, M.; et al. MTHFR gene variants and non-MALT lymphoma development in primary Sjogren’s syndrome. Sci. Rep. 2017, 7, 7354. [Google Scholar] [CrossRef] [Green Version]
- Nezos, A.; Mavragani, C.P. Contribution of Genetic Factors to Sjögren’s Syndrome and Sjögren’s Syndrome Related Lymphomagenesis. J. Immunol. Res. 2015, 2015, 754825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arvaniti, P.; Le Dantec, C.; Charras, A.; Arleevskaya, M.A.; Hedrich, C.M.; Zachou, K.; Dalekos, G.N.; Renaudineau, Y. Linking genetic variation with epigenetic profiles in Sjögren’s syndrome. Clin. Immunol. 2020, 210, 108314. [Google Scholar] [CrossRef]
- Konsta, O.D.; Thabet, Y.; Le Dantec, C.; Brooks, W.H.; Tzioufas, A.G.; Pers, J.-O.; Renaudineau, Y. The contribution of epigenetics in Sjögren’s Syndrome. Front. Genet. 2014, 5, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thabet, Y.; Le Dantec, C.; Ghedira, I.; Devauchelle, V.; Cornec, D.; Pers, J.-O.; Renaudineau, Y. Epigenetic dysregulation in salivary glands from patients with primary Sjögren’s syndrome may be ascribed to infiltrating B cells. J. Autoimmun. 2013, 41, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Cannat, A.; Seligmann, M. Induction by isoniazid and hydrallazine of antinuclear factors in mice. Clin. Exp. Immunol. 1968, 3, 99–105. [Google Scholar]
- Imgenberg-Kreuz, J.; Sandling, J.K.; Almlöf, J.C.; Nordlund, J.; Signér, L.; Norheim, K.B.; Omdal, R.; Rönnblom, L.; Eloranta, M.-L.; Syvänen, A.-C.; et al. Genome-wide DNA methylation analysis in multiple tissues in primary Sjögren’s syndrome reveals regulatory effects at interferon-induced genes. Ann. Rheum. Dis. 2016, 75, 2029–2036. [Google Scholar] [CrossRef] [Green Version]
- Toso, A.; Aluffi, P.; Capello, D.; Conconi, A.; Gaidano, G.; Pia, F. Clinical and molecular features of mucosa-associated lymphoid tissue (MALT) lymphomas of salivary glands. Head Neck 2009, 31, 1181–1187. [Google Scholar] [CrossRef]
- Altorok, N.; Coit, P.; Hughes, T.; Koelsch, K.A.; Stone, D.U.; Rasmussen, A.; Radfar, L.; Scofield, R.H.; Sivils, K.L.; Farris, A.D.; et al. Genome-wide DNA methylation patterns in naive CD4+ T cells from patients with primary Sjögren’s syndrome. Arthritis Rheumatol. 2014, 66, 731–739. [Google Scholar] [CrossRef] [Green Version]
- Alevizos, I.; Illei, G.G. MicroRNAs in Sjögren’s syndrome as a prototypic autoimmune disease. Autoimmun. Rev. 2010, 9, 618–621. [Google Scholar] [CrossRef] [Green Version]
- Mendell, J.T. miRiad roles for the miR-17-92 cluster in development and disease. Cell 2008, 133, 217–222. [Google Scholar] [CrossRef] [Green Version]
- Xiao, C.; Srinivasan, L.; Calado, D.P.; Patterson, H.C.; Zhang, B.; Wang, J.; Henderson, J.M.; Kutok, J.L.; Rajewsky, K. Lymphoproliferative disease and autoimmunity in mice with increased miR-17-92 expression in lymphocytes. Nat. Immunol. 2008, 9, 405–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zilahi, E.; Tarr, T.; Papp, G.; Griger, Z.; Sipka, S.; Zeher, M. Increased microRNA-146a/b, TRAF6 gene and decreased IRAK1 gene expressions in the peripheral mononuclear cells of patients with Sjögren’s syndrome. Immunol. Lett. 2012, 141, 165–168. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.; Tetzlaff, M.T.; Vanbelle, P.; Elder, D.; Feldman, M.; Tobias, J.W.; Sepulveda, A.R.; Xu, X. MicroRNA expression profiling outperforms mRNA expression profiling in formalin-fixed paraffin-embedded tissues. Int. J. Clin. Exp. Pathol. 2009, 2, 519–527. [Google Scholar] [PubMed]
- Howe, K. Extraction of miRNAs from Formalin-Fixed Paraffin-Embedded (FFPE) Tissues. Methods Mol. Biol. 2017, 1509, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Sun, B.; Huang, S.; Zhao, L. Roles of circular RNAs in immune regulation and autoimmune diseases. Cell Death Dis. 2019, 10, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Xia, X.; Tang, X.; Wang, S. Roles of CircRNAs in Autoimmune Diseases. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef]
- Su, L.-C.; Xu, W.-D.; Liu, X.-Y.; Fu, L.; Huang, A.-F. Altered expression of circular RNA in primary Sjögren’s syndrome. Clin. Rheumatol. 2019, 38, 3425–3433. [Google Scholar] [CrossRef]
- Roy, S.; Awasthi, A. Emerging roles of noncoding RNAs in T cell differentiation and functions in autoimmune diseases. Int. Rev. Immunol. 2019, 38, 232–245. [Google Scholar] [CrossRef]
- Dolcino, M.; Tinazzi, E.; Vitali, C.; Del Papa, N.; Puccetti, A.; Lunardi, C. Long Non-Coding RNAs Modulate Sjögren’s Syndrome Associated Gene Expression and Are Involved in the Pathogenesis of the Disease. J. Clin. Med. 2019, 8, 1349. [Google Scholar] [CrossRef] [Green Version]
- Han, S.-B.; Moratz, C.; Huang, N.-N.; Kelsall, B.; Cho, H.; Shi, C.-S.; Schwartz, O.; Kehrl, J.H. Rgs1 and Gnai2 regulate the entrance of B lymphocytes into lymph nodes and B cell motility within lymph node follicles. Immunity 2005, 22, 343–354. [Google Scholar] [CrossRef] [Green Version]
- Coca, A.; Sanz, I. Updates on B-cell immunotherapies for systemic lupus erythematosus and Sjogren’s syndrome. Curr. Opin. Rheumatol. 2012, 24, 451–456. [Google Scholar] [CrossRef] [PubMed]
- Béguelin, W.; Teater, M.; Gearhart, M.D.; Calvo Fernández, M.T.; Goldstein, R.L.; Cárdenas, M.G.; Hatzi, K.; Rosen, M.; Shen, H.; Corcoran, C.M.; et al. EZH2 and BCL6 Cooperate to Assemble CBX8-BCOR Complex to Repress Bivalent Promoters, Mediate Germinal Center Formation and Lymphomagenesis. Cancer Cell 2016, 30, 197–213. [Google Scholar] [CrossRef] [PubMed]
- Bai, M.; Skyrlas, A.; Agnantis, N.J.; Kamina, S.; Tsanou, E.; Grepi, C.; Galani, V.; Kanavaros, P. Diffuse large B-cell lymphomas with germinal center B-cell-like differentiation immunophenotypic profile are associated with high apoptotic index, high expression of the proapoptotic proteins bax, bak and bid and low expression of the antiapoptotic protein bcl-xl. Mod. Pathol. 2004, 17, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Wei, W.; He, X.; Xie, Y.; Kamal, M.A.; Li, J. Influence of Hormones on Sjögren’s Syndrome. Curr. Pharm. Des. 2018, 24, 4167–4176. [Google Scholar] [CrossRef] [PubMed]
- McCoy, S.S.; Sampene, E.; Baer, A.N. Sjögren’s Syndrome is Associated With Reduced Lifetime Sex Hormone Exposure: A Case-Control Study. Arthritis Care Res. 2019, acr.24014. [Google Scholar] [CrossRef] [PubMed]
- Harris, V.M.; Sharma, R.; Cavett, J.; Kurien, B.T.; Liu, K.; Koelsch, K.A.; Rasmussen, A.; Radfar, L.; Lewis, D.; Stone, D.U.; et al. Klinefelter’s syndrome (47,XXY) is in excess among men with Sjögren’s syndrome. Clin. Immunol. 2016, 168, 25–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seminog, O.O.; Seminog, A.B.; Yeates, D.; Goldacre, M.J. Associations between Klinefelter’s syndrome and autoimmune diseases: English national record linkage studies. Autoimmunity 2015, 48, 125–128. [Google Scholar] [CrossRef]
- Fujimoto, M.; Ikeda, K.; Nakamura, T.; Iwamoto, T.; Furuta, S.; Nakajima, H. Development of mixed connective tissue disease and Sjögren’s syndrome in a patient with trisomy X. Lupus 2015, 24, 1217–1220. [Google Scholar] [CrossRef]
- Morthen, M.K.; Tellefsen, S.; Richards, S.M.; Lieberman, S.M.; Rahimi Darabad, R.; Kam, W.R.; Sullivan, D.A. Testosterone Influence on Gene Expression in Lacrimal Glands of Mouse Models of Sjögren Syndrome. Investig. Ophthalmol. Vis. Sci. 2019, 60, 2181–2197. [Google Scholar] [CrossRef]
- Porola, P.; Laine, M.; Virtanen, I.; Pöllänen, R.; Przybyla, B.D.; Konttinen, Y.T. Androgens and Integrins in Salivary Glands in Sjögren’s Syndrome. J. Rheumatol. 2010, 37, 1181–1187. [Google Scholar] [CrossRef]
- Taiym, S.; Haghighat, N.; Al-Hashimi, I. A comparison of the hormone levels in patients with Sjogren’s syndrome and healthy controls. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 2004, 97, 579–583. [Google Scholar] [CrossRef] [PubMed]
- Bizzarro, A.; Valentini, G.; Martino, G.D.; Daponte, A.; De Bellis, A.; Iacono, G. Influence of Testosterone Therapy on Clinical and Immunological Features of Autoimmune Diseases Associated with Klinefelter’s Syndrome. J. Clin. Endocrinol. Metab. 1987, 64, 32–36. [Google Scholar] [CrossRef] [PubMed]
- Ishimaru, N.; Arakaki, R.; Watanabe, M.; Kobayashi, M.; Miyazaki, K.; Hayashi, Y. Development of autoimmune exocrinopathy resembling Sjögren’s syndrome in estrogen-deficient mice of healthy background. Am. J. Pathol. 2003, 163, 1481–1490. [Google Scholar] [CrossRef]
- Iwasa, A.; Arakaki, R.; Honma, N.; Ushio, A.; Yamada, A.; Kondo, T.; Kurosawa, E.; Kujiraoka, S.; Tsunematsu, T.; Kudo, Y.; et al. Aromatase Controls Sjögren Syndrome–Like Lesions through Monocyte Chemotactic Protein-1 in Target Organ and Adipose Tissue–Associated Macrophages. Am. J. Pathol. 2015, 185, 151–161. [Google Scholar] [CrossRef]
- Shim, G.-J.; Warner, M.; Kim, H.-J.; Andersson, S.; Liu, L.; Ekman, J.; Imamov, O.; Jones, M.E.; Simpson, E.R.; Gustafsson, J.-A. Aromatase-deficient mice spontaneously develop a lymphoproliferative autoimmune disease resembling Sjogren’s syndrome. Proc. Natl. Acad. Sci. USA 2004, 101, 12628–12633. [Google Scholar] [CrossRef] [Green Version]
- Ishimaru, N.; Arakaki, R.; Omotehara, F.; Yamada, K.; Mishima, K.; Saito, I.; Hayashi, Y. Novel Role for RbAp48 in Tissue-Specific, Estrogen Deficiency-Dependent Apoptosis in the Exocrine Glands. Mol. Cell. Biol. 2006, 26, 2924–2935. [Google Scholar] [CrossRef] [Green Version]
- Ishimaru, N.; Arakaki, R.; Yoshida, S.; Yamada, A.; Noji, S.; Hayashi, Y. Expression of the retinoblastoma protein RbAp48 in exocrine glands leads to Sjögren’s syndrome–like autoimmune exocrinopathy. J. Exp. Med. 2008, 205, 2915–2927. [Google Scholar] [CrossRef] [Green Version]
- Manoussakis, M.N.; Tsinti, M.; Kapsogeorgou, E.K.; Moutsopoulos, H.M. The salivary gland epithelial cells of patients with primary Sjögren’s syndrome manifest significantly reduced responsiveness to 17β-estradiol. J. Autoimmun. 2012, 39, 64–68. [Google Scholar] [CrossRef]
- Laroche, M.; Borg, S.; Lassoued, S.; De Lafontan, B.; Roché, H. Joint pain with aromatase inhibitors: Abnormal frequency of Sjögren’s syndrome. J. Rheumatol. 2007, 34, 2259–2263. [Google Scholar]
- Shanmugam, V.K.; McCloskey, J.; Elston, B.; Allison, S.J.; Eng-Wong, J. The CIRAS study: A case control study to define the clinical, immunologic, and radiographic features of aromatase inhibitor-induced musculoskeletal symptoms. Breast Cancer Res. Treat. 2012, 131, 699–708. [Google Scholar] [CrossRef] [Green Version]
- Guidelli, G.M.; Martellucci, I.; Galeazzi, M.; Francini, G.; Fioravanti, A. Sjögren’s syndrome and aromatase inhibitors treatment: Is there a link? Clin. Exp. Rheumatol. 2013, 31, 653–654. [Google Scholar] [PubMed]
- Laine, M.; Porola, P.; Udby, L.; Kjeldsen, L.; Cowland, J.B.; Borregaard, N.; Hietanen, J.; Ståhle, M.; Pihakari, A.; Konttinen, Y.T. Low salivary dehydroepiandrosterone and androgen-regulated cysteine-rich secretory protein 3 levels in Sjögren’s syndrome. Arthritis Rheum. 2007, 56, 2575–2584. [Google Scholar] [CrossRef] [PubMed]
- Konttinen, Y.T.; Fuellen, G.; Bing, Y.; Porola, P.; Stegaev, V.; Trokovic, N.; Falk, S.S.I.; Liu, Y.; Szodoray, P.; Takakubo, Y. Sex steroids in Sjögren’s syndrome. J. Autoimmun. 2012, 39, 49–56. [Google Scholar] [CrossRef]
- Spaan, M.; Porola, P.; Laine, M.; Rozman, B.; Azuma, M.; Konttinen, Y.T. Healthy human salivary glands contain a DHEA-sulphate processing intracrine machinery, which is deranged in primary Sjögren’s syndrome. J. Cell. Mol. Med. 2009, 13, 1261–1270. [Google Scholar] [CrossRef] [PubMed]
- Moutsopoulos, H.M.; Kordossis, T. Sjögren’s syndrome revisited: Autoimmune epithelitis. Br. J. Rheumatol. 1996, 35, 204–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spachidou, M.P.; Bourazopoulou, E.; Maratheftis, C.I.; Kapsogeorgou, E.K.; Moutsopoulos, H.M.; Tzioufas, A.G.; Manoussakis, M.N. Expression of functional Toll-like receptors by salivary gland epithelial cells: Increased mRNA expression in cells derived from patients with primary Sjögren’s syndrome. Clin. Exp. Immunol. 2007, 147, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-Q.; Szodoray, P.; Zeher, M. Toll-Like Receptor Pathways in Autoimmune Diseases. Clin. Rev. Allergy Immunol. 2016, 50, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spachidou, M.; Kapsogeorgou, E.; Bourazopoulou, E.; Moutsopoulos, H.; Manoussakis, M. Cultured salivary gland epithelial cells from patients with primary Sjögren’s syndrome and disease controls are sensitive to signaling via Toll-like receptors 2 and 3: Upregulation of intercellular adhesion molecule-1 expression. Arthritis Res. Ther. 2005, 7, P154. [Google Scholar] [CrossRef] [Green Version]
- Iwanaszko, M.; Kimmel, M. NF-κB and IRF pathways: Cross-regulation on target genes promoter level. BMC Genom. 2015, 16, 307. [Google Scholar] [CrossRef] [Green Version]
- Ichiyama, T.; Nakatani, E.; Tatsumi, K.; Hideshima, K.; Urano, T.; Nariai, Y.; Sekine, J. Expression of aquaporin 3 and 5 as a potential marker for distinguishing dry mouth from Sjögren’s syndrome. J. Oral Sci. 2018, 60, 212–220. [Google Scholar] [CrossRef] [Green Version]
- Beroukas, D.; Hiscock, J.; Gannon, B.J.; Jonsson, R.; Gordon, T.P.; Waterman, S.A. Selective down-regulation of aquaporin-1 in salivary glands in primary Sjögren’s syndrome. Lab. Investig. J. Tech. Methods Pathol. 2002, 82, 1547–1552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sisto, M.; Lorusso, L.; Ingravallo, G.; Nico, B.; Ribatti, D.; Ruggieri, S.; Lofrumento, D.D.; Lisi, S. Abnormal distribution of AQP4 in minor salivary glands of primary Sjögren’s syndrome patients. Autoimmunity 2017, 50, 202–210. [Google Scholar] [CrossRef] [PubMed]
- Ring, T.; Kallenbach, M.; Praetorius, J.; Nielsen, S.; Melgaard, B. Successful treatment of a patient with primary Sjögren’s syndrome with Rituximab. Clin. Rheumatol. 2006, 25, 891–894. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Ying, X.; Qian, Y.; Liu, H.; Lan, Y.; Xie, A.; Zhu, X. Physiological and pathological impact of AQP1 knockout in mice. Biosci. Rep. 2019, 39. [Google Scholar] [CrossRef]
- Verkman, A.S.; Yang, B.; Song, Y.; Manley, G.T.; Ma, T. Role of water channels in fluid transport studied by phenotype analysis of aquaporin knockout mice. Exp. Physiol. 2000, 85, 233S–241S. [Google Scholar] [CrossRef]
- Hosoi, K.; Yao, C.; Hasegawa, T.; Yoshimura, H.; Akamatsu, T. Dynamics of Salivary Gland AQP5 under Normal and Pathologic Conditions. Int. J. Mol. Sci. 2020, 21, 1182. [Google Scholar] [CrossRef] [Green Version]
- Delporte, C.; Bryla, A.; Perret, J. Aquaporins in Salivary Glands: From Basic Research to Clinical Applications. Int. J. Mol. Sci. 2016, 17, 166. [Google Scholar] [CrossRef] [Green Version]
- Steinfeld, S.; Cogan, E.; King, L.S.; Agre, P.; Kiss, R.; Delporte, C. Abnormal distribution of aquaporin-5 water channel protein in salivary glands from Sjögren’s syndrome patients. Lab. Investig. J. Tech. Methods Pathol. 2001, 81, 143–148. [Google Scholar] [CrossRef] [Green Version]
- Soyfoo, M.S.; De Vriese, C.; Debaix, H.; Martin-Martinez, M.D.; Mathieu, C.; Devuyst, O.; Steinfeld, S.D.; Delporte, C. Modified aquaporin 5 expression and distribution in submandibular glands from NOD mice displaying autoimmune exocrinopathy. Arthritis Rheum. 2007, 56, 2566–2574. [Google Scholar] [CrossRef] [Green Version]
- Yoshimura, S.; Nakamura, H.; Horai, Y.; Nakajima, H.; Shiraishi, H.; Hayashi, T.; Takahashi, T.; Kawakami, A. Abnormal distribution of AQP5 in labial salivary glands is associated with poor saliva secretion in patients with Sjögren’s syndrome including neuromyelitis optica complicated patients. Mod. Rheumatol. 2016, 26, 384–390. [Google Scholar] [CrossRef]
- Lee, B.H.; Gauna, A.E.; Perez, G.; Park, Y.; Pauley, K.M.; Kawai, T.; Cha, S. Autoantibodies against Muscarinic Type 3 Receptor in Sjögren’s Syndrome Inhibit Aquaporin 5 Trafficking. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Roche, J.V.; Törnroth-Horsefield, S. Aquaporin Protein-Protein Interactions. Int. J. Mol. Sci. 2017, 18, 2255. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, Y.; Tsuzaka, K.; Takeuchi, T.; Sasaki, Y.; Tsubota, K. Altered distribution of aquaporin 5 and its C-terminal binding protein in the lacrimal glands of a mouse model for Sjögren’s syndrome. Curr. Eye Res. 2008, 33, 621–629. [Google Scholar] [CrossRef]
- Soyfoo, M.S.; Konno, A.; Bolaky, N.; Oak, J.S.; Fruman, D.; Nicaise, C.; Takiguchi, M.; Delporte, C. Link between inflammation and aquaporin-5 distribution in submandibular gland in Sjögren’s syndrome? Oral Dis. 2012, 18, 568–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soyfoo, M.S.; Bolaky, N.; Depoortere, I.; Delporte, C. Relationship between aquaporin-5 expression and saliva flow in streptozotocin-induced diabetic mice? Oral Dis. 2012, 18, 501–505. [Google Scholar] [CrossRef]
- Jin, J.-O.; Yu, Q. T Cell-Associated Cytokines in the Pathogenesis of Sjögren’s Syndrome. J. Clin. Cell. Immunol. 2013, S1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, R.I.; Kang, H.I.; Ando, D.; Abrams, J.; Pisa, E. Cytokine mRNA expression in salivary gland biopsies of Sjögren’s syndrome. J. Immunol. 1950 1994, 152, 5532–5539. [Google Scholar]
- Boumba, D.; Skopouli, F.N.; Moutsopoulos, H.M. Cytokine mRNA expression in the labial salivary gland tissues from patients with primary Sjögren’s syndrome. Br. J. Rheumatol. 1995, 34, 326–333. [Google Scholar] [CrossRef]
- Sumida, T.; Tsuboi, H.; Iizuka, M.; Hirota, T.; Asashima, H.; Matsumoto, I. The role of M3 muscarinic acetylcholine receptor reactive T cells in Sjögren’s syndrome: A critical review. J. Autoimmun. 2014, 51, 44–50. [Google Scholar] [CrossRef]
- Zhou, J.; Jin, J.-O.; Kawai, T.; Yu, Q. Endogenous programmed death ligand-1 restrains the development and onset of Sjögren’s syndrome in non-obese diabetic mice. Sci. Rep. 2016, 6, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Arce-Franco, M.; Dominguez-Luis, M.; Pec, M.K.; Martínez-Gimeno, C.; Miranda, P.; Alvarez de la Rosa, D.; Giraldez, T.; García-Verdugo, J.M.; Machado, J.D.; Díaz-González, F. Functional effects of proinflammatory factors present in Sjögren’s syndrome salivary microenvironment in an in vitro model of human salivary gland. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, E.H.; Lee, Y.J.; Hyon, J.Y.; Yun, P.Y.; Song, Y.W. Salivary cytokine profiles in primary Sjögren’s syndrome differ from those in non-Sjögren sicca in terms of TNF-α levels and Th-1/Th-2 ratios. Clin. Exp. Rheumatol. 2011, 29, 970–976. [Google Scholar] [PubMed]
- Yamamura, Y.; Motegi, K.; Kani, K.; Takano, H.; Momota, Y.; Aota, K.; Yamanoi, T.; Azuma, M. TNF-α inhibits aquaporin 5 expression in human salivary gland acinar cells via suppression of histone H4 acetylation. J. Cell. Mol. Med. 2012, 16, 1766–1775. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Kawai, T.; Yu, Q. Pathogenic role of endogenous TNF-α in the development of Sjögren’s-like sialadenitis and secretory dysfunction in non-obese diabetic mice. Lab. Investig. 2017, 97, 458–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, R.I.; Adamson, T.C.; Fong, S.; Young, C.; Howell, F.V. Characterization of the phenotype and function of lymphocytes infiltrating the salivary gland in patients with primary Sjogren syndrome. Diagn. Immunol. 1983, 1, 233–239. [Google Scholar] [PubMed]
- Roescher, N.; Tak, P.P.; Illei, G.G. Cytokines in Sjögren’s syndrome. Oral Dis. 2009, 15, 519–526. [Google Scholar] [CrossRef] [Green Version]
- Fox, R.I.; Kang, H.I. Pathogenesis of Sjögren’s syndrome. Rheum. Dis. Clin. N. Am. 1992, 18, 517–538. [Google Scholar]
- Bertorello, R.; Cordone, M.P.; Contini, P.; Rossi, P.; Indiveri, F.; Puppo, F.; Cordone, G. Increased levels of interleukin-10 in saliva of Sjögren’s syndrome patients. Correlation with disease activity. Clin. Exp. Med. 2004, 4, 148–151. [Google Scholar] [CrossRef]
- Youinou, P.; Pers, J.-O. Disturbance of cytokine networks in Sjögren’s syndrome. Arthritis Res. Ther. 2011, 13, 227. [Google Scholar] [CrossRef] [Green Version]
- Ohlsson, M.; Jonsson, R.; Brokstad, K.A. Subcellular redistribution and surface exposure of the Ro52, Ro60 and La48 autoantigens during apoptosis in human ductal epithelial cells: A possible mechanism in the pathogenesis of Sjögren’s syndrome. Scand. J. Immunol. 2002, 56, 456–469. [Google Scholar] [CrossRef]
- Davies, M.L.; Taylor, E.J.; Gordon, C.; Young, S.P.; Welsh, K.; Bunce, M.; Wordsworth, B.P.; Davidson, B.; Bowman, S.J. Candidate T cell epitopes of the human La/SSB autoantigen. Arthritis Rheum. 2002, 46, 209–214. [Google Scholar] [CrossRef]
- Hasegawa, H.; Inoue, A.; Kohno, M.; Muraoka, M.; Miyazaki, T.; Terada, M.; Nakayama, T.; Yoshie, O.; Nose, M.; Yasukawa, M. Antagonist of interferon-inducible protein 10/CXCL10 ameliorates the progression of autoimmune sialadenitis in MRL/lpr mice. Arthritis Rheum. 2006, 54, 1174–1183. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Ogawa, N.; Nakabayashi, T.; Liu, G.T.; D’Souza, E.; McGuff, H.S.; Guerrero, D.; Talal, N.; Dang, H. Fas and Fas ligand expression in the salivary glands of patients with primary Sjögren’s syndrome. Arthritis Rheum. 1997, 40, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Ibrahem, H.M. B cell dysregulation in primary Sjögren’s syndrome: A review. Jpn. Dent. Sci. Rev. 2019, 55, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Verstappen, G.M.; Corneth, O.B.J.; Bootsma, H.; Kroese, F.G.M. Th17 cells in primary Sjögren’s syndrome: Pathogenicity and plasticity. J. Autoimmun. 2018, 87, 16–25. [Google Scholar] [CrossRef]
- Katsifis, G.E.; Rekka, S.; Moutsopoulos, N.M.; Pillemer, S.; Wahl, S.M. Systemic and local interleukin-17 and linked cytokines associated with Sjögren’s syndrome immunopathogenesis. Am. J. Pathol. 2009, 175, 1167–1177. [Google Scholar] [CrossRef] [Green Version]
- Sonnenberg, G.F.; Nair, M.G.; Kirn, T.J.; Zaph, C.; Fouser, L.A.; Artis, D. Pathological versus protective functions of IL-22 in airway inflammation are regulated by IL-17A. J. Exp. Med. 2010, 207, 1293–1305. [Google Scholar] [CrossRef] [Green Version]
- Lavoie, T.N.; Stewart, C.M.; Berg, K.M.; Li, Y.; Nguyen, C.Q. Expression of interleukin-22 in Sjögren’s syndrome: Significant correlation with disease parameters. Scand. J. Immunol. 2011, 74, 377–382. [Google Scholar] [CrossRef]
- Monteiro, R.; Martins, C.; Barcelos, F.; Nunes, G.; Lopes, T.; Borrego, L.-M. Follicular helper and follicular cytotoxic T cells in primary Sjögren’s Syndrome: Clues for an abnormal antiviral response as a pathogenic mechanism. Ann. Med. 2019, 51, 42. [Google Scholar] [CrossRef] [Green Version]
- Saito, M.; Otsuka, K.; Ushio, A.; Yamada, A.; Arakaki, R.; Kudo, Y.; Ishimaru, N. Unique Phenotypes and Functions of Follicular Helper T Cells and Regulatory T Cells in Sjögren’s Syndrome. Curr. Rheumatol. Rev. 2018, 14, 239–245. [Google Scholar] [CrossRef]
- Scheid, J.F.; Mouquet, H.; Kofer, J.; Yurasov, S.; Nussenzweig, M.C.; Wardemann, H. Differential regulation of self-reactivity discriminates between IgG+ human circulating memory B cells and bone marrow plasma cells. Proc. Natl. Acad. Sci. USA 2011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mouquet, H.; Nussenzweig, M.C. Polyreactive antibodies in adaptive immune responses to viruses. Cell. Mol. Life Sci. 2012, 69, 1435–1445. [Google Scholar] [CrossRef] [PubMed]
- Corsiero, E.; Sutcliffe, N.; Pitzalis, C.; Bombardieri, M. Accumulation of self-reactive naïve and memory B cell reveals sequential defects in B cell tolerance checkpoints in Sjögren’s syndrome. PLoS ONE 2014, 9, e114575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samuels, J.; Ng, Y.-S.; Coupillaud, C.; Paget, D.; Meffre, E. Impaired early B cell tolerance in patients with rheumatoid arthritis. J. Exp. Med. 2005, 201, 1659–1667. [Google Scholar] [CrossRef]
- Mietzner, B.; Tsuiji, M.; Scheid, J.; Velinzon, K.; Tiller, T.; Abraham, K.; Gonzalez, J.B.; Pascual, V.; Stichweh, D.; Wardemann, H.; et al. Autoreactive IgG memory antibodies in patients with systemic lupus erythematosus arise from nonreactive and polyreactive precursors. Proc. Natl. Acad. Sci. USA 2008, 105, 9727–9732. [Google Scholar] [CrossRef] [Green Version]
- Hayakawa, I.; Tedder, T.F.; Zhuang, Y. B-lymphocyte depletion ameliorates Sjögren’s syndrome in Id3 knockout mice. Immunology 2007, 122, 73–79. [Google Scholar] [CrossRef]
- Baff and April: A Tutorial on B Cell Survival. PubMed NCBI. Available online: https://www.ncbi.nlm.nih.gov/pubmed/12427767 (accessed on 2 April 2020).
- Pers, J.-O.; Daridon, C.; Devauchelle, V.; Jousse, S.; Saraux, A.; Jamin, C.; Youinou, P. BAFF overexpression is associated with autoantibody production in autoimmune diseases. Ann. N. Y. Acad. Sci. 2005, 1050, 34–39. [Google Scholar] [CrossRef]
- Nieuwenhuis, P.; Opstelten, D. Functional anatomy of germinal centers. Am. J. Anat. 1984, 170, 421–435. [Google Scholar] [CrossRef]
- Maeda, T.; Wakasawa, T.; Shima, Y.; Tsuboi, I.; Aizawa, S.; Tamai, I. Role of polyamines derived from arginine in differentiation and proliferation of human blood cells. Biol. Pharm. Bull. 2006, 29, 234–239. [Google Scholar] [CrossRef] [Green Version]
- Meyer-Hermann, M. A mathematical model for the germinal center morphology and affinity maturation. J. Theor. Biol. 2002, 216, 273–300. [Google Scholar] [CrossRef] [Green Version]
- Jonsson, M.V.; Skarstein, K. Follicular dendritic cells confirm lymphoid organization in the minor salivary glands of primary Sjögren’s syndrome. J. Oral Pathol. Med. 2008, 37, 515–521. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, M.V.; Skarstein, K.; Jonsson, R.; Brun, J.G. Serological implications of germinal center-like structures in primary Sjögren’s syndrome. J. Rheumatol. 2007, 34, 2044–2049. [Google Scholar]
- Salomonsson, S.; Jonsson, M.V.; Skarstein, K.; Brokstad, K.A.; Hjelmström, P.; Wahren-Herlenius, M.; Jonsson, R. Cellular basis of ectopic germinal center formation and autoantibody production in the target organ of patients with Sjögren’s syndrome. Arthritis Rheum. 2003, 48, 3187–3201. [Google Scholar] [CrossRef] [PubMed]
- Johnsen, S.J.; Brun, J.G.; Gøransson, L.G.; Småstuen, M.C.; Johannesen, T.B.; Haldorsen, K.; Harboe, E.; Jonsson, R.; Meyer, P.A.; Omdal, R. Risk of non-Hodgkin’s lymphoma in primary Sjögren’s syndrome: A population-based study. Arthritis Care Res. 2013, 65, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Theander, E.; Henriksson, G.; Ljungberg, O.; Mandl, T.; Manthorpe, R.; Jacobsson, L.T.H. Lymphoma and other malignancies in primary Sjögren’s syndrome: A cohort study on cancer incidence and lymphoma predictors. Ann. Rheum. Dis. 2006, 65, 796–803. [Google Scholar] [CrossRef] [Green Version]
- Nardi, N.; Brito-Zerón, P.; Ramos-Casals, M.; Aguiló, S.; Cervera, R.; Ingelmo, M.; Font, J. Circulating auto-antibodies against nuclear and non-nuclear antigens in primary Sjögren’s syndrome: Prevalence and clinical significance in 335 patients. Clin. Rheumatol. 2006, 25, 341–346. [Google Scholar] [CrossRef]
- Jones, B. Lacrimal and salivary precipitating antibodies in Sjögren’s syndrome. Lancet 1958, 272, 773–776. [Google Scholar] [CrossRef]
- Anderson, J.R.; Gray, K.; Beck, J.S.; Kinnear, W.F. Precipitating autoantibodies in Sjögren’s syndrome. Lancet 1961, 278, 456–460. [Google Scholar] [CrossRef]
- Espinosa, A.; Dardalhon, V.; Brauner, S.; Ambrosi, A.; Higgs, R.; Quintana, F.J.; Sjöstrand, M.; Eloranta, M.L.; Ní Gabhann, J.; Winqvist, O.; et al. Loss of the lupus autoantigen Ro52/Trim21 induces tissue inflammation and systemic autoimmunity by disregulating the IL-23-Th17 pathway. J. Exp. Med. 2009, 206, 1661–1671. [Google Scholar] [CrossRef]
- Keene, J.D. Molecular structure of the La and Ro autoantigens and their use in autoimmune diagnostics. J. Autoimmun. 1989, 2, 329–334. [Google Scholar] [CrossRef]
- Elkon, K.B.; Gharavi, A.E.; Hughes, G.R.; Moutsoupoulos, H.M. Autoantibodies in the sicca syndrome (primary Sjögren’s syndrome). Ann. Rheum. Dis. 1984, 43, 243–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baer, A.N.; McAdams DeMarco, M.; Shiboski, S.C.; Lam, M.Y.; Challacombe, S.; Daniels, T.E.; Dong, Y.; Greenspan, J.S.; Kirkham, B.W.; Lanfranchi, H.E.; et al. The SSB-positive/SSA-negative antibody profile is not associated with key phenotypic features of Sjögren’s syndrome. Ann. Rheum. Dis. 2015, 74, 1557–1561. [Google Scholar] [CrossRef] [PubMed]
- Manoussakis, M.N.; Pange, P.J.; Moutsopulos, H.M. The autoantibody profile in Sjögren’s syndrome. Ter. Arkh. 1988, 60, 17–20. [Google Scholar] [PubMed]
- Mavragani, C.P.; Tzioufas, A.G.; Moutsopoulos, H.M. Sjögren’s syndrome: Autoantibodies to cellular antigens. Clinical and molecular aspects. Int. Arch. Allergy Immunol. 2000, 123, 46–57. [Google Scholar] [CrossRef]
- Bournia, V.-K.K.; Diamanti, K.D.; Vlachoyiannopoulos, P.G.; Moutsopoulos, H.M. Anticentromere antibody positive Sjögren’s Syndrome: A retrospective descriptive analysis. Arthritis Res. Ther. 2010, 12, R47. [Google Scholar] [CrossRef] [Green Version]
- Salliot, C.; Gottenberg, J.-E.; Bengoufa, D.; Desmoulins, F.; Miceli-Richard, C.; Mariette, X. Anticentromere antibodies identify patients with Sjögren’s syndrome and autoimmune overlap syndrome. J. Rheumatol. 2007, 34, 2253–2258. [Google Scholar]
- Bournia, V.-K.; Vlachoyiannopoulos, P.G. Subgroups of Sjögren syndrome patients according to serological profiles. J. Autoimmun. 2012, 39, 15–26. [Google Scholar] [CrossRef]
- Kyriakidis, N.C.; Kapsogeorgou, E.K.; Tzioufas, A.G. A comprehensive review of autoantibodies in primary Sjögren’s syndrome: Clinical phenotypes and regulatory mechanisms. J. Autoimmun. 2014, 51, 67–74. [Google Scholar] [CrossRef]
- Takemoto, F.; Hoshino, J.; Sawa, N.; Tamura, Y.; Tagami, T.; Yokota, M.; Katori, H.; Yokoyama, K.; Ubara, Y.; Hara, S.; et al. Autoantibodies against carbonic anhydrase II are increased in renal tubular acidosis associated with Sjogren syndrome. Am. J. Med. 2005, 118, 181–184. [Google Scholar] [CrossRef]
- Nishimori, I.; Bratanova, T.; Toshkov, I.; Caffrey, T.; Mogaki, M.; Shibata, Y.; Hollingsworth, M.A. Induction of experimental autoimmune sialoadenitis by immunization of PL/J mice with carbonic anhydrase II. J. Immunol. 1950 1995, 154, 4865–4873. [Google Scholar]
- Takemoto, F.; Katori, H.; Sawa, N.; Hoshino, J.; Suwabe, T.; Sogawa, Y.; Nomura, K.; Nakanishi, S.; Higa, Y.; Kanbayashi, H.; et al. Induction of anti-carbonic-anhydrase-II antibody causes renal tubular acidosis in a mouse model of Sjogren’s syndrome. Nephron Physiol. 2007, 106, p63–p68. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.; Lee, J.; Park, S.-H.; Kim, H.-D.; Choi, Y. Associations of Anti-Aquaporin 5 Autoantibodies with Serologic and Histopathological Features of Sjögren’s Syndrome. J. Clin. Med. 2019, 8, 1863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clinical Associations of Autoantibodies to Human Muscarinic Acetylcholine Receptor 3 (213–228) in Primary Sjogren’s Syndrome. PubMed NCBI. Available online: https://www.ncbi.nlm.nih.gov/pubmed/?term=10.1093%2Frheumatology%2Fkeh672 (accessed on 2 April 2020).
- Sordet, C.; Gottenberg, J.E.; Goetz, J.; Bengoufa, D.; Humbel, R.-L.; Mariette, X.; Sibilia, J. Anti-α-fodrin autoantibodies are not useful diagnostic markers of primary Sjögren’s syndrome. Ann. Rheum. Dis. 2005, 64, 1244–1245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Applbaum, E.; Lichtbroun, A. Novel Sjögren’s autoantibodies found in fibromyalgia patients with sicca and/or xerostomia. Autoimmun. Rev. 2019, 18, 199–202. [Google Scholar] [CrossRef] [PubMed]
- Martín-Nares, E.; Hernández-Molina, G. Novel autoantibodies in Sjögren’s syndrome: A comprehensive review. Autoimmun. Rev. 2019, 18, 192–198. [Google Scholar] [CrossRef] [PubMed]
- De Langhe, E.; Bossuyt, X.; Shen, L.; Malyavantham, K.; Ambrus, J.L.; Suresh, L. Evaluation of Autoantibodies in Patients with Primary and Secondary Sjogren’s Syndrome. Open Rheumatol. J. 2017, 11, 10–15. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.; Li, J.; Chen, J.; Shao, M.; Zhang, R.; Liang, Y.; Zhang, X.; Zhang, X.; Zhang, Q.; Li, F.; et al. Tissue-Specific Autoantibodies Improve Diagnosis of Primary Sjögren’s Syndrome in the Early Stage and Indicate Localized Salivary Injury. J. Immunol. Res. 2019, 2019, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Suresh, L.; Malyavantham, K.; Shen, L.; Ambrus, J.L. Investigation of novel autoantibodies in Sjogren’s syndrome utilizing Sera from the Sjogren’s international collaborative clinical alliance cohort. BMC Ophthalmol. 2015, 15, 38. [Google Scholar] [CrossRef] [Green Version]
- Shen, L.; Suresh, L.; Lindemann, M.; Xuan, J.; Kowal, P.; Malyavantham, K.; Ambrus, J.L. Novel autoantibodies in Sjogren’s syndrome. Clin. Immunol. 2012, 145, 251–255. [Google Scholar] [CrossRef]
- Xuan, J.; Wang, Y.; Xiong, Y.; Qian, H.; He, Y.; Shi, G. Investigation of autoantibodies to SP-1 in Chinese patients with primary Sjögren’s syndrome. Clin. Immunol. 2018, 188, 58–63. [Google Scholar] [CrossRef]
- Everett, S.; Vishwanath, S.; Cavero, V.; Shen, L.; Suresh, L.; Malyavantham, K.; Lincoff-Cohen, N.; Ambrus, J.L. Analysis of novel Sjogren’s syndrome autoantibodies in patients with dry eyes. BMC Ophthalmol. 2017, 17, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubschman, S.; Rojas, M.; Kalavar, M.; Kloosterboer, A.; Sabater, A.L.; Galor, A. Association Between Early Sjögren Markers and Symptoms and Signs of Dry Eye. Cornea 2020, 39, 311–315. [Google Scholar] [CrossRef] [PubMed]
- Uchida, K.; Akita, Y.; Matsuo, K.; Fujiwara, S.; Nakagawa, A.; Kazaoka, Y.; Hachiya, H.; Naganawa, Y.; Oh-Iwa, I.; Ohura, K.; et al. Identification of specific autoantigens in Sjögren’s syndrome by SEREX. Immunology 2005, 116, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liao, X.; Wang, Y.; Chen, S.; Sun, Y.; Lin, Q.; Shi, G. Autoantibody to MDM2: A potential serological marker of primary Sjogren’s syndrome. Oncotarget 2017, 8, 14306–14313. [Google Scholar] [CrossRef] [PubMed]
- Nozawa, K.; Ikeda, K.; Satoh, M.; Reeves, W.H.; Stewart, C.M.; Li, Y.-C.; Yen, T.J.; Rios, R.M.; Takamori, K.; Ogawa, H.; et al. Autoantibody to NA14 is an independent marker primarily for Sjogren’s syndrome. Front. Biosci. Landmark Ed. 2009, 14, 3733–3739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uomori, K.; Nozawa, K.; Ikeda, K.; Doe, K.; Yamada, Y.; Yamaguchi, A.; Fujishiro, M.; Kawasaki, M.; Morimoto, S.; Takamori, K.; et al. A re-evaluation of anti-NA-14 antibodies in patients with primary Sjögren’s syndrome: Significant role of interferon-γ in the production of autoantibodies against NA-14. Autoimmunity 2016, 49, 347–356. [Google Scholar] [CrossRef]
- Duda, S.; Witte, T.; Stangel, M.; Adams, J.; Schmidt, R.E.; Baerlecken, N.T. Autoantibodies binding to stathmin-4: New marker for polyneuropathy in primary Sjögren’s syndrome. Immunol. Res. 2017, 65, 1099–1102. [Google Scholar] [CrossRef]
- Fiorentino, D.F.; Presby, M.; Baer, A.N.; Petri, M.; Rieger, K.E.; Soloski, M.; Rosen, A.; Mammen, A.L.; Christopher-Stine, L.; Casciola-Rosen, L. PUF60: A prominent new target of the autoimmune response in dermatomyositis and Sjögren’s syndrome. Ann. Rheum. Dis. 2016, 75, 1145–1151. [Google Scholar] [CrossRef]
- Tay, S.H.; Fairhurst, A.-M.; Mak, A. Clinical utility of circulating anti-N-methyl-d-aspartate receptor subunits NR2A/B antibody for the diagnosis of neuropsychiatric syndromes in systemic lupus erythematosus and Sjögren’s syndrome: An updated meta-analysis. Autoimmun. Rev. 2017, 16, 114–122. [Google Scholar] [CrossRef]
- Lauvsnes, M.B.; Beyer, M.K.; Kvaløy, J.T.; Greve, O.J.; Appenzeller, S.; Kvivik, I.; Harboe, E.; Tjensvoll, A.B.; Gøransson, L.G.; Omdal, R. Association of hippocampal atrophy with cerebrospinal fluid antibodies against the NR2 subtype of the N-methyl-D-aspartate receptor in patients with systemic lupus erythematosus and patients with primary Sjögren’s syndrome. Arthritis Rheumatol. 2014, 66, 3387–3394. [Google Scholar] [CrossRef]
- Wolska, N.; Rybakowska, P.; Rasmussen, A.; Brown, M.; Montgomery, C.; Klopocki, A.; Grundahl, K.; Scofield, R.H.; Radfar, L.; Stone, D.U.; et al. Brief Report: Patients With Primary Sjögren’s Syndrome Who Are Positive for Autoantibodies to Tripartite Motif-Containing Protein 38 Show Greater Disease Severity. Arthritis Rheumatol. 2016, 68, 724–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alunno, A.; Bistoni, O.; Carubbi, F.; Valentini, V.; Cafaro, G.; Bartoloni, E.; Giacomelli, R.; Gerli, R. Prevalence and significance of anti-saccharomyces cerevisiae antibodies in primary Sjögren’s syndrome. Clin. Exp. Rheumatol. 2018, 36, 73–79. [Google Scholar] [PubMed]
- Birnbaum, J.; Hoke, A.; Lalji, A.; Calabresi, P.; Bhargava, P.; Casciola-Rosen, L. Brief Report: Anti-Calponin 3 Autoantibodies: A Newly Identified Specificity in Patients With Sjögren’s Syndrome. Arthritis Rheumatol. 2018, 70, 1610–1616. [Google Scholar] [CrossRef] [Green Version]
- Mukaino, A.; Nakane, S.; Higuchi, O.; Nakamura, H.; Miyagi, T.; Shiroma, K.; Tokashiki, T.; Fuseya, Y.; Ochi, K.; Umeda, M.; et al. Insights from the ganglionic acetylcholine receptor autoantibodies in patients with Sjögren’s syndrome. Mod. Rheumatol. 2016, 26, 708–715. [Google Scholar] [CrossRef] [PubMed]
- Birnbaum, J.; Atri, N.M.; Baer, A.N.; Cimbro, R.; Montagne, J.; Casciola-Rosen, L. Relationship Between Neuromyelitis Optica Spectrum Disorder and Sjögren’s Syndrome: Central Nervous System Extraglandular Disease or Unrelated, Co-Occurring Autoimmunity?: Relationship Between Sjögren’s Syndrome and NMOSD. Arthritis Care Res. 2017, 69, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Tzartos, J.S.; Stergiou, C.; Daoussis, D.; Zisimopoulou, P.; Andonopoulos, A.P.; Zolota, V.; Tzartos, S.J. Antibodies to aquaporins are frequent in patients with primary Sjögren’s syndrome. Rheumatology 2017, 56, 2114–2122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.-H.; Zhou, P.-F.; Long, G.-F.; Tian, X.; Guo, Y.-F.; Pang, A.-M.; Di, R.; Shen, Y.-N.; Liu, Y.-D.; Cui, Y.-J. Elevated Plasma P-Selectin Autoantibodies in Primary Sjögren Syndrome Patients with Thrombocytopenia. Med. Sci. Monit. 2015, 21, 3690–3695. [Google Scholar] [CrossRef] [Green Version]
- Bergum, B.; Koro, C.; Delaleu, N.; Solheim, M.; Hellvard, A.; Binder, V.; Jonsson, R.; Valim, V.; Hammenfors, D.S.; Jonsson, M.V.; et al. Antibodies against carbamylated proteins are present in primary Sjögren’s syndrome and are associated with disease severity. Ann. Rheum. Dis. 2016, 75, 1494–1500. [Google Scholar] [CrossRef] [Green Version]
- Pecani, A.; Alessandri, C.; Spinelli, F.R.; Priori, R.; Riccieri, V.; Di Franco, M.; Ceccarelli, F.; Colasanti, T.; Pendolino, M.; Mancini, R.; et al. Prevalence, sensitivity and specificity of antibodies against carbamylated proteins in a monocentric cohort of patients with rheumatoid arthritis and other autoimmune rheumatic diseases. Arthritis Res. Ther. 2016, 18, 276. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Hussain, M.; Yang, X.; Chen, P.; Yang, C.; Xun, Y.; Tian, Y.; Du, H. Identification of Moesin as a Novel Autoantigen in Patients with Sjögren’s Syndrome. Protein Pept. Lett. 2018, 25, 350–355. [Google Scholar] [CrossRef]
- Cui, L.; Elzakra, N.; Xu, S.; Xiao, G.G.; Yang, Y.; Hu, S. Investigation of three potential autoantibodies in Sjogren’s syndrome and associated MALT lymphoma. Oncotarget 2017, 8, 30039–30049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nezos, A.; Cinoku, I.; Mavragani, C.P.; Moutsopoulos, H.M. Antibodies against citrullinated alpha enolase peptides in primary Sjogren’s syndrome. Clin. Immunol. 2017, 183, 300–303. [Google Scholar] [CrossRef] [PubMed]
- Segerberg-Konttinen, M.; Konttinen, Y.T.; Bergroth, V. Focus score in the diagnosis of Sjögren’s syndrome. Scand. J. Rheumatol. Suppl. 1986, 61, 47–51. [Google Scholar] [PubMed]
- Bodeutsch, C.; de Wilde, P.C.; Kater, L.; van Houwelingen, J.C.; van den Hoogen, F.H.; Kruize, A.A.; Hené, R.J.; van de Putte, L.B.; Vooijs, G.P. Quantitative immunohistologic criteria are superior to the lymphocytic focus score criterion for the diagnosis of Sjögren’s syndrome. Arthritis Rheum. 1992, 35, 1075–1087. [Google Scholar] [CrossRef]
- Barrera, M.J.; Bahamondes, V.; Sepúlveda, D.; Quest, A.F.G.; Castro, I.; Cortés, J.; Aguilera, S.; Urzúa, U.; Molina, C.; Pérez, P.; et al. Sjögren’s syndrome and the epithelial target: A comprehensive review. J. Autoimmun. 2013, 42, 7–18. [Google Scholar] [CrossRef]
- Pérez, P.; Goicovich, E.; Alliende, C.; Aguilera, S.; Leyton, C.; Molina, C.; Pinto, R.; Romo, R.; Martinez, B.; González, M.J. Differential expression of matrix metalloproteinases in labial salivary glands of patients with primary Sjögren’s syndrome. Arthritis Rheum. 2000, 43, 2807–2817. [Google Scholar] [CrossRef]
- Sun, D.; Emmert-Buck, M.R.; Fox, P.C. Differential cytokine mRNA expression in human labial minor salivary glands in primary Sjögren’s syndrome. Autoimmunity 1998, 28, 125–137. [Google Scholar] [CrossRef]
- Molina, C.; Alliende, C.; Aguilera, S.; Kwon, Y.-J.; Leyton, L.; Martínez, B.; Leyton, C.; Pérez, P.; González, M.-J. Basal lamina disorganisation of the acini and ducts of labial salivary glands from patients with Sjogren’s syndrome: Association with mononuclear cell infiltration. Ann. Rheum. Dis. 2006, 65, 178–183. [Google Scholar] [CrossRef] [Green Version]
- Ng, W.-F.; Bowman, S.J. Primary Sjogren’s syndrome: Too dry and too tired. Rheumatology 2010, 49, 844–853. [Google Scholar] [CrossRef] [Green Version]
- Hackett, K.L.; Gotts, Z.M.; Ellis, J.; Deary, V.; Rapley, T.; Ng, W.-F.; Newton, J.L.; Deane, K.H.O. An investigation into the prevalence of sleep disturbances in primary Sjögren’s syndrome: A systematic review of the literature. Rheumatology 2017, 56, 570–580. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.-C.; Chang, K.; Lin, C.-Y.; Chen, Y.-H.; Lu, P.-L. Periodic fever as the manifestation of primary Sjogren’s syndrome: A case report and literature review. Clin. Rheumatol. 2012, 31, 1517–1519. [Google Scholar] [CrossRef] [PubMed]
- Voulgarelis, M.; Moutsopoulos, H.M. Mucosa-associated lymphoid tissue lymphoma in Sjögren’s syndrome: Risks, management, and prognosis. Rheum. Dis. Clin. N. Am. 2008, 34, 921–933. [Google Scholar] [CrossRef] [PubMed]
- Kassan, S.S.; Moutsopoulos, H.M. Clinical manifestations and early diagnosis of Sjögren syndrome. Arch. Intern. Med. 2004, 164, 1275–1284. [Google Scholar] [CrossRef]
- Retamozo, S.; Acar-Denizli, N.; Rasmussen, A.; Horváth, I.F.; Baldini, C.; Priori, R.; Sandhya, P.; Hernandez-Molina, G.; Armagan, B.; Praprotnik, S.; et al. Systemic manifestations of primary Sjögren’s syndrome out of the ESSDAI classification: Prevalence and clinical relevance in a large international, multi-ethnic cohort of patients. Clin. Exp. Rheumatol. 2019, 37, 97–106. [Google Scholar] [PubMed]
- López-Pintor, R.M.; Fernández Castro, M.; Hernández, G. Oral involvement in patients with primary Sjögren’s syndrome. Multidisciplinary care by dentists and rheumatologists. Reumatol. Clin. 2015, 11, 387–394. [Google Scholar] [CrossRef] [PubMed]
- Generali, E.; Costanzo, A.; Mainetti, C.; Selmi, C. Cutaneous and Mucosal Manifestations of Sjögren’s Syndrome. Clin. Rev. Allergy Immunol. 2017, 53, 357–370. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Casals, M.; Brito-Zerón, P.; Bombardieri, S.; Bootsma, H.; De Vita, S.; Dörner, T.; Fisher, B.A.; Gottenberg, J.-E.; Hernandez-Molina, G.; Kocher, A.; et al. EULAR recommendations for the management of Sjögren’s syndrome with topical and systemic therapies. Ann. Rheum. Dis. 2020, 79, 3–18. [Google Scholar] [CrossRef] [Green Version]
- Ramos-Casals, M.; Brito-Zerón, P.; Seror, R.; Bootsma, H.; Bowman, S.J.; Dörner, T.; Gottenberg, J.-E.; Mariette, X.; Theander, E.; Bombardieri, S.; et al. Characterization of systemic disease in primary Sjögren’s syndrome: EULAR-SS Task Force recommendations for articular, cutaneous, pulmonary and renal involvements. Rheumatology 2015, 54, 2230–2238. [Google Scholar] [CrossRef] [Green Version]
- Mirouse, A.; Seror, R.; Vicaut, E.; Mariette, X.; Dougados, M.; Fauchais, A.-L.; Deroux, A.; Dellal, A.; Costedoat-Chalumeau, N.; Denis, G.; et al. Arthritis in primary Sjögren’s syndrome: Characteristics, outcome and treatment from French multicenter retrospective study. Autoimmun. Rev. 2019, 18, 9–14. [Google Scholar] [CrossRef]
- Vitali, C.; Del Papa, N. Pain in primary Sjögren’s syndrome. Best Pract. Res. Clin. Rheumatol. 2015, 29, 63–70. [Google Scholar] [CrossRef]
- Atzeni, F.; Cazzola, M.; Benucci, M.; Di Franco, M.; Salaffi, F.; Sarzi-Puttini, P. Chronic widespread pain in the spectrum of rheumatological diseases. Best Pract. Res. Clin. Rheumatol. 2011, 25, 165–171. [Google Scholar] [CrossRef]
- Alunno, A.; Carubbi, F.; Bartoloni, E.; Cipriani, P.; Giacomelli, R.; Gerli, R. The kaleidoscope of neurological manifestations in primary Sjögren’s syndrome. Clin. Exp. Rheumatol. 2019, 37, 192–198. [Google Scholar]
- Flament, T.; Bigot, A.; Chaigne, B.; Henique, H.; Diot, E.; Marchand-Adam, S. Pulmonary manifestations of Sjögren’s syndrome. Eur. Respir. Rev. 2016, 25, 110–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatron, P.-Y.; Tillie-Leblond, I.; Launay, D.; Hachulla, E.; Fauchais, A.L.; Wallaert, B. Pulmonary manifestations of Sjögren’s syndrome. Presse Med. 1983 2011, 40, e49–e64. [Google Scholar] [CrossRef] [PubMed]
- Tavoni, A.; Vitali, C.; Cirigliano, G.; Frigelli, S.; Stampacchia, G.; Bombardieri, S. Shrinking lung in primary Sjögren’s syndrome. Arthritis Rheum. 1999, 42, 2249–2250. [Google Scholar] [CrossRef]
- Singh, R.; Huang, W.; Menon, Y.; Espinoza, L.R. Shrinking lung syndrome in systemic lupus erythematosus and Sjogren’s syndrome. J. Clin. Rheumatol. 2002, 8, 340–345. [Google Scholar] [CrossRef] [PubMed]
- Langenskiöld, E.; Bonetti, A.; Fitting, J.W.; Heinzer, R.; Dudler, J.; Spertini, F.; Lazor, R. Shrinking lung syndrome successfully treated with rituximab and cyclophosphamide. Respiration 2012, 84, 144–149. [Google Scholar] [CrossRef]
- Blanco Pérez, J.J.; Pérez González, A.; Guerra Vales, J.L.; Melero Gonzalez, R.; Pego Reigosa, J.M. Shrinking Lung in Primary Sjogrën Syndrome Successfully Treated with Rituximab. Arch. Bronconeumol. 2015, 51, 475–476. [Google Scholar] [CrossRef] [PubMed]
- Baenas, D.F.; Retamozo, S.; Pirola, J.P.; Caeiro, F. Shrinking lung syndrome and pleural effusion as an initial manifestation of primary Sjögren’s syndrome. Síndrome de pulmón encogido y derrame pleural como manifestación inicial de síndrome de Sjögren primario. Rheumatol. Clin. 2020, 16, 65–68. [Google Scholar] [CrossRef]
- Uslu, S.; Köken Avşar, A.; Erez, Y.; Sarı, İ. Shrinking Lung Syndrome in Primary Sjögren Syndrome. Balk. Med. J. 2020. [Google Scholar] [CrossRef]
- Liang, M.; Bao, L.; Xiong, N.; Jin, B.; Ni, H.; Zhang, J.; Zou, H.; Luo, X.; Li, J. Cardiac arrhythmias as the initial manifestation of adult primary Sjögren’s syndrome: A case report and literature review. Int. J. Rheum. Dis. 2015, 18, 800–806. [Google Scholar] [CrossRef] [PubMed]
- Sung, M.J.; Park, S.-H.; Kim, S.-K.; Lee, Y.-S.; Park, C.-Y.; Choe, J.-Y. Complete atrioventricular block in adult Sjögren’s syndrome with anti-Ro autoantibody. Kor. J. Intern. Med. 2011, 26, 213–215. [Google Scholar] [CrossRef] [PubMed]
- Popov, Y.; Salomon-Escoto, K. Gastrointestinal and Hepatic Disease in Sjogren Syndrome. Rheum. Dis. Clin. N. Am. 2018, 44, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Ebert, E.C. Gastrointestinal and hepatic manifestations of Sjogren syndrome. J. Clin. Gastroenterol. 2012, 46, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Evans, R.; Zdebik, A.; Ciurtin, C.; Walsh, S.B. Renal involvement in primary Sjögren’s syndrome. Rheumatology 2015, 54, 1541–1548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geng, Y.; Zhao, Y.; Zhang, Z. Tubulointerstitial nephritis-induced hypophosphatemic osteomalacia in Sjögren’s syndrome: A case report and review of the literature. Clin. Rheumatol. 2018, 37, 257–263. [Google Scholar] [CrossRef]
- Gu, X.; Su, Z.; Chen, M.; Xu, Y.; Wang, Y. Acquired Gitelman syndrome in a primary Sjögren syndrome patient with a SLC12A3 heterozygous mutation: A case report and literature review. Nephrology 2017, 22, 652–655. [Google Scholar] [CrossRef] [Green Version]
- Darrieutort-Laffite, C.; André, V.; Hayem, G.; Saraux, A.; Le Guern, V.; Le Jeunne, C.; Puéchal, X. Sjögren’s syndrome complicated by interstitial cystitis: A case series and literature review. Joint Bone Spine 2015, 82, 245–250. [Google Scholar] [CrossRef]
- Manganelli, P.; Fietta, P.; Quaini, F. Hematologic manifestations of primary Sjögren’s syndrome. Clin. Exp. Rheumatol. 2006, 24, 438–448. [Google Scholar]
- Ramos-Casals, M.; Font, J.; Garcia-Carrasco, M.; Brito, M.-P.; Rosas, J.; Calvo-Alen, J.; Pallares, L.; Cervera, R.; Ingelmo, M. Primary Sjögren syndrome: Hematologic patterns of disease expression. Medicine 2002, 81, 281–292. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, H.; Takahashi, Y.; Kaneko, H.; Kano, T.; Mimori, A. Thrombotic thrombocytopenic purpura with an autoantibody to ADAMTS13 complicating Sjögren’s syndrome: Two cases and a literature review. Mod. Rheumatol. 2013, 23, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zhu, T.; Wu, D.; Zhang, L. Sjögren’s syndrome initially presented as thrombotic thrombocytopenic purpura in a male patient: A case report and literature review. Clin. Rheumatol. 2018, 37, 1421–1426. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Gu, W.; Ma, Y.; Wang, J.; Wu, M. Relapsed/refractory acquired thrombotic thrombocytopenic purpura in a patient with Sjögren syndrome: Case report and review of the literature. Medicine 2018, 97, e12989. [Google Scholar] [CrossRef]
- García-Montoya, L.; Sáenz-Tenorio, C.N.; Janta, I.; Menárguez, J.; López-Longo, F.J.; Monteagudo, I.; Naredo, E. Hemophagocytic lymphohistiocytosis in a patient with Sjögren’s syndrome: Case report and review. Rheumatol. Int. 2017, 37, 663–669. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Molina, G.; Faz-Munoz, D.; Astudillo-Angel, M.; Iturralde-Chavez, A.; Reyes, E. Coexistance of Amyloidosis and Primary Sjögren’s Syndrome: An Overview. Curr. Rheumatol. Rev. 2018, 14, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Freeman, S.R.M.; Sheehan, P.Z.; Thorpe, M.A.; Rutka, J.A. Ear, Nose, and Throat Manifestations of Sjögren’s Syndrome: Retrospective Review of a Multidisciplinary Clinic. J. Otolaryngol. 2005, 34, 20. [Google Scholar] [CrossRef] [PubMed]
- Midilli, R.; Gode, S.; Oder, G.; Kabasakal, Y.; Karci, B. Nasal and paranasal involvement in primary Sjogren‘s syndrome. Rhinol. J. 2013, 51, 265–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belafsky, P.C.; Postma, G.N. The laryngeal and esophageal manifestations of Sjögren’s syndrome. Curr. Rheumatol. Rep. 2003, 5, 297–303. [Google Scholar] [CrossRef]
- Rodriguez, M.A.; Tapanes, F.J.; Stekman, I.L.; Pinto, J.A.; Camejo, O.; Abadi, I. Auricular chondritis and diffuse proliferative glomerulonephritis in primary Sjogren’s syndrome. Ann. Rheum. Dis. 1989, 48, 683–685. [Google Scholar] [CrossRef] [Green Version]
- Tumiati, B. Hearing Loss in the Sjogren Syndrome. Ann. Intern. Med. 1997, 126, 450. [Google Scholar] [CrossRef]
- Isik, H.; Isik, M.; Aynioglu, O.; Karcaaltincaba, D.; Sahbaz, A.; Beyazcicek, T.; Harma, M.I.; Demircan, N. Are the women with Sjögren’s Syndrome satisfied with their sexual activity? Rev. Bras. Reumatol. Engl. Ed. 2017, 57, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Capone, C.; Buyon, J.P.; Friedman, D.M.; Frishman, W.H. Cardiac Manifestations of Neonatal Lupus: A Review of Autoantibody-associated Congenital Heart Block and its Impact in an Adult Population. Cardiol. Rev. 2012, 20, 72–76. [Google Scholar] [CrossRef] [Green Version]
- Picone, O.; Alby, C.; Frydman, R.; Mariette, X. Sjögren syndrome in Obstetric and Gynecology: Literature review. J. Gynecol. Obstet. Biol. Reprod. 2006, 35, 169–175. [Google Scholar] [CrossRef]
- Costedoat-Chalumeau, N.; Amoura, Z.; Villain, E.; Cohen, L.; Fermont, L.; Le Thi Huong, D.; Vauthier, D.; Georgin-Lavialle, S.; Wechsler, B.; Dommergues, M.; et al. Prise en charge obstétricale des patientes à risque de « lupus néonatal ». J. Gynécologie Obstétrique Biol. Reprod. 2006, 35, 146–156. [Google Scholar] [CrossRef]
- Upala, S.; Yong, W.C.; Sanguankeo, A. Association between primary Sjögren’s syndrome and pregnancy complications: A systematic review and meta-analysis. Clin. Rheumatol. 2016, 35, 1949–1955. [Google Scholar] [CrossRef]
- Brito-Zerón, P.; Theander, E.; Baldini, C.; Seror, R.; Retamozo, S.; Quartuccio, L.; Bootsma, H.; Bowman, S.J.; Dörner, T.; Gottenberg, J.-E.; et al. Early diagnosis of primary Sjögren’s syndrome: EULAR-SS task force clinical recommendations. Expert Rev. Clin. Immunol. 2016, 12, 137–156. [Google Scholar] [CrossRef]
- Vitali, C.; Bombardieri, S.; Jonsson, R.; Moutsopoulos, H.M.; Alexander, E.L.; Carsons, S.E.; Daniels, T.E.; Fox, P.C.; Fox, R.I.; Kassan, S.S.; et al. Classification criteria for Sjögren’s syndrome: A revised version of the European criteria proposed by the American-European Consensus Group. Ann. Rheum. Dis. 2002, 61, 554–558. [Google Scholar] [CrossRef] [Green Version]
- Shiboski, S.C.; Shiboski, C.H.; Criswell, L.A.; Baer, A.N.; Challacombe, S.; Lanfranchi, H.; Schiødt, M.; Umehara, H.; Vivino, F.; Zhao, Y.; et al. American College of Rheumatology classification criteria for Sjögren’s syndrome: A data-driven, expert consensus approach in the Sjögren’s International Collaborative Clinical Alliance cohort. Arthritis Care Res. 2012, 64, 475–487. [Google Scholar] [CrossRef]
- Shiboski, C.H.; Shiboski, S.C.; Seror, R.; Criswell, L.A.; Labetoulle, M.; Lietman, T.M.; Rasmussen, A.; Scofield, H.; Vitali, C.; Bowman, S.J.; et al. 2016 American College of Rheumatology/European League Against Rheumatism Classification Criteria for Primary Sjögren’s Syndrome: A Consensus and Data-Driven Methodology Involving Three International Patient Cohorts. Arthritis Rheumatol. 2017, 69, 35–45. [Google Scholar] [CrossRef]
- Begley, C.; Caffery, B.; Chalmers, R.; Situ, P.; Simpson, T.; Nelson, J.D. Review and analysis of grading scales for ocular surface staining. Ocul. Surf. 2019, 7, 208–220. [Google Scholar] [CrossRef]
- Baldini, C.; Zabotti, A.; Filipovic, N.; Vukicevic, A.; Luciano, N.; Ferro, F.; Lorenzon, M.; De Vita, S. Imaging in primary Sjögren’s syndrome: The “obsolete and the new”. Clin. Exp. Rheumatol. 2018, 36, 215–221. [Google Scholar] [PubMed]
- Schall, G.L.; Anderson, L.G.; Wolf, R.O.; Herdt, J.R.; Tarpley, T.M.; Cummings, N.A.; Zeiger, L.S.; Talal, N. Xerostomia in Sjögren’s syndrome. Evaluation by sequential salivary scintigraphy. JAMA 1971, 216, 2109–2116. [Google Scholar] [CrossRef]
- Vinagre, F.; Santos, M.J.; Prata, A.; da Silva, J.C.; Santos, A.I. Assessment of salivary gland function in Sjögren’s syndrome: The role of salivary gland scintigraphy. Autoimmun. Rev. 2009, 8, 672–676. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Song, S.; Wu, S.; Duan, T.; Chen, L.; Ye, J.; Xiao, J. Diagnostic accuracy of salivary gland ultrasonography with different scoring systems in Sjögren’s syndrome: A systematic review and meta-analysis. Sci. Rep. 2018, 8, 17128. [Google Scholar] [CrossRef] [PubMed]
- Jousse-Joulin, S.; Milic, V.; Jonsson, M.V.; Plagou, A.; Theander, E.; Luciano, N.; Rachele, P.; Baldini, C.; Bootsma, H.; Vissink, A.; et al. Is salivary gland ultrasonography a useful tool in Sjögren’s syndrome? A systematic review. Rheumatology 2016, 55, 789–800. [Google Scholar] [CrossRef] [Green Version]
- Nimwegen, J.F.; Mossel, E.; Delli, K.; Ginkel, M.S.; Stel, A.J.; Kroese, F.G.M.; Spijkervet, F.K.L.; Vissink, A.; Arends, S.; Bootsma, H. Incorporation of Salivary Gland Ultrasonography Into the American College of Rheumatology/European League Against Rheumatism Criteria for Primary Sjögren’s Syndrome. Arthritis Care Res. 2020, 72, 583–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, B.A.; Everett, C.C.; Rout, J.; O’Dwyer, J.L.; Emery, P.; Pitzalis, C.; Ng, W.-F.; Carr, A.; Pease, C.T.; Price, E.J.; et al. Effect of rituximab on a salivary gland ultrasound score in primary Sjögren’s syndrome: Results of the TRACTISS randomised double-blind multicentre substudy. Ann. Rheum. Dis. 2018, 77, 412–416. [Google Scholar] [CrossRef] [PubMed]
- Jousse-Joulin, S.; Devauchelle-Pensec, V.; Cornec, D.; Marhadour, T.; Bressollette, L.; Gestin, S.; Pers, J.O.; Nowak, E.; Saraux, A. Brief Report: Ultrasonographic Assessment of Salivary Gland Response to Rituximab in Primary Sjögren’s Syndrome: Ultrasonographic response to Rituximab in primary SS. Arthritis Rheumatol. 2015, 67, 1623–1628. [Google Scholar] [CrossRef]
- Varela-Centelles, P.; Seoane-Romero, J.-M.; Sánchez-Sánchez, M.; González-Mosquera, A.; Diz-Dios, P.; Seoane, J. Minor salivary gland biopsy in Sjögren’s syndrome: A review and introduction of a new tool to ease the procedure. Med. Oral Patol. Oral Cirugia Bucal 2014, 19, e20–e23. [Google Scholar] [CrossRef]
- Spijkervet, F.K.L.; Haacke, E.; Kroese, F.G.M.; Bootsma, H.; Vissink, A. Parotid Gland Biopsy, the Alternative Way to Diagnose Sjögren Syndrome. Rheum. Dis. Clin. N. Am. 2016, 42, 485–499. [Google Scholar] [CrossRef]
- Fisher, B.A.; Jonsson, R.; Daniels, T.; Bombardieri, M.; Brown, R.M.; Morgan, P.; Bombardieri, S.; Ng, W.-F.; Tzioufas, A.G.; Vitali, C.; et al. Standardisation of labial salivary gland histopathology in clinical trials in primary Sjögren’s syndrome. Ann. Rheum. Dis. 2017, 76, 1161–1168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chisholm, D.M.; Mason, D.K. Labial salivary gland biopsy in Sjogren’s disease. J. Clin. Pathol. 1968, 21, 656–660. [Google Scholar] [CrossRef] [PubMed]
- Greenspan, J.S.; Daniels, T.E.; Talal, N.; Sylvester, R.A. The histopathology of Sjögren’s syndrome in labial salivary gland biopsies. Oral Surg. Oral Med. Oral Pathol. 1974, 37, 217–229. [Google Scholar] [CrossRef]
- Guellec, D.; Cornec, D.; Jousse-Joulin, S.; Marhadour, T.; Marcorelles, P.; Pers, J.-O.; Saraux, A.; Devauchelle-Pensec, V. Diagnostic value of labial minor salivary gland biopsy for Sjögren’s syndrome: A systematic review. Autoimmun. Rev. 2013, 12, 416–420. [Google Scholar] [CrossRef]
- Campos, J.; Hillen, M.R.; Barone, F. Salivary Gland Pathology in Sjögren’s Syndrome. Rheum. Dis. Clin. N. Am. 2016, 42, 473–483. [Google Scholar] [CrossRef]
- Barone, F.; Campos, J.; Bowman, S.; Fisher, B.A. The value of histopathological examination of salivary gland biopsies in diagnosis, prognosis and treatment of Sjögren’s Syndrome. Swiss Med. Wkly. 2015, 145, w14168. [Google Scholar] [CrossRef]
- Pijpe, J.; Kalk, W.W.I.; van der Wal, J.E.; Vissink, A.; Kluin, P.M.; Roodenburg, J.L.N.; Bootsma, H.; Kallenberg, C.G.M.; Spijkervet, F.K.L. Parotid gland biopsy compared with labial biopsy in the diagnosis of patients with primary Sjogren’s syndrome. Rheumatology 2006, 46, 335–341. [Google Scholar] [CrossRef] [Green Version]
- Marx, R.E.; Hartman, K.S.; Rethman, K.V. A prospective study comparing incisional labial to incisional parotid biopsies in the detection and confirmation of sarcoidosis, Sjögren’s disease, sialosis and lymphoma. J. Rheumatol. 1988, 15, 621–629. [Google Scholar]
- Franceschini, F.; Cavazzana, I. Anti-Ro/SSA and La/SSB antibodies. Autoimmunity 2005, 38, 55–63. [Google Scholar] [CrossRef]
- Tzioufas, A.G.; Tatouli, I.P.; Moutsopoulos, H.M. Autoantibodies in Sjögren’s syndrome: Clinical presentation and regulatory mechanisms. Presse Med. 1983 2012, 41, e451–e460. [Google Scholar] [CrossRef]
- Trevisani, V.F.M.; Pasoto, S.G.; Fernandes, M.L.M.S.; Lopes, M.L.L.; de Magalhães Souza Fialho, S.C.; Pinheiro, A.C.; dos Santos, L.C.; Appenzeller, S.; Fidelix, T.; Ribeiro, S.L.E.; et al. Recommendations from the Brazilian society of rheumatology for the diagnosis of Sjögren’s syndrome (Part I): Glandular manifestations (systematic review). Adv. Rheumatol. 2019, 59, 58. [Google Scholar] [CrossRef] [Green Version]
- Robbins, A.; Hentzien, M.; Toquet, S.; Didier, K.; Servettaz, A.; Pham, B.-N.; Giusti, D. Diagnostic Utility of Separate Anti-Ro60 and Anti-Ro52/TRIM21 Antibody Detection in Autoimmune Diseases. Front. Immunol. 2019, 10, 444. [Google Scholar] [CrossRef] [Green Version]
- Kontny, E.; Lewandowska-Poluch, A.; Chmielińska, M.; Olesińska, M. Subgroups of Sjögren’s syndrome patients categorised by serological profiles: Clinical and immunological characteristics. Reumatologia 2018, 56, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Brito-Zerón, P.; Retamozo, S.; Ramos-Casals, M. Phenotyping Sjögren’s syndrome: Towards a personalised management of the disease. Clin. Exp. Rheumatol. 2018, 36, 198–209. [Google Scholar]
- Cornec, D.; Saraux, A.; Jousse-Joulin, S.; Pers, J.O.; Boisramé-Gastrin, S.; Renaudineau, Y.; Gauvin, Y.; Roguedas-Contios, A.M.; Genestet, S.; Chastaing, M.; et al. The Differential Diagnosis of Dry Eyes, Dry Mouth, and Parotidomegaly: A Comprehensive Review. Clin. Rev. Allergy. Immunol. 2015, 49, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Moutsopoulos, H.M.; Chused, T.M.; Mann, D.L.; Klippel, J.H.; Fauci, A.S.; Frank, M.M.; Lawley, T.J.; Hamburger, M.I. Sjögren’s syndrome (Sicca syndrome): Current issues. Ann. Intern. Med. 1980, 92, 212–226. [Google Scholar] [CrossRef] [PubMed]
- Fragoulis, G.E.; Fragkioudaki, S.; Reilly, J.H.; Kerr, S.C.; McInnes, I.B.; Moutsopoulos, H.M. Analysis of the cell populations composing the mononuclear cell infiltrates in the labial minor salivary glands from patients with rheumatoid arthritis and sicca syndrome. J. Autoimmun. 2016, 73, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Manoussakis, M.N.; Georgopoulou, C.; Zintzaras, E.; Spyropoulou, M.; Stavropoulou, A.; Skopouli, F.N.; Moutsopoulos, H.M. Sjögren’s syndrome associated with systemic lupus erythematosus: Clinical and laboratory profiles and comparison with primary Sjögren’s syndrome. Arthritis Rheum. 2004, 50, 882–891. [Google Scholar] [CrossRef]
- Salliot, C.; Mouthon, L.; Ardizzone, M.; Sibilia, J.; Guillevin, L.; Gottenberg, J.E.; Mariette, X. Sjogren’s syndrome is associated with and not secondary to systemic sclerosis. Rheumatology 2007, 46, 321–326. [Google Scholar] [CrossRef] [Green Version]
- Rojas-Villarraga, A.; Amaya-Amaya, J.; Rodriguez-Rodriguez, A.; Mantilla, R.D.; Anaya, J.M. Introducing polyautoimmunity: Secondary autoimmune diseases no longer exist. Autoimmune Dis. 2012, 2012, 254319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kollert, F.; Fisher, B.A. Equal rights in autoimmunity: Is Sjögren’s syndrome ever ’secondary’? Rheumatology 2020, 59, 1218–1225. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.G.; Singh, S.; Matteson, E.L. Rate, risk factors and causes of mortality in patients with Sjögren’s syndrome: A systematic review and meta-analysis of cohort studies. Rheumatology 2016, 55, 450–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, Y.; Yang, Z.; Qin, B.; Zhong, R. Primary Sjogren’s syndrome and malignancy risk: A systematic review and meta-analysis. Ann. Rheum. Dis. 2014, 73, 1151–1156. [Google Scholar] [CrossRef] [PubMed]
- Zintzaras, E.; Voulgarelis, M.; Moutsopoulos, H.M. The risk of lymphoma development in autoimmune diseases: A meta-analysis. Arch. Intern. Med. 2005, 165, 2337–2344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jonsson, M.V.; Theander, E.; Jonsson, R. Predictors for the development of non-Hodgkin lymphoma in primary Sjögren’s syndrome. Presse Med. 1983 2012, 41, e511–e516. [Google Scholar] [CrossRef] [PubMed]
- Nishishinya, M.B.; Pereda, C.A.; Muñoz-Fernández, S.; Pego-Reigosa, J.M.; Rúa-Figueroa, I.; Andreu, J.-L.; Fernández-Castro, M.; Rosas, J.; Loza Santamaría, E. Identification of lymphoma predictors in patients with primary Sjögren’s syndrome: A systematic literature review and meta-analysis. Rheumatol. Int. 2015, 35, 17–26. [Google Scholar] [CrossRef]
- Papageorgiou, A.; Voulgarelis, M.; Tzioufas, A.G. Clinical picture, outcome and predictive factors of lymphoma in Sjögren syndrome. Autoimmun. Rev. 2015, 14, 641–649. [Google Scholar] [CrossRef]
- Hernandez-Molina, G.; Michel-Peregrina, M.; Bermúdez-Bermejo, P.; Sánchez-Guerrero, J. Early and late extraglandular manifestations in primary Sjögren’s syndrome. Clin. Exp. Rheumatol. 2012, 30, 455. [Google Scholar]
- Ter Borg, E.J.; Kelder, J.C. Development of new extra-glandular manifestations or associated auto-immune diseases after establishing the diagnosis of primary Sjögren’s syndrome: A long-term study of the Antonius Nieuwegein Sjögren (ANS) cohort. Rheumatol. Int. 2017, 37, 1153–1158. [Google Scholar] [CrossRef]
- Seror, R.; Meiners, P.; Baron, G.; Bootsma, H.; Bowman, S.J.; Vitali, C.; Gottenberg, J.-E.; Theander, E.; Tzioufas, A.; De Vita, S.; et al. Development of the ClinESSDAI: A clinical score without biological domain. A tool for biological studies. Ann. Rheum. Dis. 2016, 75, 1945–1950. [Google Scholar] [CrossRef]
- Seror, R.; Ravaud, P.; Bowman, S.J.; Baron, G.; Tzioufas, A.; Theander, E.; Gottenberg, J.-E.; Bootsma, H.; Mariette, X.; Vitali, C. EULAR Sjögren’s syndrome disease activity index: Development of a consensus systemic disease activity index for primary Sjögren’s syndrome. Ann. Rheum. Dis. 2010, 69, 1103–1109. [Google Scholar] [CrossRef] [PubMed]
- Seror, R.; Ravaud, P.; Mariette, X.; Bootsma, H.; Theander, E.; Hansen, A.; Ramos-Casals, M.; Dörner, T.; Bombardieri, S.; Hachulla, E.; et al. EULAR Sjögren’s Syndrome Patient Reported Index (ESSPRI): Development of a consensus patient index for primary Sjögren’s syndrome. Ann. Rheum. Dis. 2011, 70, 968–972. [Google Scholar] [CrossRef] [PubMed]
- Vitali, C.; Palombi, G.; Baldini, C.; Benucci, M.; Bombardieri, S.; Covelli, M.; Del Papa, N.; De Vita, S.; Epis, O.; Franceschini, F.; et al. Sjögren’s syndrome disease damage index and disease activity index: Scoring systems for the assessment of disease damage and disease activity in Sjögren’s syndrome, derived from an analysis of a cohort of Italian patients. Arthritis Rheum. 2007, 56, 2223–2231. [Google Scholar] [CrossRef] [PubMed]
- Barry, R.J.; Sutcliffe, N.; Isenberg, D.A.; Price, E.; Goldblatt, F.; Adler, M.; Canavan, A.; Hamburger, J.; Richards, A.; Regan, M.; et al. The Sjogren’s Syndrome Damage Index—A damage index for use in clinical trials and observational studies in primary Sjogren’s syndrome. Rheumatology 2008, 47, 1193–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quartuccio, L.; Baldini, C.; Bartoloni, E.; Priori, R.; Carubbi, F.; Corazza, L.; Alunno, A.; Colafrancesco, S.; Luciano, N.; Giacomelli, R.; et al. Anti-SSA/SSB-negative Sjögren’s syndrome shows a lower prevalence of lymphoproliferative manifestations, and a lower risk of lymphoma evolution. Autoimmun. Rev. 2015, 14, 1019–1022. [Google Scholar] [CrossRef] [PubMed]
- Fauchais, A.L.; Martel, C.; Gondran, G.; Lambert, M.; Launay, D.; Jauberteau, M.O.; Hachulla, E.; Vidal, E.; Hatron, P.Y. Immunological profile in primary Sjögren syndrome: Clinical significance, prognosis and long-term evolution to other auto-immune disease. Autoimmun. Rev. 2010, 9, 595–599. [Google Scholar] [CrossRef] [PubMed]
- Krylova, L.; Isenberg, D. Assessment of patients with primary Sjogren’s syndrome--outcome over 10 years using the Sjogren’s Syndrome Damage Index. Rheumatology 2010, 49, 1559–1562. [Google Scholar] [CrossRef] [Green Version]
- Baldini, C.; Ferro, F.; Pepe, P.; Luciano, N.; Sernissi, F.; Cacciatore, C.; Martini, D.; Tavoni, A.; Mosca, M.; Bombardieri, S. Damage Accrual In a Single Centre Cohort Of Patients With Primary Sjögren’s Syndrome Followed Up For Over 10 Years. In Sjögren’s Syndrome: Clinical Aspects, Proceedings of the 2013 ACR/ARHP Annual Meeting, San Diego, CA, USA, 25–30 October 2013; WILEY: Hoboken, NJ, USA, 2013. [Google Scholar]
- Cho, H.J.; Yoo, J.J.; Yun, C.Y.; Kang, E.H.; Lee, H.-J.; Hyon, J.Y.; Song, Y.W.; Lee, Y.J. The EULAR Sjogren’s syndrome patient reported index as an independent determinant of health-related quality of life in primary Sjogren’s syndrome patients: In comparison with non-Sjogren’s sicca patients. Rheumatology 2013, 52, 2208–2217. [Google Scholar] [CrossRef] [Green Version]
- Hackett, K.L.; Newton, J.L.; Frith, J.; Elliott, C.; Lendrem, D.; Foggo, H.; Edgar, S.; Mitchell, S.; Ng, W.-F. Impaired functional status in primary Sjögren’s syndrome. Arthritis Care Res. 2012, 64, 1760–1764. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, X.; Chen, H.; Shen, B. Sjögren’s syndrome is associated with negatively variable impacts on domains of health-related quality of life: Evidence from Short Form 36 questionnaire and a meta-analysis. Patient Prefer. Adherence 2017, 11, 905–911. [Google Scholar] [CrossRef] [Green Version]
- Haldorsen, K.; Moen, K.; Jacobsen, H.; Jonsson, R.; Brun, J.G. Exocrine function in primary Sjögren syndrome: Natural course and prognostic factors. Ann. Rheum. Dis. 2008, 67, 949–954. [Google Scholar] [CrossRef]
- Al-Ezzi, M.Y.; Pathak, N.; Tappuni, A.R.; Khan, K.S. Primary Sjögren’s syndrome impact on smell, taste, sexuality and quality of life in female patients: A systematic review and meta-analysis. Mod. Rheumatol. 2017, 27, 623–629. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, S.T.; Valim, V.; Fisher, B.A. Health-related quality of life and costs in Sjögren’s syndrome. Rheumatology 2019. [Google Scholar] [CrossRef] [PubMed]
- Ter Borg, E.J.; Kelder, J.C. Lower prevalence of extra-glandular manifestations and anti-SSB antibodies in patients with primary Sjögren’s syndrome and widespread pain: Evidence for a relatively benign subset. Clin. Exp. Rheumatol. 2014, 32, 349–353. [Google Scholar] [PubMed]
- Ostuni, P.; Botsios, C.; Sfriso, P.; Punzi, L.; Chieco-Bianchi, F.; Semerano, L.; Grava, C.; Todesco, S. Fibromyalgia in Italian patients with primary Sjögren’s syndrome. Joint Bone Spine 2002, 69, 51–57. [Google Scholar] [CrossRef]
- Champey, J.; Corruble, E.; Gottenberg, J.; Buhl, C.; Meyer, T.; Caudmont, C.; Bergé, E.; Pellet, J.; Hardy, P.; Mariette, X. Quality of life and psychological status in patients with primary Sjögren’s syndrome and sicca symptoms without autoimmune features. Arthritis Rheum. 2006, 55, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Mariette, X. Dry eyes and mouth syndrome or sicca, asthenia and polyalgia syndrome? Rheumatology 2003, 42, 914–915. [Google Scholar] [CrossRef] [Green Version]
- Mavragani, C.P.; Skopouli, F.N.; Moutsopoulos, H.M. Increased Prevalence of Antibodies to Thyroid Peroxidase in Dry Eyes and Mouth Syndrome or Sicca Asthenia Polyalgia Syndrome. J. Rheumatol. 2009, 36, 1626–1630. [Google Scholar] [CrossRef]
- Price, E.J. Dry eyes and mouth syndrome—A subgroup of patients presenting with sicca symptoms. Rheumatology 2002, 41, 416–422. [Google Scholar] [CrossRef] [Green Version]
- Mandl, T.; Jørgensen, T.S.; Skougaard, M.; Olsson, P.; Kristensen, L.-E. Work Disability in Newly Diagnosed Patients with Primary Sjögren Syndrome. J. Rheumatol. 2017, 44, 209–215. [Google Scholar] [CrossRef] [Green Version]
- Pertovaara, M.; Korpela, M. ESSPRI and other patient-reported indices in patients with primary Sjogren’s syndrome during 100 consecutive outpatient visits at one rheumatological clinic. Rheumatology 2014, 53, 927–931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lendrem, D.; Mitchell, S.; McMeekin, P.; Gompels, L.; Hackett, K.; Bowman, S.; Price, E.; Pease, C.T.; Emery, P.; Andrews, J.; et al. Do the EULAR Sjogren’s syndrome outcome measures correlate with health status in primary Sjogren’s syndrome? Rheumatology 2015, 54, 655–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koh, J.; Kwok, S.; Lee, J.; Son, C.; Kim, J.-M.; Kim, H.; Park, S.; Sung, Y.; Choe, J.; Lee, S.; et al. Pain, xerostomia, and younger age are major determinants of fatigue in Korean patients with primary Sjögren’s syndrome: A cohort study. Scand. J. Rheumatol. 2017, 46, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Cornec, D.; Devauchelle-Pensec, V.; Mariette, X.; Jousse-Joulin, S.; Berthelot, J.; Perdriger, A.; Puéchal, X.; Le Guern, V.; Sibilia, J.; Gottenberg, J.; et al. Severe Health-Related Quality of Life Impairment in Active Primary Sjögren’s Syndrome and Patient-Reported Outcomes: Data From a Large Therapeutic Trial. Arthritis Care Res. 2017, 69, 528–535. [Google Scholar] [CrossRef]
- Price, E.J.; Rauz, S.; Tappuni, A.R.; Sutcliffe, N.; Hackett, K.L.; Barone, F.; Granata, G.; Ng, W.-F.; Fisher, B.A.; Bombardieri, M.; et al. The British Society for Rheumatology guideline for the management of adults with primary Sjögren’s Syndrome. Rheumatology 2017. [Google Scholar] [CrossRef] [Green Version]
- Valim, V.; Trevisani, V.F.M.; Pasoto, S.G.; Serrano, E.V.; Ribeiro, S.L.E.; de Fidelix, T.S.A.; Vilela, V.S.; do Prado, L.L.; Tanure, L.A.; Libório-Kimura, T.N.; et al. Recommendations for the treatment of Sjögren’s syndrome. Rev. Bras. Reumatol. 2015, 55, 446–457. [Google Scholar] [CrossRef]
- Sumida, T.; Azuma, N.; Moriyama, M.; Takahashi, H.; Asashima, H.; Honda, F.; Abe, S.; Ono, Y.; Hirota, T.; Hirata, S.; et al. Clinical practice guideline for Sjögren’s syndrome 2017. Mod. Rheumatol. 2018, 28, 383–408. [Google Scholar] [CrossRef] [Green Version]
- Vivino, F.B.; Carsons, S.E.; Foulks, G.; Daniels, T.E.; Parke, A.; Brennan, M.T.; Forstot, S.L.; Scofield, R.H.; Hammitt, K.M. New Treatment Guidelines for Sjögren’s Disease. Rheum. Dis. Clin. N. Am. 2016, 42, 531–551. [Google Scholar] [CrossRef] [Green Version]
- The Dry Eye Assessment and Management Study Research Group. n−3 Fatty Acid Supplementation for the Treatment of Dry Eye Disease. N. Engl. J. Med. 2018, 378, 1681–1690. [Google Scholar] [CrossRef]
- Hussain, M.; Shtein, R.M.; Pistilli, M.; Maguire, M.G.; Oydanich, M.; Asbell, P.A. The Dry Eye Assessment and Management (DREAM) extension study—A randomized clinical trial of withdrawal of supplementation with omega-3 fatty acid in patients with dry eye disease. Ocul. Surf. 2020, 18, 47–55. [Google Scholar] [CrossRef]
- Asbell, P.A.; Maguire, M.G. Why DREAM should make you think twice about recommending Omega-3 supplements. Ocul. Surf. 2019, 17, 617–618. [Google Scholar] [CrossRef]
- Skopouli, F.N.; Jagiello, P.; Tsifetaki, N.; Moutsopoulos, H.M. Methotrexate in primary Sjögren’s syndrome. Clin. Exp. Rheumatol. 1996, 14, 555–558. [Google Scholar] [PubMed]
- Nakayamada, S.; Saito, K.; Umehara, H.; Ogawa, N.; Sumida, T.; Ito, S.; Minota, S.; Nara, H.; Kondo, H.; Okada, J.; et al. Efficacy and safety of mizoribine for the treatment of Sjögren’s syndrome: A multicenter open-label clinical trial. Mod. Rheumatol. 2007, 17, 464–469. [Google Scholar] [CrossRef] [PubMed]
- Nakayamada, S.; Fujimoto, T.; Nonomura, A.; Saito, K.; Nakamura, S.; Tanaka, Y. Usefulness of initial histological features for stratifying Sjogren’s syndrome responders to mizoribine therapy. Rheumatology 2009, 48, 1279–1282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacFarlane, G.J.; Kronisch, C.; Dean, L.E.; Atzeni, F.; Häuser, W.; Fluß, E.; Choy, E.; Kosek, E.; Amris, K.; Branco, J.; et al. EULAR revised recommendations for the management of fibromyalgia. Ann. Rheum. Dis. 2017, 76, 318–328. [Google Scholar] [CrossRef] [PubMed]
- Dass, S.; Bowman, S.J.; Vital, E.M.; Ikeda, K.; Pease, C.T.; Hamburger, J.; Richards, A.; Rauz, S.; Emery, P. Reduction of fatigue in Sjogren syndrome with rituximab: Results of a randomised, double-blind, placebo-controlled pilot study. Ann. Rheum. Dis. 2008, 67, 1541–1544. [Google Scholar] [CrossRef] [PubMed]
- Devauchelle-Pensec, V.; Mariette, X.; Jousse-Joulin, S.; Berthelot, J.-M.; Perdriger, A.; Puéchal, X.; Le Guern, V.; Sibilia, J.; Gottenberg, J.-E.; Chiche, L.; et al. Treatment of Primary Sjögren Syndrome With Rituximab: A Randomized Trial. Ann. Intern. Med. 2014, 160, 233–242. [Google Scholar] [CrossRef]
- Carubbi, F.; Cipriani, P.; Marrelli, A.; Benedetto, P.; Ruscitti, P.; Berardicurti, O.; Pantano, I.; Liakouli, V.; Alvaro, S.; Alunno, A.; et al. Efficacy and safety of rituximab treatment in early primary Sjögren’s syndrome: A prospective, multi-center, follow-up study. Arthritis Res. Ther. 2013, 15, R172. [Google Scholar] [CrossRef] [Green Version]
- Norheim, K.B.; Harboe, E.; Gøransson, L.G.; Omdal, R. Interleukin-1 Inhibition and Fatigue in Primary Sjögren’s Syndrome—A Double Blind, Randomised Clinical Trial. PLoS ONE 2012, 7, e30123. [Google Scholar] [CrossRef] [Green Version]
- Van der Heijden, E.H.M.; Kruize, A.A.; Radstake, T.R.D.J.; van Roon, J.A.G. Optimizing conventional DMARD therapy for Sjögren’s syndrome. Autoimmun. Rev. 2018, 17, 480–492. [Google Scholar] [CrossRef]
- Gottenberg, J.-E.; Ravaud, P.; Puéchal, X.; Le Guern, V.; Sibilia, J.; Goeb, V.; Larroche, C.; Dubost, J.-J.; Rist, S.; Saraux, A.; et al. Effects of Hydroxychloroquine on Symptomatic Improvement in Primary Sjögren Syndrome: The JOQUER Randomized Clinical Trial. JAMA 2014, 312, 249. [Google Scholar] [CrossRef] [PubMed]
- Yoon, C.H.; Lee, H.J.; Lee, E.Y.; Lee, E.B.; Lee, W.-W.; Kim, M.K.; Wee, W.R. Effect of Hydroxychloroquine Treatment on Dry Eyes in Subjects with Primary Sjögren’s Syndrome: A Double-Blind Randomized Control Study. J. Kor. Med. Sci. 2016, 31, 1127. [Google Scholar] [CrossRef] [PubMed]
- Gottenberg, J.-E.; Dörner, T.; Bootsma, H.; Devauchelle-Pensec, V.; Bowman, S.J.; Mariette, X.; Bartz, H.; Oortgiesen, M.; Shock, A.; Koetse, W.; et al. Efficacy of Epratuzumab, an Anti-CD22 Monoclonal IgG Antibody, in Systemic Lupus Erythematosus Patients with Associated Sjögren’s Syndrome: Post Hoc Analyses From the EMBODY Trials. Arthritis Rheumatol. 2018, 70, 763–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mariette, X.; Seror, R.; Quartuccio, L.; Baron, G.; Salvin, S.; Fabris, M.; Desmoulins, F.; Nocturne, G.; Ravaud, P.; De Vita, S. Efficacy and safety of belimumab in primary Sjögren’s syndrome: Results of the BELISS open-label phase II study. Ann. Rheum. Dis. 2015, 74, 526–531. [Google Scholar] [CrossRef]
- De Vita, S.; Quartuccio, L.; Seror, R.; Salvin, S.; Ravaud, P.; Fabris, M.; Nocturne, G.; Gandolfo, S.; Isola, M.; Mariette, X. Efficacy and safety of belimumab given for 12 months in primary Sjögren’s syndrome: The BELISS open-label phase II study. Rheumatology 2015. [Google Scholar] [CrossRef] [Green Version]
- Jakez-Ocampo, J.; Atisha-Fregoso, Y.; Llorente, L. Refractory Primary Sjögren Syndrome Successfully Treated With Bortezomib. JCR J. Clin. Rheumatol. 2015, 21, 31–32. [Google Scholar] [CrossRef]
- Shah, U. Pilot Trial of Ustekinumab for Primary Sjögren’s Syndrome. 2020. Available online: clinicaltrials.gov (accessed on 9 July 2020).
- Gilead Sciences. Study to Assess Safety and Efficacy of Filgotinib, Lanraplenib and Tirabrutinib in Adults With Active Sjogren’s Syndrome; Clinical Trial Registration; Gilead Sciences, Inc.: Foster City, CA, USA, 2020. [Google Scholar]
- Meijer, J.M.; Meiners, P.M.; Vissink, A.; Spijkervet, F.K.L.; Abdulahad, W.; Kamminga, N.; Brouwer, E.; Kallenberg, C.G.M.; Bootsma, H. Effectiveness of rituximab treatment in primary Sjögren’s syndrome: A randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2010, 62, 960–968. [Google Scholar] [CrossRef]
- Bowman, S.J.; Everett, C.C.; O’Dwyer, J.L.; Emery, P.; Pitzalis, C.; Ng, W.-F.; Pease, C.T.; Price, E.J.; Sutcliffe, N.; Gendi, N.S.T.; et al. Randomized Controlled Trial of Rituximab and Cost-Effectiveness Analysis in Treating Fatigue and Oral Dryness in Primary Sjögren’s Syndrome: Rituximab for symptomatic fatigue and oral dryness in primary SS. Arthritis Rheumatol. 2017, 69, 1440–1450. [Google Scholar] [CrossRef] [Green Version]
- RemeGen A Phase II Study of RC18, a Recombinant Human B Lymphocyte Stimulator Receptor: Immunoglobulin G (IgG) Fc Fusion Protein for Injection for the Treatment of Subjects with Primary Sjögren’s Syndrome. 2019. Available online: clinicaltrials.gov (accessed on 20 May 2020).
- GlaxoSmithKline A Randomized, Double Blind (Sponsor Open), Comparative, Multicenter Study to Evaluate the Safety and Efficacy of Subcutaneous Belimumab (GSK1550188) and Intravenous Rituximab Co-administration in Subjects With Primary Sjögren’s Syndrome. 2020. Available online: clinicaltrials.gov (accessed on 9 July 2020).
- Eli Lilly and Company A Multiple Ascending Dose Study to Evaluate the Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of LY3090106 in Subjects With Sjögren’s Syndrome. 2018. Available online: clinicaltrials.gov (accessed on 20 May 2020).
- Dörner, T.; Posch, M.G.; Li, Y.; Petricoul, O.; Cabanski, M.; Milojevic, J.M.; Kamphausen, E.; Valentin, M.-A.; Simonett, C.; Mooney, L.; et al. Treatment of primary Sjögren’s syndrome with ianalumab (VAY736) targeting B cells by BAFF receptor blockade coupled with enhanced, antibody-dependent cellular cytotoxicity. Ann. Rheum. Dis. 2019, 78, 641–647. [Google Scholar] [CrossRef]
- Novartis Pharmaceuticals Study of Safety and Efficacy of Multiple VAY736 Doses in Patients with Moderate to Severe Primary Sjogren’s Syndrome (pSS). 2020. Available online: clinicaltrials.gov (accessed on 9 July 2020).
- Novartis Pharmaceuticals An Adaptive Phase 2 Randomized Double-blind, Placebo-controlled Multi-center Study to Evaluate the Safety and Efficacy of Multiple LOU064 Doses in Patients With Moderate to Severe Sjögren’s Syndrome (LOUiSSe). 2020. Available online: clinicaltrials.gov (accessed on 9 July 2020).
- Bristol-Myers Squibb A Phase II, Randomized, Multi-Center, Double-Blind, Placebo Controlled Study to Evaluate the Efficacy and Safety of BMS-931699 (Lulizumab) or BMS-986142 in Subjects With Moderate to Severe Primary Sjögren’s Syndrome. 2018. Available online: clinicaltrials.gov (accessed on 20 May 2020).
- Bristol-Myers Squibb A Randomized, Placebo-Controlled, Double-Blind, Multicenter Study to Assess the Efficacy and Safety of Branebrutinib Treatment in Subjects With Active Systemic Lupus Erythematosus or Primary Sjögren’s Syndrome, or Branebrutinib Treatment Followed by Open-label Abatacept Treatment in Subjects With Active Rheumatoid Arthritis. 2020. Available online: clinicaltrials.gov (accessed on 9 July 2020).
- St.Clair, E.W.; Baer, A.N.; Wei, C.; Noaiseh, G.; Parke, A.; Coca, A.; Utset, T.O.; Genovese, M.C.; Wallace, D.J.; McNamara, J.; et al. Clinical Efficacy and Safety of Baminercept, a Lymphotoxin β Receptor Fusion Protein, in Primary Sjögren’s Syndrome: Results From a Phase II Randomized, Double-Blind, Placebo-Controlled Trial. Arthritis Rheumatol. 2018, 70, 1470–1480. [Google Scholar] [CrossRef]
- Incyte Corporation An Open-Label Phase 2 Study of INCB050465 in Participants With Primary Sjögren’s Syndrome. 2020. Available online: clinicaltrials.gov (accessed on 20 May 2020).
- Juarez, M.; Diaz, N.; Johnston, G.I.; Nayar, S.; Payne, A.; Helmer, E.; Cain, D.; Williams, P.; Ng, W.F.; Fisher, B.; et al. AB0458 a phase II randomised double-blind, placebo-controlled, proof of concept study of oral Seletalisib in patients with priary Sjögren’s syndrome (PSS). Ann. Rheum. Dis. 2019, 78, 1692–1693. [Google Scholar]
- Dörner, T.; Zeher, M.; Laessing, U.; Chaperon, F.; De Buck, S.; Hasselberg, A.; Valentin, M.-A.; Ma, S.; Cabanski, M.; Kalis, C.; et al. OP0250 A randomised, double-blind study to assess the safety, tolerability and preliminary efficacy of leniolisib (CDZ173) in patients with primary sjÖgren’s syndrome. Ann. Rheum. Dis. 2018, 77, 174. [Google Scholar]
- Baer, A.; Gottenberg, J.-E.; St.Clair, W.E.; Sumida, T.; Takeuchi, T.; Seror, R.; Foulks, G.; Nys, M.; Johnsen, A.; Wong, R.; et al. OP0039 Efficacy and safety of Abatacept in active primary Sjögren’s syndrome: Results of a randomised placebo-controlled phase III trial. Ann. Rheum. Dis. 2019, 78, 89–90. [Google Scholar]
- Meiners, P.M.; Vissink, A.; Kroese, F.G.M.; Spijkervet, F.K.L.; Smitt-Kamminga, N.S.; Abdulahad, W.H.; Bulthuis-Kuiper, J.; Brouwer, E.; Arends, S.; Bootsma, H. Abatacept treatment reduces disease activity in early primary Sjogren’s syndrome (open-label proof of concept ASAP study). Ann. Rheum. Dis. 2014, 73, 1393–1396. [Google Scholar] [CrossRef] [PubMed]
- Van Nimwegen, J.F.; Mossel, E.; van Zuiden, G.S.; Wijnsma, R.F.; Delli, K.; Stel, A.J.; van der Vegt, B.; Haacke, E.A.; Olie, L.; Los, L.I.; et al. Abatacept treatment for patients with early active primary Sjögren’s syndrome: A single-centre, randomised, double-blind, placebo-controlled, phase 3 trial (ASAP-III study). Lancet Rheumatol. 2020, 2, e153–e163. [Google Scholar] [CrossRef]
- Fisher, B.A.; Szanto, A.; Ng, W.-F.; Bombardieri, M.; Posch, M.G.; Papas, A.S.; Gergely, P. Assessment of the anti-CD40 antibody iscalimab in patients with primary Sjögren’s syndrome: A multicentre, randomised, double-blind, placebo-controlled, proof-of-concept study. Lancet Rheumatol. 2020, 2. [Google Scholar] [CrossRef]
- Novartis Pharmaceuticals A 48-week, 6-arm, Randomized, Double-blind, Placebo-controlled Multicenter Trial to Assess the Safety and Efficacy of Multiple CFZ533 Doses Administered Subcutaneously in Two Distinct Populations of Patients with Sjögren’s Syndrome (TWINSS). 2020. Available online: clinicaltrials.gov (accessed on 9 July 2020).
- Viela Bio A Phase 2 Randomized, Double-blind, Placebo-controlled, Proof of Concept Study to Evaluate the Efficacy and Safety of VIB4920 in Subjects With Sjögren’s Syndrome (SS). 2020. Available online: clinicaltrials.gov (accessed on 9 July 2020).
- Mariette, X.; Bombardieri, M.; Alevizos, I.; Moate, R.; Sullivan, B.; Noaiseh, G.; Kvarnström, M.; Rees, W.; Wang, L.; Illei, G. A Phase 2a Study of MEDI5872 (AMG557), a Fully Human Anti-ICOS Ligand Monoclonal Antibody in Patients with Primary Sjögren’s Syndrome. In Sjögrenʼs Syndrome—Basic & Clinical Science Poster I, Proceedings of the 2019 ACR/ARP Annual Meeting, Atlanta, GA, USA, 8–13 November 2019; WILEY: Hoboken, NJ, USA, 2019. [Google Scholar]
- Hoffmann-La Roche A Multi-Center, Randomized, Double-Blind, Placebo-Controlled, Parallel Group Phase 2A Study to Assess the Efficacy of RO5459072 in Patients With Primary Sjogren’s Syndrome. 2018. Available online: clinicaltrials.gov (accessed on 20 May 2020).
- Gabor Illei, M. D A Randomized, Placebo Controlled, Proof of Concept, Study of Raptiva, a Humanized Anti-CD-11a Monoclonal Antibody, in Patients With Sjogren’s Syndrome. 2015. Available online: clinicaltrials.gov (accessed on 20 May 2020).
- Cacoub, P.; Felten, R.; Devauchelle-Pensec, V.; Duffau, P.; Hachulla, E.; Hatron, P.Y.; Salliot, C.; Perdriger, A.; Morel, J.; Mekinian, A.; et al. Inhibition du récepteur de l’interleukine-6 au cours du syndrome de Gougerot-Sjögren primaire: Essai randomisé multicentrique académique en double aveugle tocilizumab versus placebo (ETAP study). Rev. Médecine Interne 2019, 40, A33. [Google Scholar] [CrossRef]
- Mariette, X.; Ravaud, P.; Steinfeld, S.; Baron, G.; Goetz, J.; Hachulla, E.; Combe, B.; Puéchal, X.; Pennec, Y.; Sauvezie, B.; et al. Inefficacy of infliximab in primary Sjögren’s syndrome: Results of the randomized, controlled trial of remicade in primary Sjögren’s syndrome (TRIPSS): Infliximab in Primary Sjögren’s Syndrome. Arthritis Rheum. 2004, 50, 1270–1276. [Google Scholar] [CrossRef]
- Sankar, V.; Brennan, M.T.; Kok, M.R.; Leakan, R.A.; Smith, J.A.; Manny, J.; Baum, B.J.; Pillemer, S.R. Etanercept in Sjögren’s syndrome: A twelve-week randomized, double-blind, placebo-controlled pilot clinical trial: Randomized Controlled Pilot Study of Etanercept in SS. Arthritis Rheum. 2004, 50, 2240–2245. [Google Scholar] [CrossRef]
- GlaxoSmithKline A Two Part Phase IIa Study, to Evaluate the Safety and Tolerability, Pharmacokinetics, Proof of Mechanism and Potential for Efficacy of an Anti-IL-7 Receptor-α Monoclonal Antibody (GSK2618960) in the Treatment of Primary Sjögren’s Syndrome. 2018. Available online: clinicaltrials.gov (accessed on 20 May 2020).
- Viela Bio A Phase 1 Randomized, Placebo-Controlled, Blinded, Multiple Ascending Dose Study to Evaluate VIB7734 in Systemic Lupus Erythematosus, Cutaneous Lupus Erythematosus, Sjogren’s Syndrome, Systemic Sclerosis, Polymyositis, and Dermatomyositis. 2020. Available online: clinicaltrials.gov (accessed on 20 May 2020).
- Fisher, B.; Barone, F.; Jobling, K.; Gallagher, P.; Macrae, V.; Filby, A.; Hulmes, G.; Milne, P.; Traianos, E.; Iannizzotto, V.; et al. OP0202 Effects of RSLV-132 on fatigue in patients with primary Sjögren’s syndrome—Results of a phase II randomised double-blind, placebo-controlled proof of concept study. Ann. Rheum. Dis. 2019, 78, 177. [Google Scholar] [CrossRef] [Green Version]
- Assistance Publique—Hôpitaux de Paris Induction of Regulatory t Cells by Low Dose il2 in Autoimmune and Inflammatory Diseases. 2020. Available online: clinicaltrials.gov (accessed on 9 July 2020).





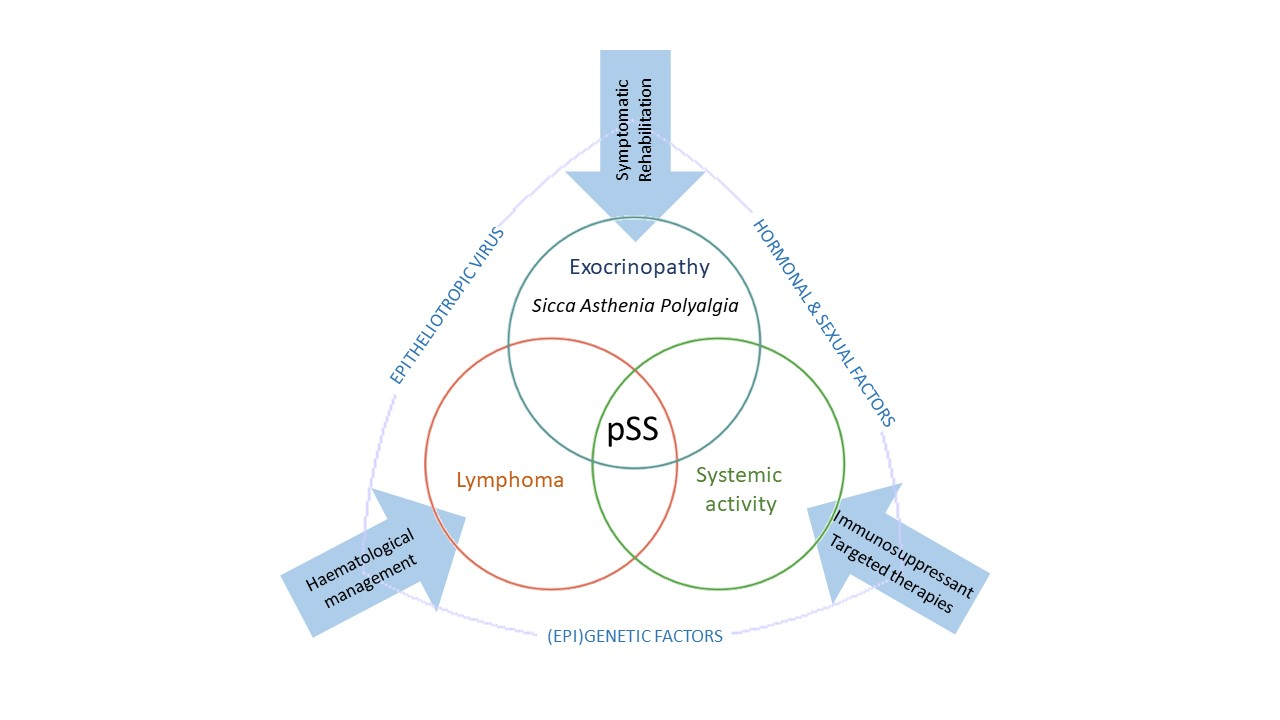{kind=link}
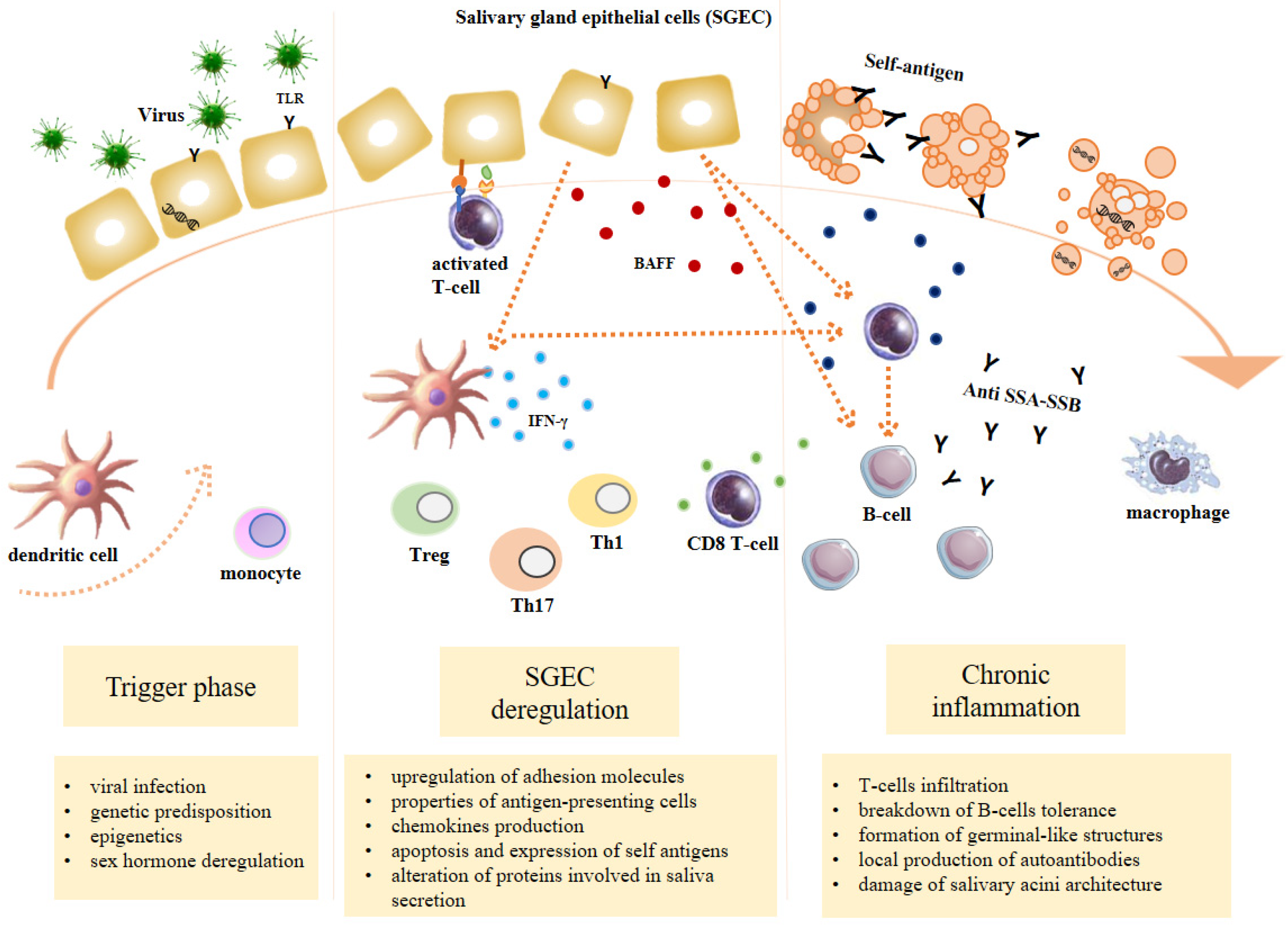{kind=link}
{kind=link}
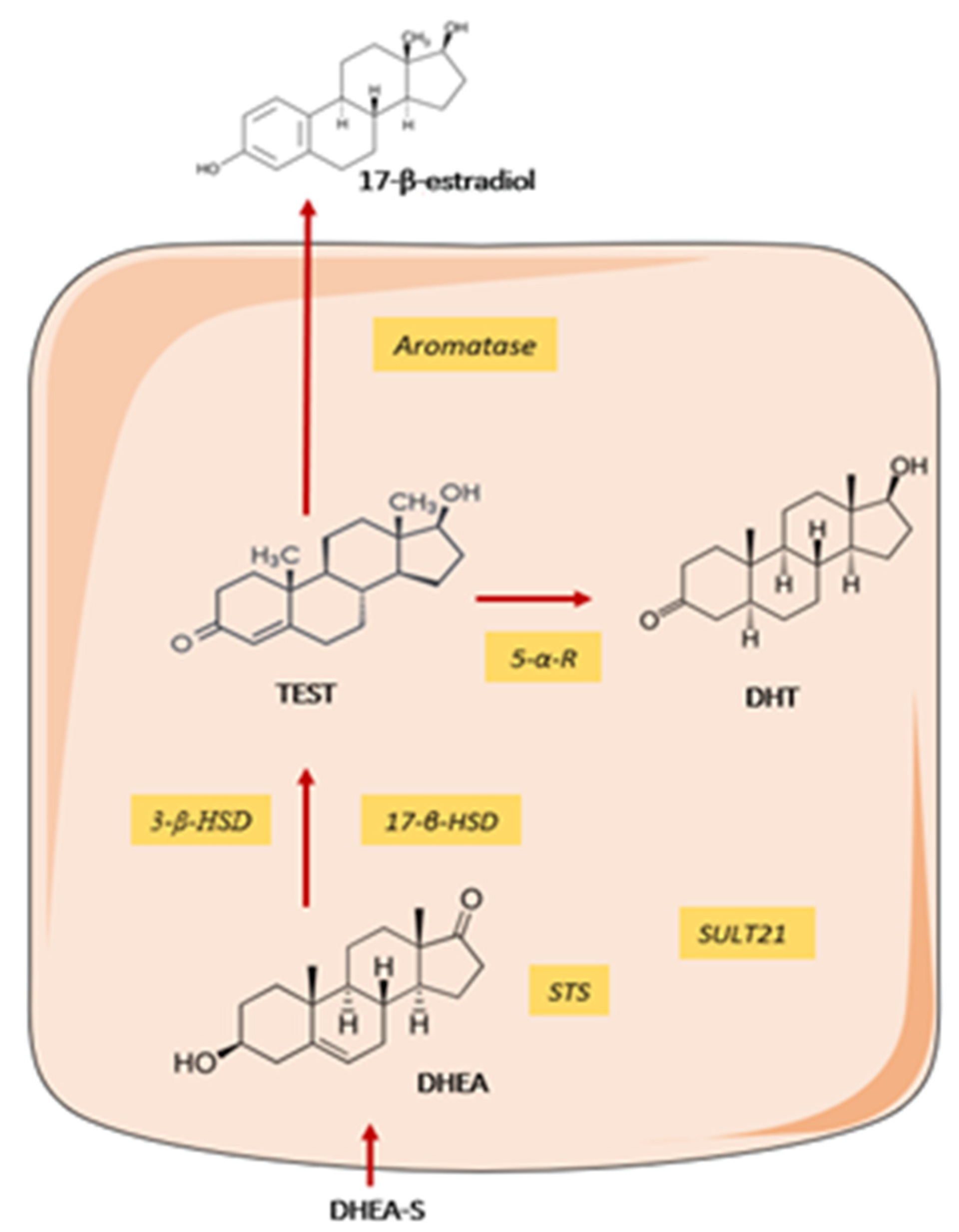{kind=link}
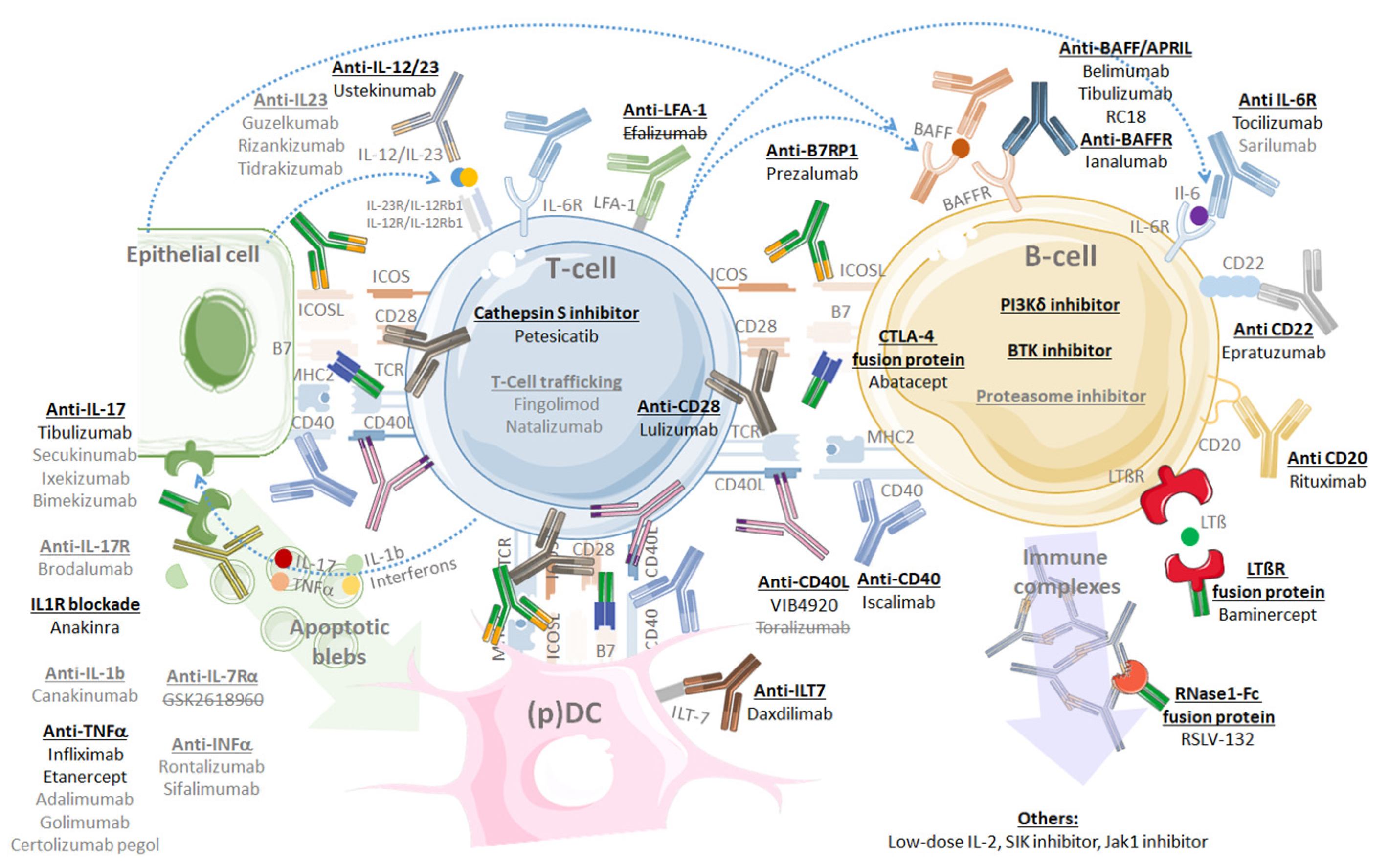{kind=link}
| Autoantigen Targeted by Autoantibody | Number of Patients (N Total/Pooled) | Autoantibody Prevalence (% of Total) | Clinical Associations | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| pSS | pSS MALT | Sicca | FM Sicca | Crtl | pSS | pSS MALT | Sicca | FM Sicca | Crtl | ||
| Salivary protein 1 (SP1) | 270 | _ | 29 | 151 | 148 | 46.3 | _ | 75.9 | 45.7 | 27 | Early disease, low focus-score, SSA−/SSB− [169,170,171,172,173] Found in non-pSS dry eye and fibromyalgia with sicca syndrome [167,174,175] |
| Carbonic anhydrase 6 (CA6) | 13 | _ | _ | 151 | 23 | 53.8 | _ | _ | 7.3 | 4.3 | |
| Parotid secretory protein (PSP) | 13 | _ | _ | 151 | 23 | 15.4 | _ | _ | 11.3 | 4.3 | |
| Interferon-inducible protein-16 | 250 | _ | _ | _ | 255 | 37.2 | _ | _ | _ | 2.7 | High focus-score and GC, hyperγ, ANA > 1:320 [176] |
| Mouse double minute 2 (MDM2) | 100 | _ | _ | _ | 74 | 21 | _ | _ | _ | 5.4 | ⇧ disease duration, ESSDAI, ⇧ focus-score, anaemia, thrombocytopenia, SSB+ [177] |
| Nuclear autoantigen 14 kDa (NA-14) | 204 | _ | _ | _ | 144 | 12.7 | _ | _ | _ | 0 | ⇧ IgA level, ANA < 1:320, ANA−, shorter disease duration [178,179] |
| Stathmin-4 | 72 | _ | _ | _ | 128 | 15 | _ | _ | _ | 5 | Polyneuropathy, vasculitis [180] |
| Poly(U)-binding splicing factor 60 kDa | 84 | _ | _ | _ | 38 | 30 | _ | _ | _ | 5.3 | Asian or African descent, ANA+, RF+, hyperγ, SSA+, SSB+ [181] |
| NR2 | 66 | _ | _ | _ | 99 | 20 | _ | _ | _ | 7.6 | ⇩ memory function, ⇧ depression rate [182] ⇩ hippocampal grey matter [183] |
| 50 | _ | _ | _ | _ | 12 * | _ | _ | _ | _ | ||
| TRIM38 | 235 | _ | _ | _ | 50 | 10 | _ | _ | _ | 4 | ⇧ ocular stain scores, ⇩ Schirmer’s test, focus-score ≥ 3, SSA+, RF+, hyperγ [184] |
| Saccharomyces cerevisiae | 104 | _ | _ | _ | _ | 5 | _ | _ | _ | _ | Triple Ro52+/Ro60+/La+, hypocomplementemia, cutaneous involvement [185] |
| Calponin-3 | 209 | _ | _ | _ | 46 | 11 | _ | _ | _ | 2.2 | Peripheral neuropathy [186] |
| Ganglionic acetylcholine receptor | 39 | _ | _ | _ | 39 | 23 | _ | _ | _ | 0 | Autonomic neuropathy [187] |
| Aquaporin-4 | 109 | _ | _ | _ | _ | 10 | _ | _ | _ | _ | NMOSD overlap [188] |
| Aquaporin-5 | 112 | _ | _ | _ | 53 | 73 | _ | _ | _ | 32 | Low resting salivary flow [164] |
| Other aquaporins (1, 3, 8, 9) | 34 | _ | _ | _ | _ | 38 | _ | _ | _ | _ | ⇧ ocular stain scores [189] |
| P-selectin | 70 | _ | _ | _ | 35 | 21 | _ | _ | 0 | Low platelet count [190] | |
| Carbamylated proteins | 123 | _ | _ | _ | 172 | 28.5 | _ | _ | _ | 3.5 | ⇧ total IgG, IgM, RF+, β2-microglobulin, ⇧ focus-score and GC [191,192] |
| Moesin | 50 | _ | _ | _ | 50 | 42 | _ | _ | _ | 4 | [193] |
| Cofilin-1 | 50 | 20 | _ | _ | 50 | 76 | 80 | _ | _ | 18 | Association with pSS lymphoma [194] IgA isotype of anti-Ro/SSA ACPA+ and high urine pH for anti-alpha-enolase [195] |
| Alpha-enolase | 50 | 20 | _ | _ | 50 | 82 | 90 | _ | _ | 26 | |
| Rho GDP-dissociation inhibitor 2 | 50 | 20 | _ | _ | 50 | 86 | 90 | _ | _ | 26 | |
| AECG Classification Criteria (2002) [250] | SICCA Classification Criteria (2012) [251] | ACR-EULAR Classification Criteria (2016) [252] | ||||
|---|---|---|---|---|---|---|
| Domain | Item Definition | Value | Item Definition | Value | Item Definition | Value |
| Subjective eye dryness | ≥1/3 specific questions | minor | / | _ | / | _ |
| Subjective oral dryness | ≥1/3 specific questions | minor | / | _ | / | _ |
| Ocular signs | Schirmer (≤5 mm/5 min) OR Van Bijsterveld ≥ 4 | minor | OSS ≥3 | 1 | Schirmer (<5 mm/5 min) | 1 |
| OSS ≥ 5 OR Van Bijsterveld ≥ 4 | 1 | |||||
| SG dysfunction | UWSF (≤1.5 mL/15 min) OR Compatible parotid sialography OR Anormal salivary scintigraphy | minor | / | _ | UWSF (≤0.1 mL/min) | 1 |
| MSGB | Focus-score ≥ 1 | Major | Focus-score ≥ 1 | 1 | Focus-score ≥ 1 | 3 |
| Auto antibodies | Anti-Ro/SSA or Anti-La/SSB | Major | Anti-Ro/SSA or Anti-La/SSB OR RF(+) with ANA(+) ≥1:320 | 1 | Anti-Ro/SSA | 3 |
| pSS definition | 4 out of 6 with ≥ 1 Major (or 3 out of 4 objectives findings) | pSS signs and/or symptoms with ≥2/3 criteria | Sicca or ESSDAI manifestation with a total score ≥ 4 | |||
| Exclusions criteria |
|
|
| |||
| Sensitivity | 93.5% | 92.5% | 96% | |||
| Specificity | 94.0% | 95.4% | 95% | |||
| Sicca Symptoms Complex | Glandular Involvement | Articular Involvement | Systemic Involvement | |
|---|---|---|---|---|
| Xerogenic medications | X | _ | _ | _ |
| Aromatase inhibitors | (X) | _ | X | (X) pSS-like |
| Age-related dryness | X | _ | _ | _ |
| Metabolic sialadenosis | _ | X | _ | _ |
| Non-SS dry eye diseases | X | _ | _ | _ |
| Head and neck irradiation | X | _ | _ | _ |
| Sarcoïdosis | X | X | X | X |
| Hyperlipoproteinemia (II, IV, V type) | X | X | (X) | _ |
| Chronic Graft vs. Host disease | X | X | X | X |
| Primary lymphoma | X | X | _ | (X) |
| Amyloïdosis | X | X | (X) | (X) Renal, purpura |
| Viral chronic sialadenitis (HCV, HIV, HTLV-1) | X | (X) | X | X |
| Other chronic Non-specific sialadenitis | X | X Usually unilateral | _ | _ |
| Diabetes Mellitus | X | (X) Sialadenosis | (X) Cheiroarthropathy | (X) Neuropathy |
| Haemochromatosis | X | (X) | X CPPD | (X) |
| Other connective tissue disease | X | _ | X | X |
| Rheumatoid arthritis | (X) | _ | X | (X) |
| Granulomatosis with polyangiitis | X | (X) | X | X |
| IgG4-related disease (Mikulicz syndrome) | X | X | (X) | (X) |
| Anxiety, fibromyalgia | X | _ | (X) | _ |
| Checkpoint inhibitors | X | (X) | X | X |
| EULAR Sjögren’s Syndrome Disease Activity Index | EULAR Sjögren’s Syndrome Patient Reported Index | Sjögren’s Syndrome Disease Damage Index | Sjögren’s Syndrome Damage Index | |
|---|---|---|---|---|
| Abbreviation | ESSDAI | ESSPRI | SSDDI | SSDI |
| First description | Seror et al. [294] | Seror et al. [295] | Vitali et al. [296] | Barry et al. [297] |
| Year | 2010 | 2011 | 2007 | 2008 |
| Type | Activity index | PRO | Damage index | Damage index |
| Domains (n) | 12 | 1 | 6 | 9 |
| Items (n) | 44 | 3 | 9 | 27 |
| Items scoring | 0 to 3 | VAS (0–10) | 1, 2 or 5 | 1 |
| Domain weight | 1 to 6 | 1 | 1 | 1 |
| Calculation | Sum | Mean | Sum | Sum |
| Score range | 0–123 | 0–10 | 0–16 | 0–27 |
| Clinically significant threshold | <5 Low ≥5, ≤13 moderate ≥14 high | ≥5/10 is an unsatisfactory symptom state | - | - |
| Minimal clinically important difference | ≥3 points improvement | ≥1 point or ≥15% improvement | - | - |
| Salivary Gland Involvement | Lachrymal Gland Involvement | Skin and Vaginal Mucosa Involvement | |
|---|---|---|---|
| Self-Care |
|
| |
| Conserve |
| ||
| Replace |
|
|
|
| Stimulate |
|
|
|
| Complications Prevention and Management |
|
|
| DRUG | TRIAL (Reference) | Inclusion Criteria | Number of Subjects | Age (Years) | Disease Duration (Years) | Mean ESSDAI | Primary Outcome | Results | Effects (Statistically Significant) | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Drg | Ctrl | Sicca Syndrome | Fibro-Like | Systemic | ||||||||
| Rituximab Anti-CD20 | NCT00363350 Phase I/II [343] | AECG criteria RF+ and SSa and/or SSb+ SWS >0.15 mL/min | 20 | 10 | 43 ± 11 | 5.25 ± 4.17 | 8 (4–13) | SWS ⇧ at 48w | met | SWS/UWS ⇧ LG test ⇧ Schirmer = BUT = | SF36 ⇧ MFI ⇩ | Vasculitis ⇩ |
| Phase III [330] | AECG criteria SSa and/or SSb+ F-VAS ≥5/10 | 8 | 9 | 51 (22–64) | 7.25 (1–18) | na | ⇩ > 20% of F-VAS at 24w; ⇩ F-VAS at 24w; ⇩ >30% of F-VAS at 24w | not met not met not met | UWS = Schirmer = | F-VAS ⇩ PROFAD ⇩ P-VAS = Soc-SF36 ⇧ | Glandular ⇩ | |
| Phase III [332] | AECG criteria SSa and/or SSb+ Disease duration ≤ 2y 2/5 of [PhGA >50 mm or ESSDAI ≥ 6 or subESSPRI ≥ 5] | 19 | 22 | 40 (27–53) | 1 (1–2) | 20 (6–41) | ΔESSDAI until 120W | met from 24w to 120w | D-VAS ⇩ Schirmer ⇧ UWS ⇧ | P-VAS = F-VAS ⇩ | ESSDAI ⇩ | |
| NCT00740948 Phase III TEARS [331] | AECG criteria with 2/4 VAS ≥ 5/10 for PhGA, pain, fatigue and dryness AND biologically active OR 1 extra-glandular manifestation or parotid gland enlargement. | 63 | 57 | 52.9 ± 13.3 | 4.6 ± 4.8 | 10 ± 6.9 | ⇩ 30% of at least 2/4 VAS at 6-16-24w | met at 6w not met at 16-24w | D-VAS ⇩ Schirmer = | P-VAS = F-VAS ⇩ | ESSDAI = Glandular = Articular = | |
| Phase III TRACTISS [344] | pSS with SSa+ UWS >0 mL/min F-VAS and D-VAS >5/10 | 67 | 66 | 54 ± 11.5 | 5.7 ± 5.4 | 5.7 ± 4.5 | ⇩ 30% D-VAS and F-VAS at 48w | not met | UWS ⇧ ESSPRI = D-VAS = | F-VAS = SF36 = PROFAD = | ESSDAI ⇩ | |
| Epratuzumab Anti-CD22 | Post-hoc Phase I/II EMBODY [337] | SLE with SSa+ and SS diagnosis | 31 + 41 | 40 | 46.4 ± 12.3 | 5.1 (0–34) | na | BICLA at 48w ΔBILAG at 48w ΔSLEDAI at 48w ΔPhGA at 48w | met met not met not met | na | na | BILAG ⇩ |
| DRUG | TRIAL (References) | Inclusion Criteria | Number of Subjects | Age (Years) | Disease Duration (Years) | Mean ESSDAI | Primary Outcome | Results | Effects (Statistically Significant) | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Drg | Ctrl | Sicca Syndrome | Fibro-Like | Systemic | ||||||||
| Belimumab Anti-BAFF | NCT01160666 NCT01008982 Phase II BELISS [338,339] | AECG criteria SSa and/or SSb+ AND systemic complication OR B cell activation OR early disease (≤5 years) | 30 | - | 49.5 ± 6.5 | 5.7 ± 5.6 | 8.8 ± 7.4 | ⇩ of 2/5 VAS at 28w
| 60% response | ESSPRI ⇩ D-VAS ⇩ UWS = Schirmer = | ESSPRI ⇩ P-VAS = F-VAS = SF36 = | ESSDAI ⇩ Glandular ⇩ |
| Follow-up of previous study | 15 | - | 40.2 ± 11.8 | 5.9 ± 5.7 | 3.8 ± 3.1 | Idem between 28–52w | 86.7% Stable response | ESSPRI ⇩ D-VAS ⇩ UWS = Schirmer = | ESSPRI ⇩ P-VAS = F-VAS = Phys-SF36⇧ | ESSDAI ⇩ Glandular ⇩ Articular ⇩ Biologic ⇩ | ||
| RC18 TACI-Igfusion protein | NCT04078386 Phase II [345] | AECG criteria SSa+ ESSDAI ≥ 5 | 30 | ? | ? | ? | ΔESSDAI at 24w | December 2020 | Secondary endpoint | Secondary endpoint | Primary endpoint | |
| Rituximab Anti-CD20 + Belimumab Anti-BAFF | NCT02631538 Phase II [346] | AECG criteria SSa and/or SSb+ ESSDAI ≥ 5 UWS >0 mL/min D-VAS ≥ 5/10 | 70 | ? | ? | ? | SAEs at 104w AESIs at 104w | Study completed on June 2020 | Secondary endpoint | na | Secondary endpoint | |
| Tibulizumab (LY3090106) Anti-BAFF + Anti-IL-17 | NCT02614716 Phase I [347] | AECG criteria SSa and/or SSb+ | 32 | ? | ? | ? | SAEs at 197d | Not published | na | na | na | |
| Ianalumab (VAY736) Anti-BAFFR | NCT02149420 Phase II [348] | AECG criteria ANA ≥1:160 SSa and/or SSb+ ESSDAI ≥ 6 UWS >0 mL/min | 6+12 | 9 | 50.5 ± 12.16 | ? | 12.5 (6, 31) | ΔESSDAI at 12w | not met | D-VAS ⇩ | SF-36 = MFI ⇩ F-VAS ⇩ | ESSDAI = Articular ⇩ |
| NCT02962895 Phase II [349] | AECG criteria SSa+ ESSDAI ≥ 6 (from 7 domains only) | 195 | ? | ? | ? | Change in multi-dimensional disease activity at 24w | Study completed on June 2020 | Secondary endpoint | Secondary endpoint | Primary endpoint | ||
| DRUG | TRIAL (Reference) | Inclusion Criteria | Number of Subjects | Age (Years) | Disease Duration (Years) | Mean ESSDAI | Primary Outcome | Results | Effects (Statistically Significant) | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Drg | Ctrl | Sicca Syndrome | Fibro-Like | Systemic | ||||||||
| LOU064 BTK inhibitor | NCT04035668 Phase II LOUISSe [350] | 2016 ACR/EULAR criteria SSa and/or SSb+ ESSDAI ≥ 6 UWS >0 mL/min | 252 | ? | ? | ? | ΔESSDAI at 24w | Estimated Study Completion on January 2023 | Secondary endpoint | Secondary endpoint | Secondary endpoint | |
| Tirabrutinib (GS-4059) BTK inhibitor | NCT03100942 Phase II [342] | AECG criteria SSa and/or SSb+ ESSDAI ≥ 4 | 38 | 37 | 55.8 ± 10.06 | ? | 10.4 ± 5.36 | Protocol-Specified Response Criteria at 12w | not met | ESSPRI = | ESSPRI = | ESSDAI = |
| BMS-986142 BTK inhibitor | NCT02843659 Phase II [351] | 2016 ACR/EULAR criteria SSa and/or SSb+ ESSDAI ≥ 6 UWS >0 mL/min | 5+6 | 7 | 51.2 ± 11.41 | ? | ? | ΔESSDAI at 12w | Not published | Secondary endpoint | Secondary endpoint | Secondary endpoint |
| Branebrutinib BTK inhibitor | NCT04186871 Phase II [352] | 2016 ACR/EULAR criteria Moderate to severe pSS | ? | ? | ? | ? | ? | Protocol-Specified Response Criteria at 24w | Estimated Study Completion on June 2022 | na | na | Primary endpoint |
| Baminercept LTβ-R fusion protein | NCT01552681 Phase II [353] | 2016 ACR/EULAR criteria UWS >0.1 mL/min ≥ 1 non-life-threatening systemic manifestation(s) | 33 | 19 | 52.0 ± 11.0 | ? | 3.1 ± 3.4 | ΔSWS at 24w | not met | D-VAS = Schirmer ⇧ UWS = | F-VAS = | ESSDAI = |
| Parsaclisib (INCB050465) PI3Kδ inhibitor | NCT03627065 Phase II [354] | AECG criteria SGUS score > 2 SSa and/or SSb+ ESSDAI ≥ 6 Oral dryness score ≥ 5. | 10 | ? | ? | ? | ΔSGUS score at 12w | Not published | na | na | na | |
| Seletalisib (UCB5857) PI3Kδ inhibitor | NCT02610543 Phase II [355] | AECG criteria FAN ≥ 1:160 SSa and/or SSb+ ESSDAI ≥ 6 | 13 | 14 | ? | ? | ? | ΔESSDAI at 12w | not met | ESSPRI = SWSF = Schirmer = | na | ESSDAI = |
| Leniolisib (CDZ173) PI3Kδ inhibitor | NCT02775916 Phase II [356] | pSS diagnosis SSa and/or SSb+ ESSDAI ≥ 6, ESSPRI ≥ 5 SWS > 0.1 mL/min | 20 | 10 | 47.3 ± 13.07 | ? | ? | ΔESSDAI at 12w SAEs at 12w | not met | ESSPRI = | SF-36 = MFI = | na |
| DRUG | TRIAL | Inclusion Criteria | Number of Subjects | Age (Years) | Disease Duration (Years) | Mean ESSDAI | Primary Outcome | Results | Effects (Statistically Significant) | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Drg | Ctrl | Sicca Syndrome | Fibro-Like | Systemic | ||||||||
| Abatacept CTLA-4 Ig fusion protein | NCT02915159 Phase III [357] | 2016 ACR/EULAR criteria SSa+ ESSDAI ≥ 5 | 92 | 95 | 52 ± 12.9 | ? | 9.4 ± 4.3 | ΔESSDAI at 169d | Not met | ESSPRI = SWS = | ESSPRI = | ESSDAI = DAS28 ⇩ |
| Phase I/II ASAP [358] | AECG criteria and ESSDAI ≥ 6 Disease duration ≤ 5 years SWS ≥ 0.10 mL/min SSa and/or SSb+ or FR+ Proven by parotid gland biopsy. | 15 | - | 43 (32–51) | 11 (7–36) | 11 (8–14) | ΔESSDAI at 24-48w | met | ESSPRI ⇩ SWS/UWS = Schirmer = BUT ⇧ | ESSPRI ⇩ | ESSDAI ⇩ | |
| NCT02067910 Phase III ASAPIII [359] | AECG criteria and ESSDAI ≥ 5 Time from diagnosis ≤ 7 years | 40 | 39 | 49 ± 16 | 8 (4–14) | ? | ΔESSDAI at 24w | Not met | ESSPRI ⇩ FSFI ⇧ DVAS = UWS = Schirmer = | Fatigue = | ESSDAI = Articular ⇩ | |
| Iscalimab (CFZ533) Anti-CD40 | NCT02291029 Phase IIa [360] | AECG criteria and ESSDAI ≥ 6 SSA+ OR FR+ and FAN ≥ 1:320 SWS ≥ 0 mL/min | 8+21 | 4+11 | 51.3 ± 13.5 | ? | 10.7 ± 4.6 | SAEs at 12w | safe | ESSPRI ⇩ UWS = Schirmer = | MFI = SF-36 = | ESSDAI ⇩ Articular ⇩ |
| NCT03905525 Phase II TWINSS [361] | 2016 ACR/EULAR criteria SSa+ SWS > 0.01 mL/min, P1: ESSDAI ≥ 5 or P2 ESSPRI ≥ 5. | 260 | ? | ? | ? | ΔESSDAI at 24w in P1 ΔESSPRI at 24w in P2 | Estimated Study Completion on June 2022 | Included endpoint | Included endpoint | Included endpoint | ||
| VIB4920 MEDI4920 Anti-CD40L | NCT04129164 Phase II [362] | P1: ESSDAI ≥ 5 P2: ESSDAI < 5 et ESSPRI ≥ 5 | 174 | ? | ? | ? | ΔESSDAI at 169d in P1 ΔESSPRI at 169d in P2 | Estimated Study Completion on April 2022 | Included endpoint | Included endpoint | Included endpoint | |
| Prezalumab (AMG557) (MEDI5872) Anti-B7RP1 | NCT02334306 Phase IIa [363] | AECG criteria and ESSDAI ≥ 5 SSa and/or SSb+ FR+, cryoglobulinemia or hypergammaglobulinemia | 16 | 16 | 50.7 ± 13 | ? | ? | ΔESSDAI at 99d | Not met | ESSPRI = | ESSPRI = | ESSDAI = |
| Lulizumab (BMS-931699) Anti-CD28 | NCT02843659 Phase II [351] | 2016 ACR/EULAR criteria SSa and/or SSb+ ESSDAI ≥ 5 USW > 0.01 mL/min | 5+6 | 7 | 51.2 ± 11.41 | ? | ? | ΔESSDAI at 12w | Not published | Secondary endpoint | Secondary endpoint | Primary endpoint |
| DRUG | TRIAL (Reference) | Inclusion Criteria | Number of Subjects | Age (Years) | Disease Duration (Years) | Mean ESSDAI | Primary Outcome | Results | Effects (Statistically Significant) | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Drg | Ctrl | Sicca Syndrome | Fibro-Like | Systemic | ||||||||
| Petesicatib RO5459072 Cathepsin S Inhibitor | NCT02701985 Phase IIa [364] | AECG criteria SSa and/or SSb+ ESSDAI ≥ 5 ESSPRI ≥ 5 USW > 0.0 mL/min Oral D-VAS ≥ 5/10 | 38 | 37 | 52.2 ± 12.5 | ? | ? | ΔESSDAI ≥ 3 at 12w | Not met | ESSPRI = | ESSPRI = SF36 = | ESSDAI = |
| Efalizumab Anti-LFA-1 | NCT00344448 Phase II [365] | AECG criteria SSa and/or SSb+ | 6 | 3 | 53 ± 11.2 | ? | ? | Protocol-specified composite score at 12w | Early termination due to serious side effect in other trial | |||
| DRUG | TRIAL | Inclusion Criteria | Number of Subjects | Age (Years) | Disease Duration (Years) | Mean ESSDAI | Primary Outcome | Results | Effects (Statistically Significant) | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Drg | Ctrl | Sicca Syndrome | Fibro-Like | Systemic | ||||||||
| Anakinra IL1R antagonist protein | NCT00683345 Phase II [333] | AECG criteria 18–80 years Western European descent No depression or comorbidity | 13 | 13 | 55 (36–80) | 5 (1–17) | ? | Group-wise comparison of the fatigue scores at 4w | not met | na | F-VAS ⇩ | na |
| Tocilizumab Anti-IL-6R | NCT01782235 Phase I/II ETAP [366] | AECG criteria ESSDAI ≥ 5 | 55 | 55 | 50.9 (26–76) | ? | 11.5 (5–25) | ΔESSDAI ≥ 3 at 12W without new item without ⇧ ≥1/10 PGA | not met | ESSPRI = Schirmer = | ESSPRI = | ESSDAI = Articular ⇩ |
| Infliximab Anti-TNF | Phase III TRIPSS [367] | AECG criteria 2/3 D-VAS, F-VAS, P-VAS ≥ 5/10 | 54 | 49 | 54.4 ± 10.4 | 4.0 ± 5.5 | na | ⇧ 30% in 2/3 D-VAS, F-VAS, P-VAS at 10–22w | not met | SWS = Schirmer = | SF-36 = | SJC = TJC = |
| Etanercept TNFR-Ig fusion protein | NCT00001954 Phase II [368] | 1986 and AECG criteria Elevated ESR or IgG levels | 14 | 14 | 55.5 (46, 59) | ? | na | ⇧ 20% in 2/3 pSS domains (protocol-specified) | not met | D-VAS = Schirmer = VB = SWS = | na | na |
| Ustekinumab Anti- IL-12/IL-23 (p40 subunit) | NCT04093531 Phase I [341] | 2016 ACR/EULAR criteria | 15 | - | ? | ? | ? | ΔESSDAI at 24W | Estimated Study Completion on December 2021 | na | Secondary endpoint | Primary endpoint |
| GSK2618960 anti–IL-7Rα | NCT03239600 Phase II [369] | AECG criteria SWS >0.1 mL/min ⇧ Ig or FR+ or ANA ≥ 1:320 D-VAS ≥ 5/10 or Schirmer < 10 mm | 0 | - | - | - | SAEs at 27w | Withdraw The study is stopped for Portfolio prioritization | ||||
| DRUG | TRIAL (Reference) | Inclusion Criteria | Number of Subjects | Age (years) | Disease Duration (Years) | Mean ESSDAI | Primary Outcome | Results | Effects (Statistically Significant) | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Drg | Ctrl | Sicca Syndrome | Fibro-Like | Systemic | ||||||||
| Daxdilimab VIB7734 Anti-ILT7 | NCT03817424 Phase I [370] | Unspecified | ? | ? | ? | ? | SAEs at 169d AESIs at 169d | June 2020 | na | na | na | na |
| Filgotinib Jak1 inhibitor | NCT03100942 Phase II [342] | AECG criteria SSa and/or SSb+ ESSDAI ≥ 4 | 38 | 37 | 52.2 ± 10.54 | ? | 10.2 ± 6.23 | Protocol-Specified Response Criteria at 12w | not met | ESSPRI = | ESSPRI = | ESSDAI = |
| Lanraplenib (GS-9876) SIK inhibitor | NCT03100942 Phase II [342] | AECG criteria SSa and/or SSb+ ESSDAI ≥ 4 | 38 | 37 | 56.2 ± 9.72 | ? | 10.5 ± 4.89 | Protocol-Specified Response Criteria at 12w | not met | ESSPRI = | ESSPRI = | ESSDAI = |
| RSLV-132 RNase1-Fc fusion protein | NCT03247686 Phase II [371] | AECG criteria SSA+ Interferon signature | 22 | 8 | ? | ? | ? | Interferon gene expression at day99 | Not published | ESSPRI = | mPRO-F ⇧ DSST ⇩ FACIT-F = ESSPRI = | ESSDAI = |
| Low-dose IL-2 T-reg induction | NCT01988506 Phase II Transreg [372] | pSS diagnosis | 84-132 | ? | ? | ? | T-reg percentage | Estimated Study Completion on February 2022 | na | na | na | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parisis, D.; Chivasso, C.; Perret, J.; Soyfoo, M.S.; Delporte, C. Current State of Knowledge on Primary Sjögren’s Syndrome, an Autoimmune Exocrinopathy. J. Clin. Med. 2020, 9, 2299. https://doi.org/10.3390/jcm9072299
Parisis D, Chivasso C, Perret J, Soyfoo MS, Delporte C. Current State of Knowledge on Primary Sjögren’s Syndrome, an Autoimmune Exocrinopathy. Journal of Clinical Medicine. 2020; 9(7):2299. https://doi.org/10.3390/jcm9072299
Chicago/Turabian StyleParisis, Dorian, Clara Chivasso, Jason Perret, Muhammad Shahnawaz Soyfoo, and Christine Delporte. 2020. "Current State of Knowledge on Primary Sjögren’s Syndrome, an Autoimmune Exocrinopathy" Journal of Clinical Medicine 9, no. 7: 2299. https://doi.org/10.3390/jcm9072299
APA StyleParisis, D., Chivasso, C., Perret, J., Soyfoo, M. S., & Delporte, C. (2020). Current State of Knowledge on Primary Sjögren’s Syndrome, an Autoimmune Exocrinopathy. Journal of Clinical Medicine, 9(7), 2299. https://doi.org/10.3390/jcm9072299