NIR Photodynamic Destruction of PDAC and HNSCC Nodules Using Triple-Receptor-Targeted Photoimmuno-Nanoconjugates: Targeting Heterogeneity in Cancer

 , ,
, ,
Abstract
:1. Introduction
2. Experimental Section
2.1. Materials
2.2. Cell Culture
2.3. NHS-PEG4-N3 and AF488-NHS Conjugation to Cetuximab, Holo-Transferrin, and Trastuzumab
2.4. Synthesis of Photosensitizing-Nanoconstructs (Untargeted-PSN)
2.5. Preparation of Photoimmuno-Nanoconjugates (PIN)
2.6. Physical Characterizations
2.7. Singlet Oxygen Measurements
2.8. Cellular Binding of PINs
2.9. In vitro PINs Internalization Studies
2.10. Photodynamic Treatment of PDAC and HNSCC Monocellular and Heterocellular 3D Nodules and Image Analysis
3. Results
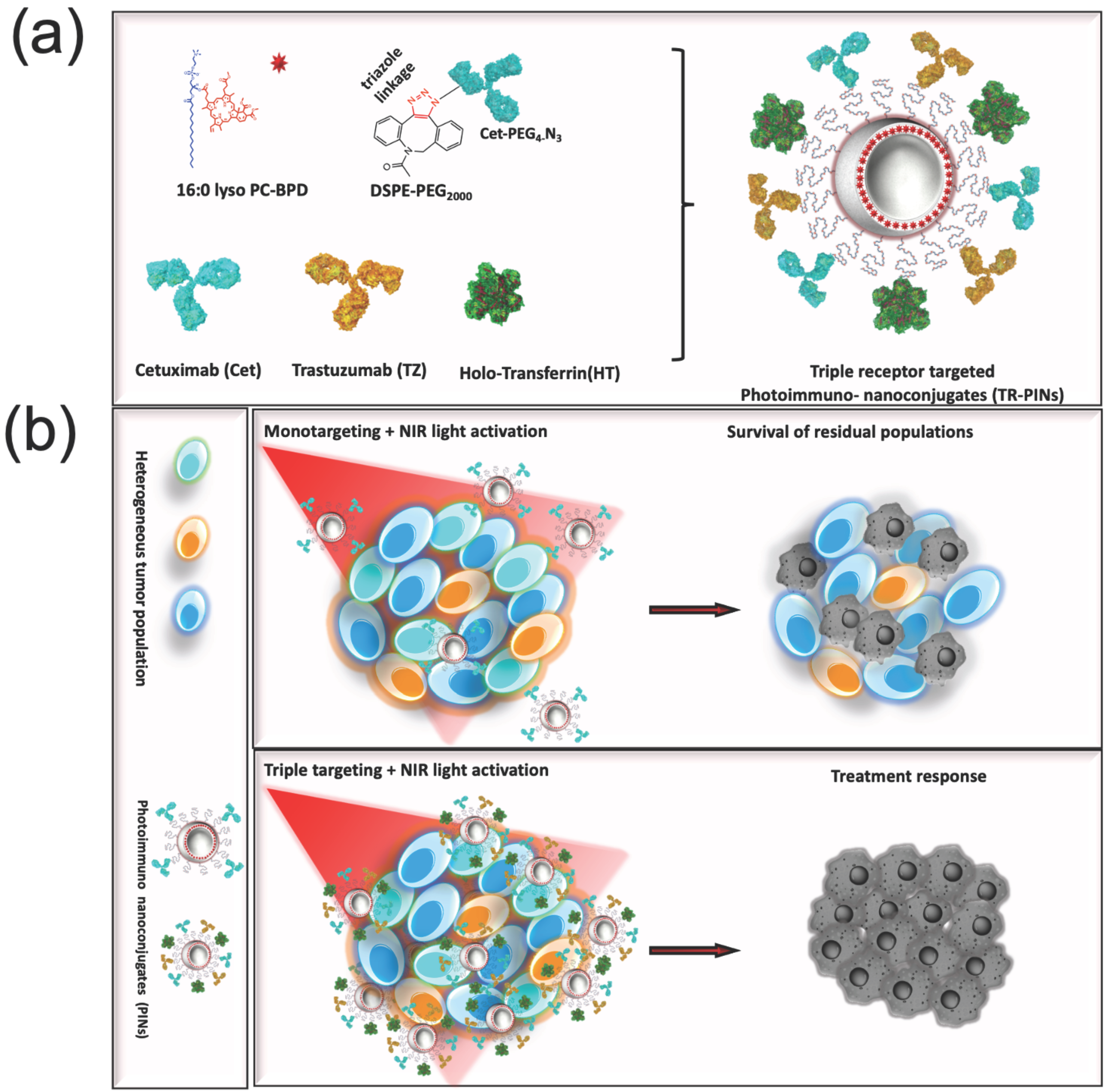
3.1. Design, Preparation, and Characterization of Photoimmuno-Nanoconjugates (PINs)
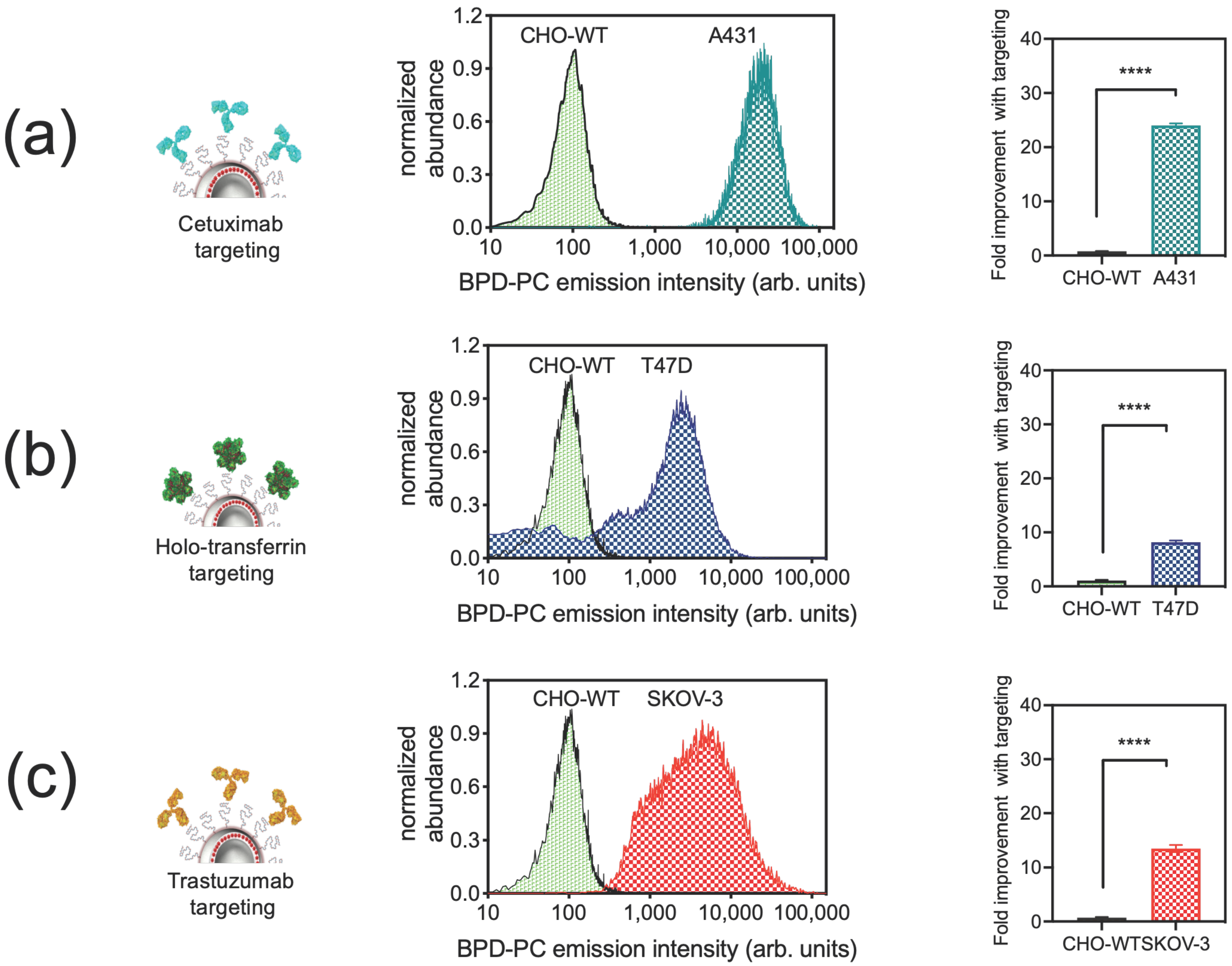
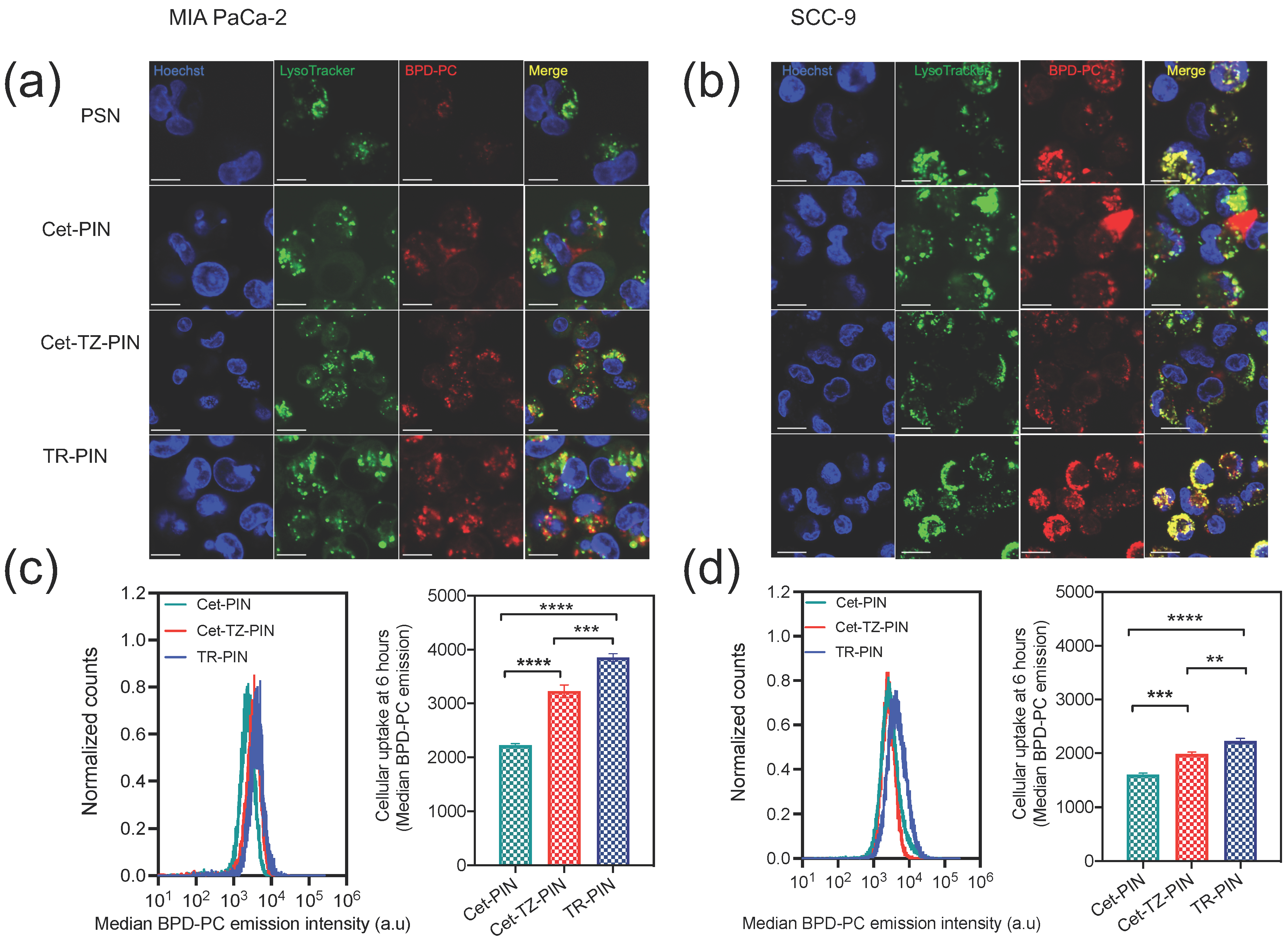
3.2. Cellular Binding Specificity of Photoimmuno-Nanoconjugates (PINs)
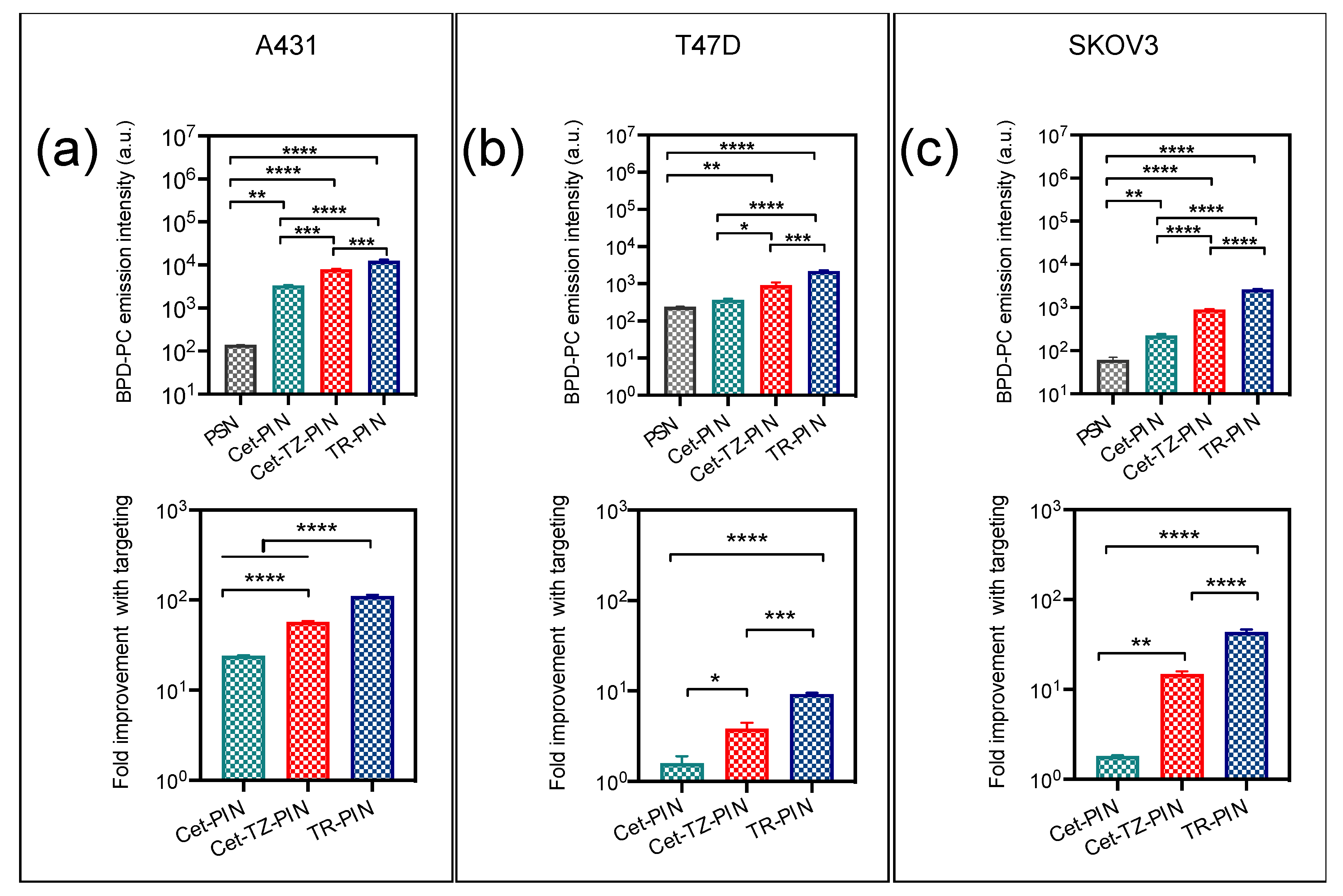
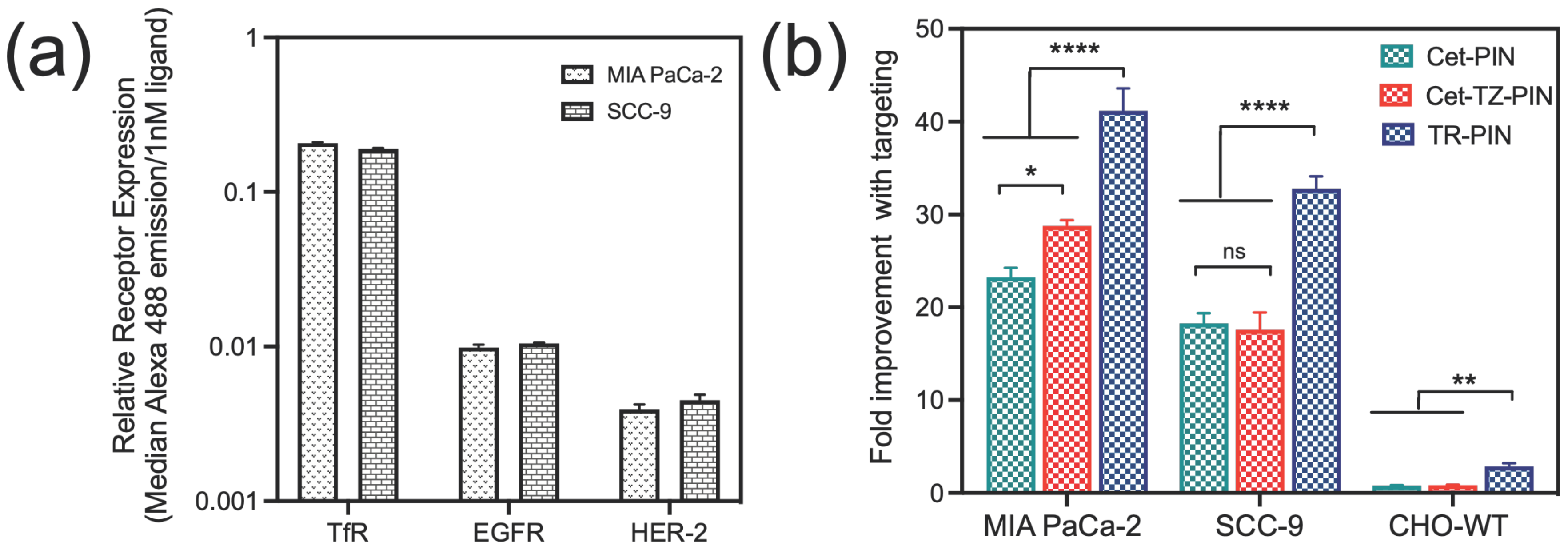
3.3. Triple Receptor Targeting Enhances PIN Binding and Cellular Uptake in MIA PaCa-2 PDAC Cells and SCC-9 HNSCC Cells
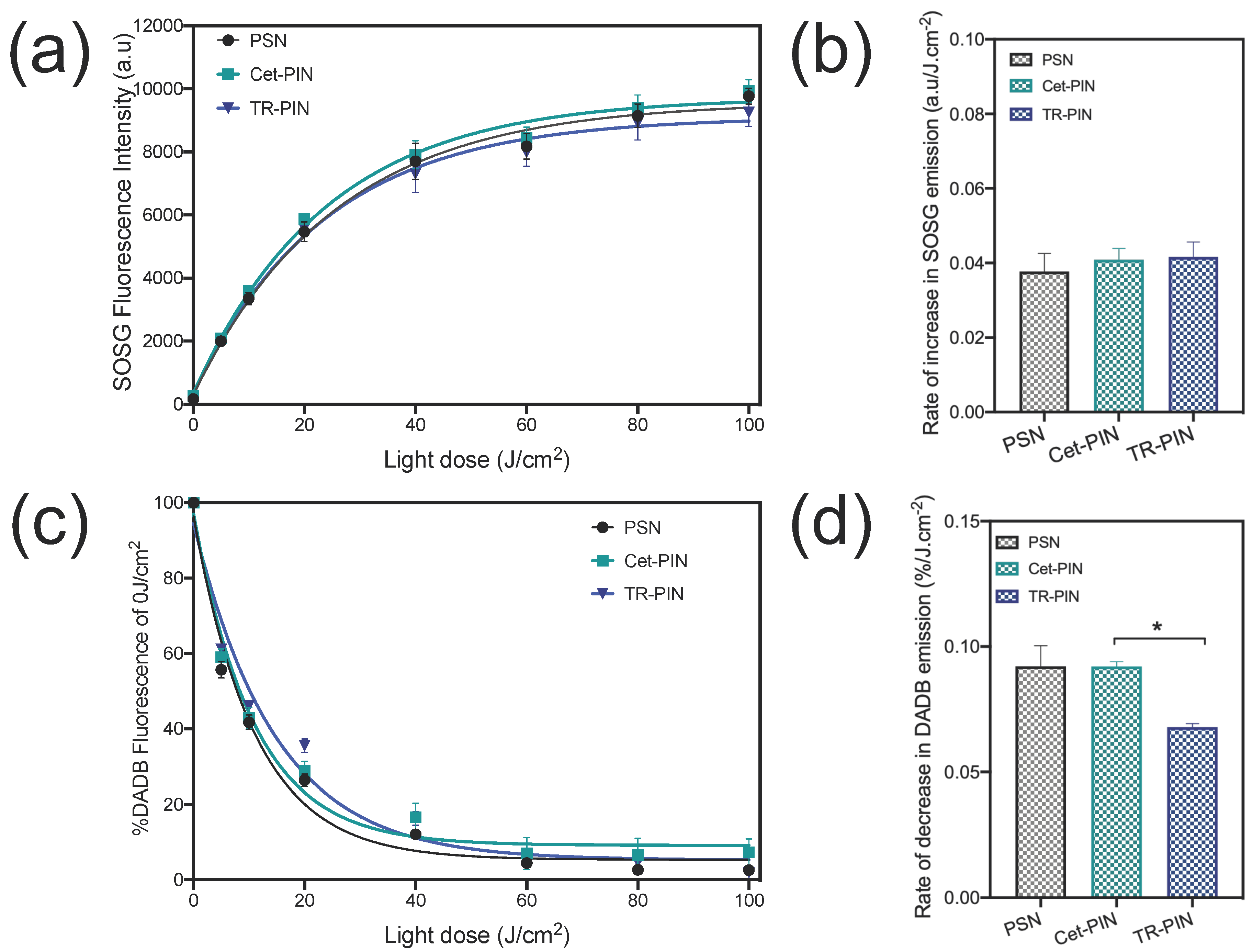
3.4. Singlet Oxygen Measurements
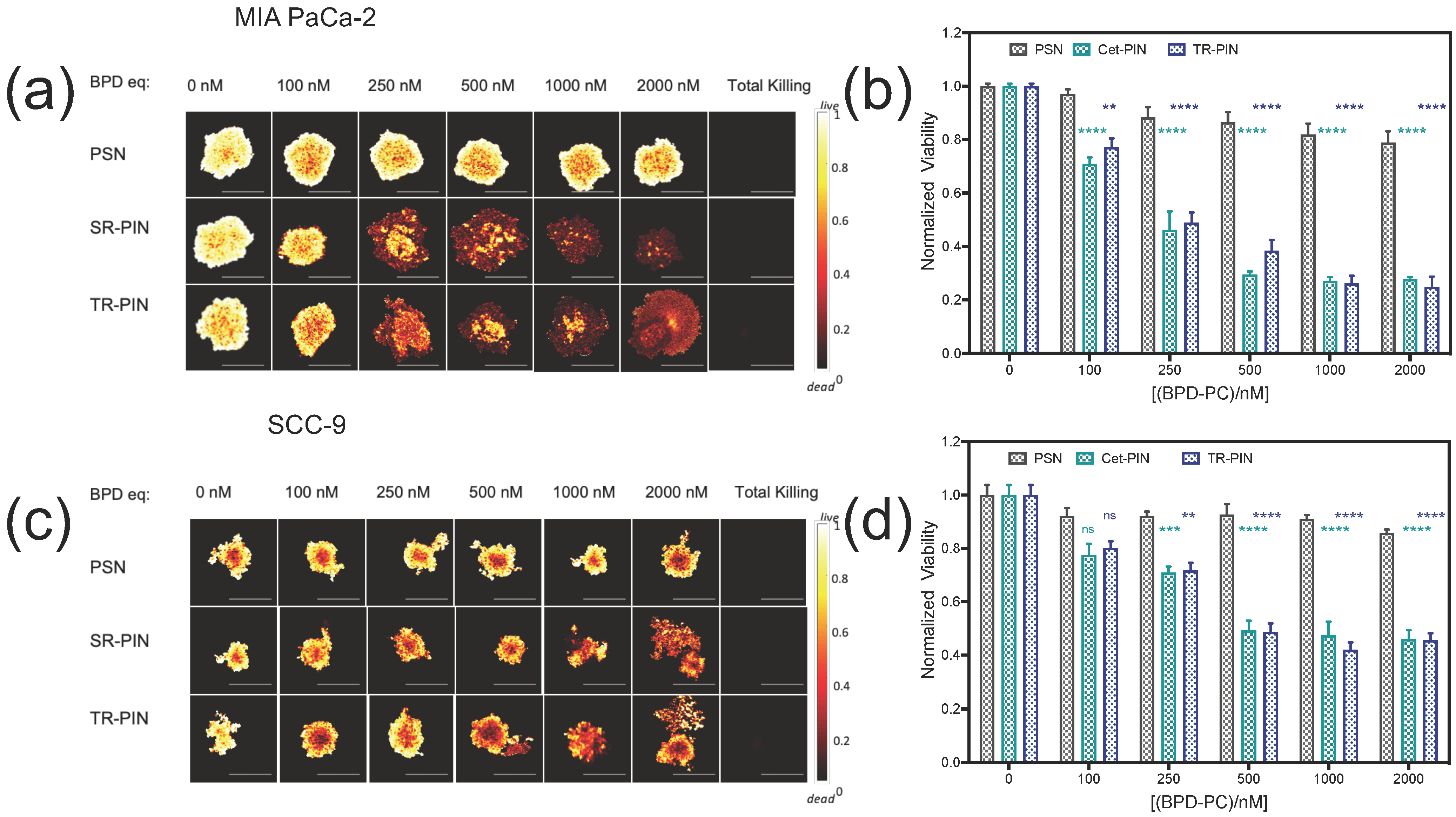
3.5. NIR Light-Mediated Photodynamic Treatment of PDAC and HNSCC Monocellular and Heterocellular 3D Models of Heterogeneity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Celli, J.P.; Spring, B.Q.; Rizvi, I.; Evans, C.L.; Samkoe, K.S.; Verma, S.; Pogue, B.W.; Hasan, T. Imaging and photodynamic therapy: Mechanisms, monitoring, and optimization. Chem. Rev. 2010, 110, 2795–2838. [Google Scholar] [CrossRef] [Green Version]
- Baskaran, R.; Lee, J.; Yang, S.-G. Clinical development of photodynamic agents and therapeutic applications. Biomater. Res. 2018, 22, 25. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, S.R.; Fernandes, R.; Sarmento, B.; Pereira, P.M.; Tomé, J.P. Photoimmunoconjugates: Novel synthetic strategies to target and treat cancer by photodynamic therapy. Org. Biomol. Chem. 2019, 17, 2579–2593. [Google Scholar] [CrossRef]
- Mew, D.; Wat, C.-K.; Towers, G.; Levy, J. Photoimmunotherapy: Treatment of animal tumors with tumor-specific monoclonal antibody-hematoporphyrin conjugates. J. Immunol. 1983, 130, 1473–1477. [Google Scholar]
- Vrouenraets, M.B.; Visser, G.W.; Stewart, F.A.; Stigter, M.; Oppelaar, H.; Postmus, P.E.; Snow, G.B.; Van Dongen, G.A. Development of meta-tetrahydroxyphenylchlorin-monoclonal antibody conjugates for photoimmunotherapy. Cancer Res. 1999, 59, 1505–1513. [Google Scholar]
- Hudson, R.; Carcenac, M.; Smith, K.; Madden, L.; Clarke, O.; Pelegrin, A.; Greenman, J.; Boyle, R. The development and characterisation of porphyrin isothiocyanate–monoclonal antibody conjugates for photoimmunotherapy. Br. J. Cancer 2005, 92, 1442–1449. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, S.; Wagner, U.; Oehr, P.; Krebs, D. Clinical use of photodynamic therapy in gynecologic tumor patients–antibody-targeted photodynamic laser therapy as a new oncologic treatment procedure. Zent. Gynakol. 1992, 114, 307–311. [Google Scholar]
- Schmidt, S.; Wagner, U.; Schultes, B.; Oehr, P.; Decleer, W.; Ertmer, W.; Lubaschowski, H.; Biersack, H.; Krebs, D. Photodynamic laser therapy with antibody-bound dyes. A new procedure in therapy of gynecologic malignancies. Fortschr. Med. 1992, 110, 298–301. [Google Scholar]
- Duska, L.R.; Hamblin, M.R.; Miller, J.L.; Hasan, T. Combination photoimmunotherapy and cisplatin: Effects on human ovarian cancer ex vivo. J. Natl. Cancer. Inst. 1999, 91, 1557–1563. [Google Scholar] [CrossRef] [Green Version]
- Soukos, N.S.; Hamblin, M.R.; Keel, S.; Fabian, R.L.; Deutsch, T.F.; Hasan, T. Epidermal growth factor receptor-targeted immunophotodiagnosis and photoimmunotherapy of oral precancer in vivo. Cancer Res. 2001, 61, 4490–4496. [Google Scholar]
- Spring, B.Q.; Abu-Yousif, A.O.; Palanisami, A.; Rizvi, I.; Zheng, X.; Mai, Z.; Anbil, S.; Sears, R.B.; Mensah, L.B.; Goldschmidt, R. Selective treatment and monitoring of disseminated cancer micrometastases in vivo using dual-function, activatable immunoconjugates. Proc. Natl. Acad. Sci. USA 2014, 111, E933–E942. [Google Scholar] [CrossRef] [Green Version]
- Savellano, M.D.; Pogue, B.W.; Hoopes, P.J.; Vitetta, E.S.; Paulsen, K.D. Multiepitope HER2 targeting enhances photoimmunotherapy of HER2-overexpressing cancer cells with pyropheophorbide-a immunoconjugates. Cancer Res. 2005, 65, 6371–6379. [Google Scholar] [CrossRef] [Green Version]
- Del Governatore, M.; Hamblin, M.R.; Shea, C.R.; Rizvi, I.; Molpus, K.G.; Tanabe, K.K.; Hasan, T. Experimental photoimmunotherapy of hepatic metastases of colorectal cancer with a 17.1 A chlorine6 immunoconjugate. Cancer Res. 2000, 60, 4200–4205. [Google Scholar]
- Molpus, K.L.; Hamblin, M.R.; Rizvi, I.; Hasan, T. Intraperitoneal photoimmunotherapy of ovarian carcinoma xenografts in nude mice using charged photoimmunoconjugates. Gynecol. Oncol. 2000, 76, 397–404. [Google Scholar] [CrossRef]
- Gillenwater, A.M.; Cognetti, D.; Johnson, J.M.; Curry, J.; Kochuparambil, S.T.; McDonald, D.; Fidler, M.J.; Stenson, K.; Vasan, N.; Razaq, M. RM-1929 photo-immunotherapy in patients with recurrent head and neck cancer: Results of a multicenter phase 2a open-label clinical trial. Am. Soc. Clin. Oncol. 2018, 36, 6039. [Google Scholar] [CrossRef]
- Rybinski, B.; Yun, K. Addressing intra-tumoral heterogeneity and therapy resistance. Oncotarget 2016, 7, 72322–72342. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Dang, H.; Wang, X.W. The significance of intertumor and intratumor heterogeneity in liver cancer. Exp. Mol. Med. 2018, 50, e416. [Google Scholar] [CrossRef]
- Diaz-Cano, S.J. Tumor heterogeneity: Mechanisms and bases for a reliable application of molecular marker design. Int. J. Mol. Sci. 2012, 13, 1951–2011. [Google Scholar] [CrossRef]
- Huang, M.; Shen, A.; Ding, J.; Geng, M. Molecularly targeted cancer therapy: Some lessons from the past decade. Trends Pharmacol. Sci. 2014, 35, 41–50. [Google Scholar] [CrossRef]
- Alizadeh, A.A.; Aranda, V.; Bardelli, A.; Blanpain, C.; Bock, C.; Borowski, C.; Caldas, C.; Califano, A.; Doherty, M.; Elsner, M.; et al. Toward understanding and exploiting tumor heterogeneity. Nat. Med. 2015, 21, 846–853. [Google Scholar] [CrossRef]
- Pribluda, A.; Cecile, C.; Jackson, E.L. Intratumoral heterogeneity: From diversity comes resistance. Clin. Cancer Res. 2015, 21, 2916–2923. [Google Scholar] [CrossRef] [Green Version]
- Troiani, T.; Martinelli, E.; Capasso, A.; Morgillo, F.; Orditura, M.; De Vita, F.; Ciardiello, F. Targeting EGFR in pancreatic cancer treatment. Curr. Drug Targets 2012, 13, 802–810. [Google Scholar] [CrossRef]
- Nedaeinia, R.; Avan, A.; Manian, M.; Salehi, R.; Ghayour-Mobarhan, M. EGFR as a potential target for the treatment of pancreatic cancer: Dilemma and controversies. Curr. Drug Targets 2014, 15, 1293–1301. [Google Scholar] [CrossRef]
- Jeong, S.M.; Hwang, S.; Seong, R.H. Transferrin receptor regulates pancreatic cancer growth by modulating mitochondrial respiration and ROS generation. Biochem. Biophys. Res. Commun. 2016, 471, 373–379. [Google Scholar] [CrossRef]
- Pollock, N.I.; Grandis, J.R. HER2 as a therapeutic target in head and neck squamous cell carcinoma. Clin. Cancer Res. 2015, 21, 526–533. [Google Scholar] [CrossRef] [Green Version]
- Del Campo, J.; Hitt, R.; Sebastian, P.; Carracedo, C.; Lokanatha, D.; Bourhis, J.; Temam, S.; Cupissol, D.; De Raucourt, D.; Maroudias, N. Effects of lapatinib monotherapy: Results of a randomised phase II study in therapy-naive patients with locally advanced squamous cell carcinoma of the head and neck. Br. J. Cancer 2011, 105, 618–627. [Google Scholar] [CrossRef]
- Williams, M.D.; Roberts, D.B.; Kies, M.S.; Mao, L.; Weber, R.S.; El-Naggar, A.K. Genetic and expression analysis of HER-2 and EGFR genes in salivary duct carcinoma: Empirical and therapeutic significance. Clin. Cancer Res. 2010, 16, 2266–2274. [Google Scholar] [CrossRef] [Green Version]
- Nardi, V.; Sadow, P.M.; Juric, D.; Zhao, D.; Cosper, A.K.; Bergethon, K.; Scialabba, V.L.; Batten, J.M.; Borger, D.R.; Iafrate, A.J. Detection of novel actionable genetic changes in salivary duct carcinoma helps direct patient treatment. Clin. Cancer Res. 2013, 19, 480–490. [Google Scholar] [CrossRef] [Green Version]
- Hendler, F.; Ozanne, B. Human squamous cell lung cancers express increased epidermal growth factor receptors. J. Clin. Investig. 1984, 74, 647–651. [Google Scholar] [CrossRef]
- Hanken, H.; Gaudin, R.; Gröbe, A.; Fraederich, M.; Eichhorn, W.; Smeets, R.; Simon, R.; Sauter, G.; Grupp, K.; Izbicki, J.R. Her2 expression and gene amplification is rarely detectable in patients with oral squamous cell carcinomas. J. Oral Pathol. Med. 2014, 43, 304–308. [Google Scholar] [CrossRef]
- Falchook, G.S.; Lippman, S.M.; Bastida, C.C.; Kurzrock, R. Human epidermal receptor 2-amplified salivary duct carcinoma: Regression with dual human epidermal receptor 2 inhibition and anti-vascular endothelial growth factor combination treatment. Head Neck 2014, 36, E25–E27. [Google Scholar] [CrossRef] [Green Version]
- Kearsley, J.; Furlong, K.; Cooke, R.; Waters, M. An immunohistochemical assessment of cellular proliferation markers in head and neck squamous cell cancers. Br. J. Cancer 1990, 61, 821–827. [Google Scholar] [CrossRef] [Green Version]
- Masuda, H.; Zhang, D.; Bartholomeusz, C.; Doihara, H.; Hortobagyi, G.N.; Ueno, N.T. Role of epidermal growth factor receptor in breast cancer. Breast Cancer Res. Treat. 2012, 136, 331–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iqbal, N.; Iqbal, N. Human epidermal growth factor receptor 2 (HER2) in cancers: Overexpression and therapeutic implications. Mol. Biol. Int. 2014, 2014, 852748. [Google Scholar] [CrossRef] [PubMed]
- Rychtarcikova, Z.; Lettlova, S.; Tomkova, V.; Korenkova, V.; Langerova, L.; Simonova, E.; Zjablovskaja, P.; Alberich-Jorda, M.; Neuzil, J.; Truksa, J. Tumor-initiating cells of breast and prostate origin show alterations in the expression of genes related to iron metabolism. Oncotarget 2017, 8, 6376. [Google Scholar] [CrossRef] [Green Version]
- Teplinsky, E.; Muggia, F. EGFR and HER2: Is there a role in ovarian cancer? Transl. Cancer Res. 2015, 4, 107–117. [Google Scholar]
- Basuli, D.; Tesfay, L.; Deng, Z.; Paul, B.; Yamamoto, Y.; Ning, G.; Xian, W.; McKeon, F.; Lynch, M.; Crum, C.P.; et al. Iron addiction: A novel therapeutic target in ovarian cancer. Oncogene 2017, 36, 4089–4099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scagliotti, G.V.; Selvaggi, G.; Novello, S.; Hirsch, F.R. The biology of epidermal growth factor receptor in lung cancer. Clin. Cancer Res. 2004, 10, 4227s–4232s. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.; Parker, B.A.; Schwab, R.; Kurzrock, R. HER2 aberrations in cancer: Implications for therapy. Cancer Treat. Rev. 2014, 40, 770–780. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Zhang, J.; Song, F.; Tian, M.; Shi, B.; Jiang, H.; Xu, W.; Wang, H.; Zhou, M.; Pan, X. EGFR regulates iron homeostasis to promote cancer growth through redistribution of transferrin receptor 1. Cancer Lett. 2016, 381, 331–340. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Zhang, H.; Lin, Y.; Chen, P.; Min, J.; Wang, Z.; Xiao, W.; Chen, B. Mechanisms of gambogic acid-induced apoptosis in non-small cell lung cancer cells in relation to transferrin receptors. J. Chemother. 2009, 21, 666–672. [Google Scholar] [CrossRef]
- Chaux, A.; Cohen, J.S.; Schultz, L.; Albadine, R.; Jadallah, S.; Murphy, K.M.; Sharma, R.; Schoenberg, M.P.; Netto, G.J. High epidermal growth factor receptor immunohistochemical expression in urothelial carcinoma of the bladder is not associated with EGFR mutations in exons 19 and 21: A study using formalin-fixed, paraffin-embedded archival tissues. Hum. Pathol. 2012, 43, 1590–1595. [Google Scholar] [CrossRef] [Green Version]
- Rahman, S.A.; Yokoyama, M.; Nishio, S.; Takeuchi, M. Flow cytometric evaluation of transferrin receptor in transitional cell carcinoma. Urol. Res. 1997, 25, 325–329. [Google Scholar] [CrossRef]
- Baselga, J.; Swain, S.M. Novel anticancer targets: Revisiting ERBB2 and discovering ERBB3. Nat. Rev. Cancer 2009, 9, 463–475. [Google Scholar] [CrossRef]
- Seshacharyulu, P.; Ponnusamy, M.P.; Haridas, D.; Jain, M.; Ganti, A.K.; Batra, S.K. Targeting the EGFR signaling pathway in cancer therapy. Expert Opin. Ther. Targets 2012, 16, 15–31. [Google Scholar] [CrossRef] [Green Version]
- Tebbutt, N.; Pedersen, M.W.; Johns, T.G. Targeting the ERBB family in cancer: Couples therapy. Nat. Rev. Cancer 2013, 13, 663–673. [Google Scholar] [CrossRef]
- Kol, A.; van Scheltinga, A.G.T.; Timmer-Bosscha, H.; Lamberts, L.E.; Bensch, F.; de Vries, E.G.; Schröder, C.P. HER3, serious partner in crime: Therapeutic approaches and potential biomarkers for effect of HER3-targeting. Pharmacol. Ther. 2014, 143, 1–11. [Google Scholar] [CrossRef]
- Szekeres, T.; Sedlak, J.; Novotny, L. Benzamide riboside, a recent inhibitor of inosine 5′-monophosphate dehydrogenase induces transferrin receptors in cancer cells. Curr. Med. Chem. 2002, 9, 759–764. [Google Scholar] [CrossRef]
- Ryschich, E.; Huszty, G.; Knaebel, H.; Hartel, M.; Büchler, M.; Schmidt, J. Transferrin receptor is a marker of malignant phenotype in human pancreatic cancer and in neuroendocrine carcinoma of the pancreas. Eur. J. Cancer 2004, 40, 1418–1422. [Google Scholar] [CrossRef]
- Daniels-Wells, T.R.; Penichet, M.L. Transferrin receptor 1: A target for antibody-mediated cancer therapy. Immunotherapy 2016, 8, 991–994. [Google Scholar] [CrossRef] [Green Version]
- García-Foncillas, J.; Sunakawa, Y.; Aderka, D.; Wainberg, Z.; Ronga, P.; Witzler, P.; Stintzing, S. Distinguishing Features of Cetuximab and Panitumumab in Colorectal Cancer and Other Solid Tumors. Front. Oncol. 2019, 9, 849. [Google Scholar] [CrossRef]
- Vermorken, J.B.; Mesia, R.; Rivera, F.; Remenar, E.; Kawecki, A.; Rottey, S.; Erfan, J.; Zabolotnyy, D.; Kienzer, H.-R.; Cupissol, D. Platinum-based chemotherapy plus cetuximab in head and neck cancer. N. Engl. J. Med. 2008, 359, 1116–1127. [Google Scholar] [CrossRef] [Green Version]
- Thatcher, N.; Hirsch, F.R.; Luft, A.V.; Szczesna, A.; Ciuleanu, T.E.; Dediu, M.; Ramlau, R.; Galiulin, R.K.; Bálint, B.; Losonczy, G. Necitumumab plus gemcitabine and cisplatin versus gemcitabine and cisplatin alone as first-line therapy in patients with stage IV squamous non-small-cell lung cancer (SQUIRE): An open-label, randomised, controlled phase 3 trial. Lancet Oncol. 2015, 16, 763–774. [Google Scholar] [CrossRef]
- Maximiano, S.; Magalhaes, P.; Guerreiro, M.P.; Morgado, M. Trastuzumab in the Treatment of Breast Cancer. BioDrugs 2016, 30, 75–86. [Google Scholar] [CrossRef]
- Ross, J.S.; Mulcahy, M. HER2 Testing in Gastric/Gastroesophageal Junction Adenocarcinomas: Unique Features of a Familiar Test. Gastrointest. Cancer Res. 2011, 4, 62–66. [Google Scholar] [CrossRef]
- Daniels, T.R.; Bernabeu, E.; Rodríguez, J.A.; Patel, S.; Kozman, M.; Chiappetta, D.A.; Holler, E.; Ljubimova, J.Y.; Helguera, G.; Penichet, M.L. The transferrin receptor and the targeted delivery of therapeutic agents against cancer. Biochim. Biophys Acta 2012, 1820, 291–317. [Google Scholar] [CrossRef] [Green Version]
- Jiang, N.; Wang, D.; Hu, Z.; Shin, H.J.; Qian, G.; Rahman, M.A.; Zhang, H.; Amin, A.R.; Nannapaneni, S.; Wang, X.; et al. Combination of anti-HER3 antibody MM-121/SAR256212 and cetuximab inhibits tumor growth in preclinical models of head and neck squamous cell carcinoma. Mol. Cancer Ther. 2014, 13, 1826–1836. [Google Scholar] [CrossRef] [Green Version]
- Vermorken, J.B.; Trigo, J.; Hitt, R.; Koralewski, P.; Diaz-Rubio, E.; Rolland, F.; Knecht, R.; Amellal, N.; Schueler, A.; Baselga, J. Open-label, uncontrolled, multicenter phase II study to evaluate the efficacy and toxicity of cetuximab as a single agent in patients with recurrent and/or metastatic squamous cell carcinoma of the head and neck who failed to respond to platinum-based therapy. J. Clin. Oncol. 2007, 25, 2171–2177. [Google Scholar] [CrossRef]
- Saxby, A.J.; Nielsen, A.; Scarlett, C.J.; Clarkson, A.; Morey, A.; Gill, A.; Smith, R.C. Assessment of HER-2 status in pancreatic adenocarcinoma: Correlation of immunohistochemistry, quantitative real-time RT-PCR, and FISH with aneuploidy and survival. Am. J. Surg. Pathol. 2005, 29, 1125–1134. [Google Scholar] [CrossRef]
- Larbouret, C.; Robert, B.; Navarro-Teulon, I.; Thèzenas, S.; Ladjemi, M.-Z.; Morisseau, S.; Campigna, E.; Bibeau, F.; Mach, J.-P.; Pèlegrin, A. In vivo therapeutic synergism of anti–epidermal growth factor receptor and anti-HER2 monoclonal antibodies against pancreatic carcinomas. Clin. Cancer Res. 2007, 13, 3356–3362. [Google Scholar] [CrossRef] [Green Version]
- Day, J.D.; Digiuseppe, J.A.; Yeo, C.; Lai-Goldman, M.; Anderson, S.M.; Goodman, S.N.; Kern, S.E.; Hruban, R.H. Immunohistochemical evaluation of HER-2/neu expression in pancreatic adenocarcinoma and pancreatic intraepithelial neoplasms. Hum. Pathol. 1996, 27, 119–124. [Google Scholar] [CrossRef]
- von Roemeling, C.; Jiang, W.; Chan, C.K.; Weissman, I.L.; Kim, B.Y. Breaking down the barriers to precision cancer nanomedicine. Trends Biotechnol. 2017, 35, 159–171. [Google Scholar] [CrossRef]
- Obaid, G.; Broekgaarden, M.; Bulin, A.-L.; Huang, H.-C.; Kuriakose, J.; Liu, J.; Hasan, T. Photonanomedicine: A convergence of photodynamic therapy and nanotechnology. Nanoscale 2016, 8, 12471–12503. [Google Scholar] [CrossRef]
- Lucky, S.S.; Soo, K.C.; Zhang, Y. Nanoparticles in photodynamic therapy. Chem. Rev. 2015, 115, 1990–2042. [Google Scholar] [CrossRef]
- Obaid, G.; Bano, S.; Mallidi, S.; Broekgaarden, M.; Kuriakose, J.; Silber, Z.; Bulin, A.L.; Wang, Y.; Mai, Z.; Jin, W.; et al. Impacting Pancreatic Cancer Therapy in Heterotypic in Vitro Organoids and in Vivo Tumors with Specificity-Tuned, NIR-Activable Photoimmunonanoconjugates: Towards Conquering Desmoplasia? Nano Lett. 2019. [Google Scholar] [CrossRef]
- Stefanick, J.F.; Omstead, D.T.; Kiziltepe, T.; Bilgicer, B. Dual-receptor targeted strategy in nanoparticle design achieves tumor cell selectivity through cooperativity. Nanoscale 2019, 11, 4414–4427. [Google Scholar] [CrossRef]
- Dixit, S.; Miller, K.; Zhu, Y.; McKinnon, E.; Novak, T.; Kenney, M.E.; Broome, A.-M. Dual Receptor-Targeted Theranostic Nanoparticles for Localized Delivery and Activation of Photodynamic Therapy Drug in Glioblastomas. Mol. Pharm. 2015, 12, 3250–3260. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharyya, S.; Khan, J.A.; Curran, G.L.; Robertson, J.D.; Bhattacharya, R.; Mukherjee, P. Efficient delivery of gold nanoparticles by dual receptor targeting. Adv. Mater. 2011, 23, 5034–5038. [Google Scholar] [CrossRef]
- Li, Q.; Li, W.; Di, H.; Luo, L.; Zhu, C.; Yang, J.; Yin, X.; Yin, H.; Gao, J.; Du, Y. A photosensitive liposome with NIR light triggered doxorubicin release as a combined photodynamic-chemo therapy system. J. Control. Release 2018, 277, 114–125. [Google Scholar] [CrossRef]
- Sneider, A.; Jadia, R.; Piel, B.; VanDyke, D.; Tsiros, C.; Rai, P. Engineering Remotely Triggered Liposomes to Target Triple Negative Breast Cancer. Oncomedicine 2017, 2, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Paszko, E.; Vaz, G.M.; Ehrhardt, C.; Senge, M.O. Transferrin conjugation does not increase the efficiency of liposomal Foscan during in vitro photodynamic therapy of oesophageal cancer. Eur. J. Pharm. Sci. 2013, 48, 202–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, P.; Yang, J.; Liu, D.; Huang, L.; Fell, G.; Huang, J.; Moses, M.A.; Auguste, D.T. Dual complementary liposomes inhibit triple-negative breast tumor progression and metastasis. Sci. Adv. 2019, 5, eaav5010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kontermann, R.E. Dual targeting strategies with bispecific antibodies. MAbs 2012, 4, 182–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.-Q.; Lv, Q.; Li, L.-M.; Tang, X.-J.; Li, F.-Z.; Hu, Y.-L.; Han, M. Glioma targeting and blood–brain barrier penetration by dual-targeting doxorubincin liposomes. Biomaterials 2013, 34, 5628–5639. [Google Scholar] [CrossRef]
- Eniola, A.O.; Hammer, D.A. In vitro characterization of leukocyte mimetic for targeting therapeutics to the endothelium using two receptors. Biomaterials 2005, 26, 7136–7144. [Google Scholar] [CrossRef]
- Saul, J.M.; Annapragada, A.V.; Bellamkonda, R.V. A dual-ligand approach for enhancing targeting selectivity of therapeutic nanocarriers. J. Control. Release 2006, 114, 277–287. [Google Scholar] [CrossRef]
- Vaidya, T.; Straubinger, R.M.; Ait-Oudhia, S. Development and evaluation of tri-functional immunoliposomes for the treatment of HER2 positive breast cancer. Pharm. Res. 2018, 35, 95. [Google Scholar] [CrossRef]
- Lörz, M.; Meyer-Breiting, E. Transferrin receptors in squamous epithelial cancers of the head and neck. Laryngo-Rhino-Otol. 1991, 70, 36–40. [Google Scholar] [CrossRef]
- Heitner, T.; Moor, A.; Garrison, J.L.; Marks, C.; Hasan, T.; Marks, J.D. Selection of cell binding and internalizing epidermal growth factor receptor antibodies from a phage display library. J. Immunol. Methods 2001, 248, 17–30. [Google Scholar] [CrossRef]
- Obaid, G.; Jin, W.; Bano, S.; Kessel, D.; Hasan, T. Nanolipid Formulations of Benzoporphyrin Derivative: Exploring the Dependence of Nanoconstruct Photophysics and Photochemistry on Their Therapeutic Index in Ovarian Cancer Cells. Photochem. Photobiol. 2019, 95, 364–377. [Google Scholar] [CrossRef]
- Bulin, A.L.; Broekgaarden, M.; Hasan, T. Comprehensive high-throughput image analysis for therapeutic efficacy of architecturally complex heterotypic organoids. Sci. Rep. 2017, 7, 16645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gadella, T., Jr.; Jovin, T.M. Oligomerization of epidermal growth factor receptors on A431 cells studied by time-resolved fluorescence imaging microscopy. A stereochemical model for tyrosine kinase receptor activation. J. Cell Biol. 1995, 129, 1543–1558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abe, K.; Zhao, L.; Periasamy, A.; Intes, X.; Barroso, M. Non-invasive in vivo imaging of near infrared-labeled transferrin in breast cancer cells and tumors using fluorescence lifetime FRET. PLoS ONE 2013, 8, e80269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shailaja, K.; Katayoun, A.J.; Sergei, M.; Jean Yu, W.; Nicole, M.E.; John, D.; Ling, Q.; Shannon, P.A.; Anthony, E.P.; Nilantha, S.; et al. A Role for Transferrin Receptor in Triggering Apoptosis When Targeted with Gambogic Acid. Proc. Natl. Acad. Sci. USA 2005, 102, 12095–12100. [Google Scholar]
- Bijman, M.N.; van Berkel, M.P.; Kok, M.; Janmaat, M.L.; Boven, E. Inhibition of functional HER family members increases the sensitivity to docetaxel in human ovarian cancer cell lines. Anti-Cancer Drugs 2009, 20, 450–460. [Google Scholar] [CrossRef]
- Tolmachev, V.; Tran, T.A.; Rosik, D.; Sjöberg, A.; Abrahmsén, L.; Orlova, A. Tumor targeting using affibody molecules: Interplay of affinity, target expression level, and binding site composition. J. Nucl. Med. 2012, 53, 953–960. [Google Scholar] [CrossRef] [Green Version]
- Abu-Yousif, A.O.; Moor, A.C.; Zheng, X.; Savellano, M.D.; Yu, W.; Selbo, P.K.; Hasan, T. Epidermal growth factor receptor-targeted photosensitizer selectively inhibits EGFR signaling and induces targeted phototoxicity in ovarian cancer cells. Cancer Lett. 2012, 321, 120–127. [Google Scholar] [CrossRef] [Green Version]
- Rutledge, E.A.; Green, F.A.; Enns, C.A. Generation of the soluble transferrin receptor requires cycling through an endosomal compartment. J. Biol. Chem. 1994, 269, 31864–31868. [Google Scholar]
- Beerli, R.R.; Graus-Porta, D.; Woods-Cook, K.; Chen, X.; Yarden, Y.; Hynes, N.E. Neu differentiation factor activation of ErbB-3 and ErbB-4 is cell specific and displays a differential requirement for ErbB-2. Mol. Cell. Biol. 1995, 15, 6496–6505. [Google Scholar] [CrossRef] [Green Version]
- Modjtahedi, H.; Komurasaki, T.; Toyoda, H.; Dean, C. Anti-EGFR monoclonal antibodies which act as EGF, TGFα, HB-EGF and BTC antagonists block the binding of epiregulin to EGFR-expressing tumours. Int. J. Cancer 1998, 75, 310–316. [Google Scholar] [CrossRef]
- Bjorkelund, H.; Gedda, L.; Barta, P.; Malmqvist, M.; Andersson, K. Gefitinib Induces Epidermal Growth Factor Receptor Dimers Which Alters the Interaction Characteristics with. sup. 125I-EGF. PLoS ONE 2011, 6, e24739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hopkins, C.R.; Trowbridge, I.S. Internalization and processing of transferrin and the transferrin receptor in human carcinoma A431 cells. J. Cell Biol. 1983, 97, 508–521. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.J.; Derynck, R.; Korc, M. Production of transforming growth factor alpha in human pancreatic cancer cells: Evidence for a superagonist autocrine cycle. Proc. Natl. Acad. Sci. USA 1987, 84, 7567–7570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizvi, I.; Obaid, G.; Bano, S.; Hasan, T.; Kessel, D. Photodynamic therapy: Promoting in vitro efficacy of photodynamic therapy by liposomal formulations of a photosensitizing agent. Lasers Surg. Med. 2018, 50, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Kessel, D.; Price, M. Evaluation of Diethyl-3-3′-(9,10-anthracenediyl) bis Acrylate as a Probe for Singlet Oxygen Formation during Photodynamic Therapy. Photochem. Photobiol. 2012, 88, 717–720. [Google Scholar] [CrossRef]
- Michaeli, A.; Feitelson, J. Reactivity of singlet oxygen toward amino acids and peptides. Photochem. Photobiol. 1994, 59, 284–289. [Google Scholar] [CrossRef]
- Bossi, P.; Resteghini, C.; Paielli, N.; Licitra, L.; Pilotti, S.; Perrone, F. Prognostic and predictive value of EGFR in head and neck squamous cell carcinoma. Oncotarget 2016, 7, 74362–74379. [Google Scholar] [CrossRef] [Green Version]
- Khademi, B.; Shirazi, F.M.; Vasei, M.; Doroudchi, M.; Gandomi, B.; Modjtahedi, H.; Pezeshki, A.M.; Ghaderi, A. The expression of p53, c-erbB-1 and c-erbB-2 molecules and their correlation with prognostic markers in patients with head and neck tumors. Cancer Lett. 2002, 184, 223–230. [Google Scholar] [CrossRef]
- Azemar, M.; Schmidt, M.; Arlt, F.; Kennel, P.; Brandt, B.; Papadimitriou, A.; Groner, B.; Wels, W. Recombinant antibody toxins specific for ErbB2 and EGF receptor inhibit the in vitro growth of human head and neck cancer cells and cause rapid tumor regression in vivo. Int. J. Cancer 2000, 86, 269–275. [Google Scholar] [CrossRef]
- Dancer, J.; Takei, H.; Ro, J.Y.; Lowery-Nordberg, M. Coexpression of EGFR and HER-2 in pancreatic ductal adenocarcinoma: A comparative study using immunohistochemistry correlated with gene amplification by fluorescencent in situ hybridization. Oncol. Rep. 2007, 18, 151–155. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Seung, E.; Xu, L.; Rao, E.; Lord, D.M.; Wei, R.R.; Cortez-Retamozo, V.; Ospina, B.; Posternak, V.; Ulinski, G. Trispecific antibodies enhance the therapeutic efficacy of tumor-directed T cells through T cell receptor co-stimulation. Nat. Cancer 2020, 1, 86–98. [Google Scholar] [CrossRef]
- Runcie, K.; Budman, D.R.; John, V.; Seetharamu, N. Bi-specific and tri-specific antibodies-the next big thing in solid tumor therapeutics. Mol. Med. 2018, 24, 50. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Shi, W.; Freund, L.B. Mechanics of receptor-mediated endocytosis. Proc. Natl. Acad. Sci. USA 2005, 102, 9469–9474. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nanoliposomes | Average Diameter (nm) | Polydispersity Index (PDI) | ζ-Potential (mV) | ** Ligand Density on PINs |
|---|---|---|---|---|
| Cet-PIN | 123.4 ± 0.2 | 0.05 ± 0.03 | −16.7 ± 0.55 | * 27.6 ± 1.6 |
| Cet-TZ-PIN | 144.1 ± 1.2 | 0.06 ± 0.01 | −17.4 ± 1.04 | 36.3 ± 3.5 |
| TR-PIN | 126.9 ± 1.7 | 0.06 ± 0.00 | −18.6 ± 1.01 | 89.6 ± 16.8 |
| Untargeted-PSN | 127.9 ± 1.3 | 0.08 ± 0.03 | −18.5 ± 0.95 | NA |
| Tumor Cell Lines | EGFR/Cell | TfR/Cell | HER-2/Cell |
|---|---|---|---|
| A431 | 2–4 × 106 [82] | 1.2 × 105 [92] | 1–2 × 105 [91] |
| T47D | 7.0 × 103 [89] | NA | 3 × 104 [89] |
| SKOV-3 | 6.3 × 104 | 5.6 × 105 | 1.6 × 106 [86] |
| MIA PaCa-2 | 1.7 × 105 [93] | 3.5 × 106 | 6.7 × 104 |
| SCC-9 | 1.8 × 105 | 3.2 × 106 | 0.7 × 105 |
| Tumor Cell Lines | Fold Improvement with Cet-PINs | Fold Improvement with TZ-PINs | Fold Improvement with Cet-TZ-PINs | Fold Improvement with HT-PINs | Fold Improvement with TR-PINs |
|---|---|---|---|---|---|
| A431 | 24 | 1.7 | 57.1 | 4.8 | 111 |
| T47D | 1.5 | 1.7 | 3.8 | 8 | 9.2 |
| SKOV-3 | 1.8 | 13.5 | 19.2 | 3.08 | 43.6 |
| MIA PaCa-2 | 23.2 | 2.05 | 29 | 1.7 | 41.1 |
| SCC-9 | 18.2 | 2.1 | 18 | 1.3 | 33 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bano, S.; Obaid, G.; Swain, J.W.R.; Yamada, M.; Pogue, B.W.; Wang, K.; Hasan, T. NIR Photodynamic Destruction of PDAC and HNSCC Nodules Using Triple-Receptor-Targeted Photoimmuno-Nanoconjugates: Targeting Heterogeneity in Cancer. J. Clin. Med. 2020, 9, 2390. https://doi.org/10.3390/jcm9082390
Bano S, Obaid G, Swain JWR, Yamada M, Pogue BW, Wang K, Hasan T. NIR Photodynamic Destruction of PDAC and HNSCC Nodules Using Triple-Receptor-Targeted Photoimmuno-Nanoconjugates: Targeting Heterogeneity in Cancer. Journal of Clinical Medicine. 2020; 9(8):2390. https://doi.org/10.3390/jcm9082390
Chicago/Turabian StyleBano, Shazia, Girgis Obaid, Joseph W. R. Swain, Marina Yamada, Brian W. Pogue, Kenneth Wang, and Tayyaba Hasan. 2020. "NIR Photodynamic Destruction of PDAC and HNSCC Nodules Using Triple-Receptor-Targeted Photoimmuno-Nanoconjugates: Targeting Heterogeneity in Cancer" Journal of Clinical Medicine 9, no. 8: 2390. https://doi.org/10.3390/jcm9082390
APA StyleBano, S., Obaid, G., Swain, J. W. R., Yamada, M., Pogue, B. W., Wang, K., & Hasan, T. (2020). NIR Photodynamic Destruction of PDAC and HNSCC Nodules Using Triple-Receptor-Targeted Photoimmuno-Nanoconjugates: Targeting Heterogeneity in Cancer. Journal of Clinical Medicine, 9(8), 2390. https://doi.org/10.3390/jcm9082390