Platelet δ-Storage Pool Disease: An Update


Abstract
:1. Introduction
2. Platelet Dense-Granules: Origin, Structure, and Function
3. δ-Storage Pool Disorders
4. Tools for the Diagnosis of δ-SPD
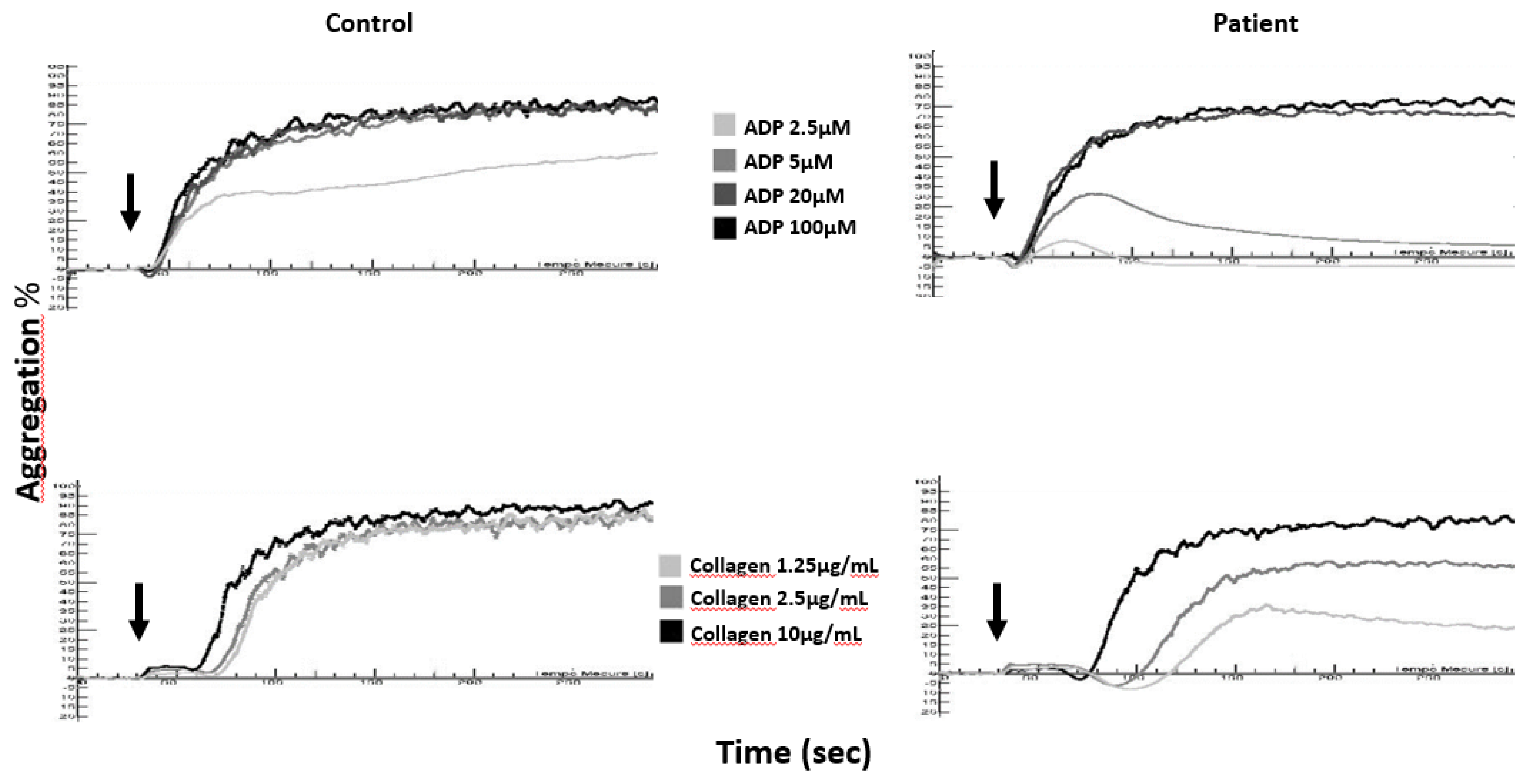
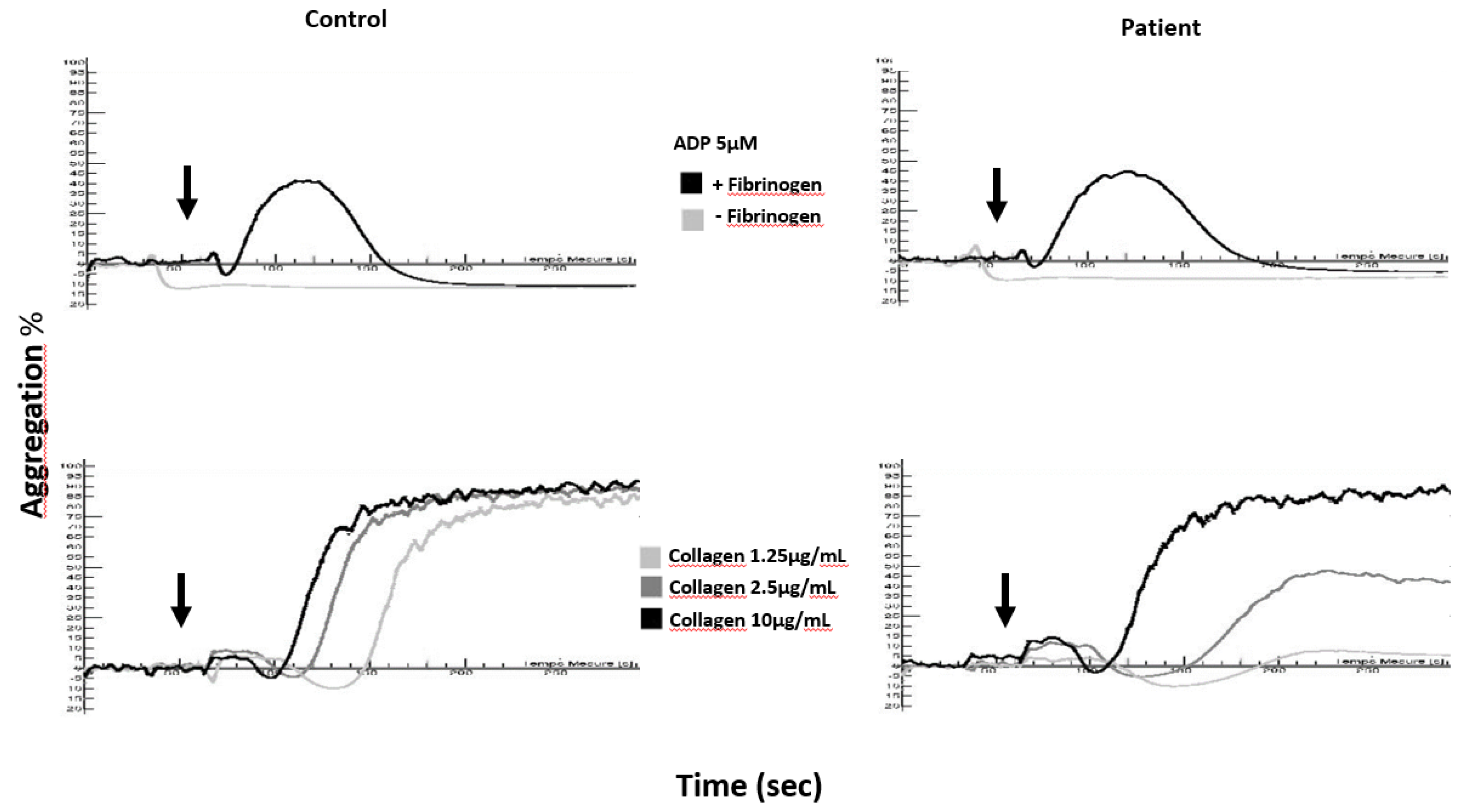
4.1. Aggregation Tests
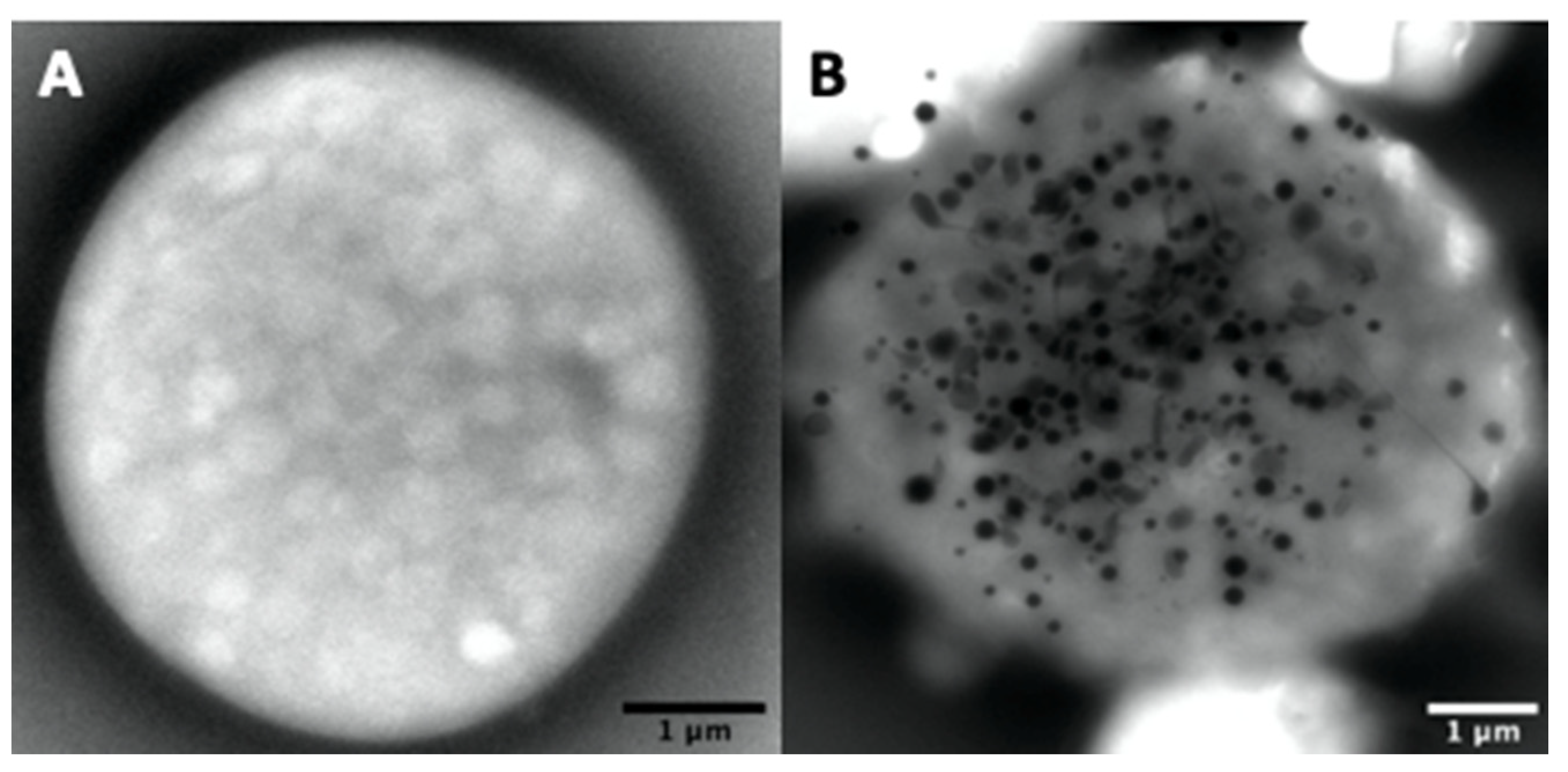
4.2. Ultrastructural Evaluation of the Dense Granules Using a Whole Platelet Mount
4.2.1. Sample Preparation
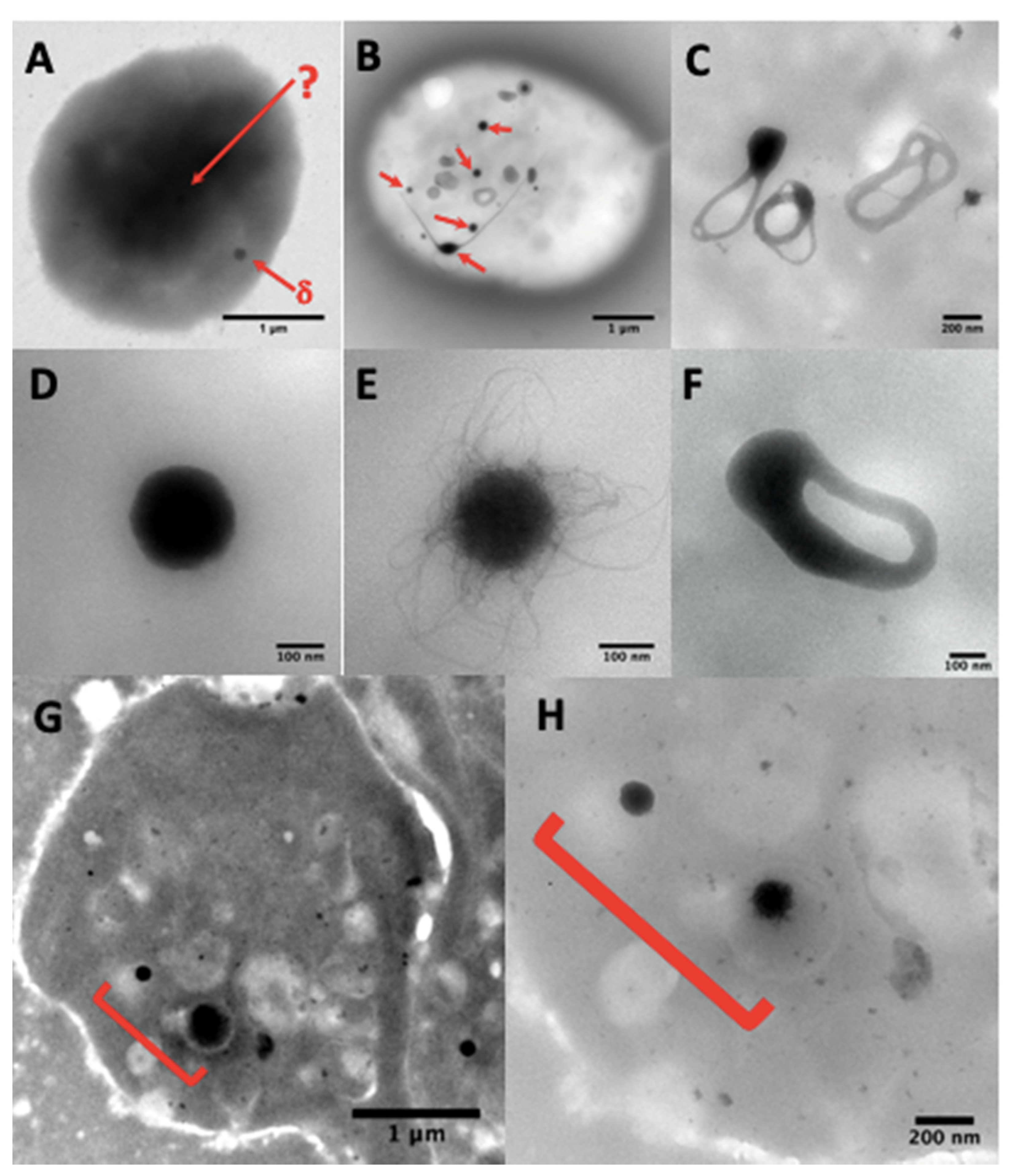
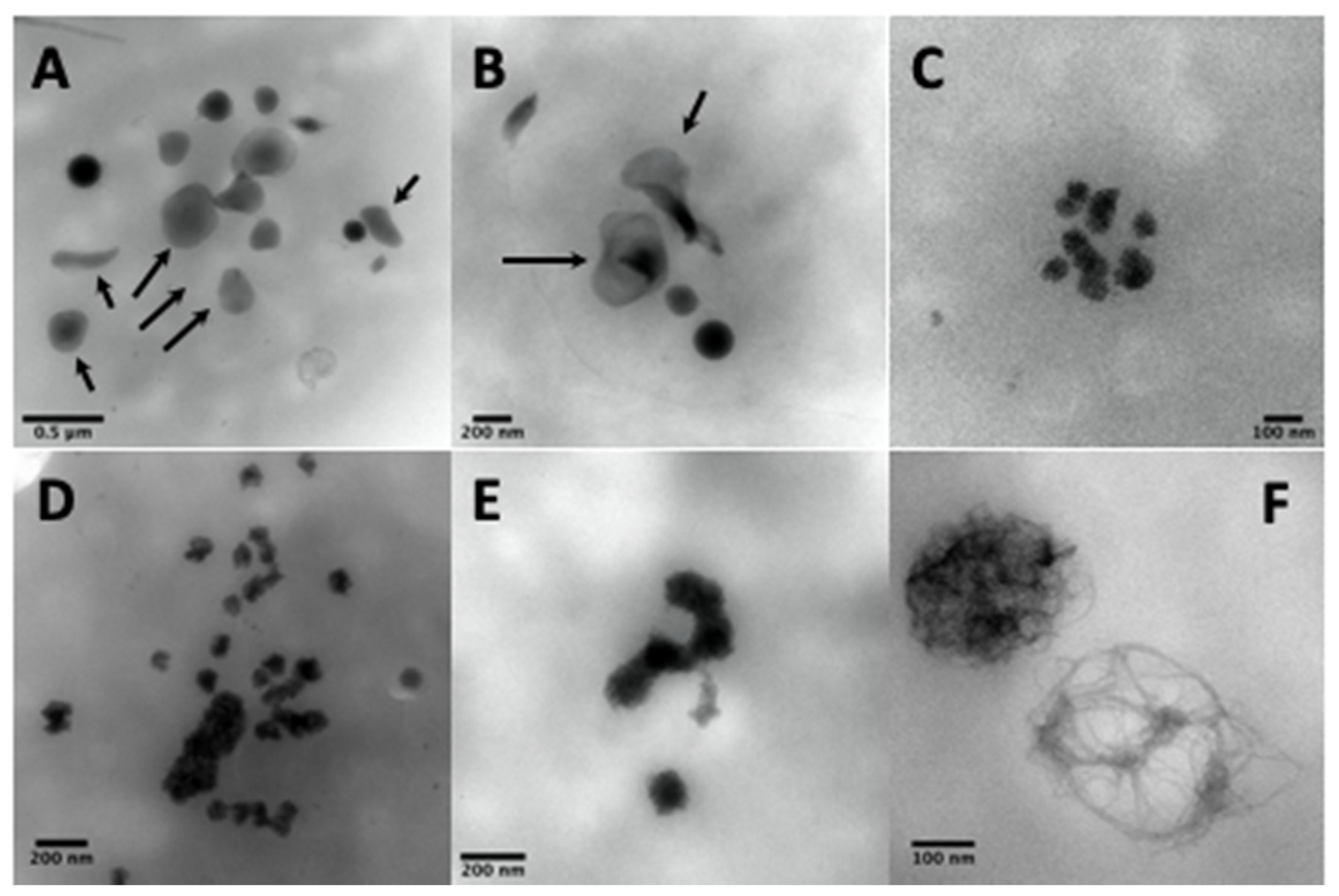
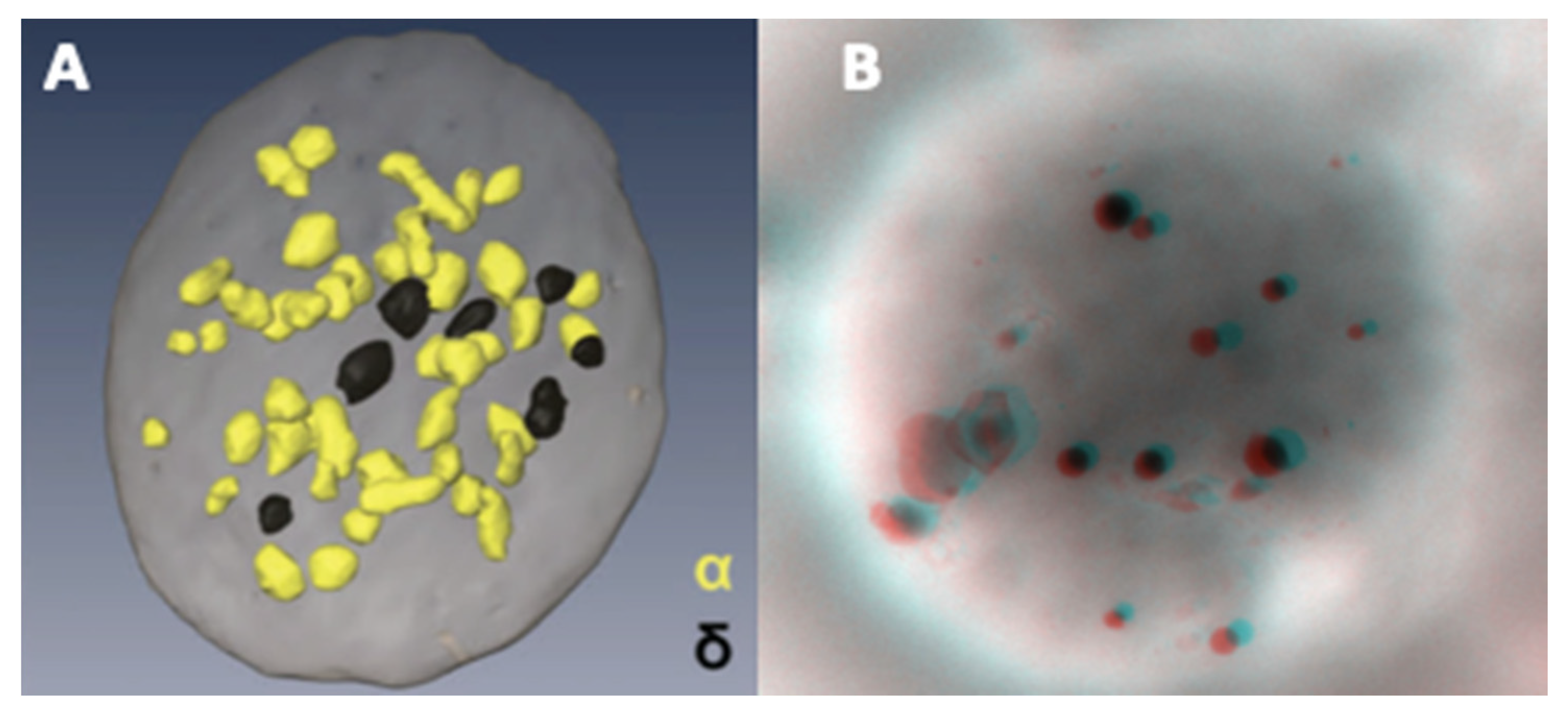
4.2.2. Ultrastructural Examination of Dense-Granules
4.2.3. Counting of Dense-Granules
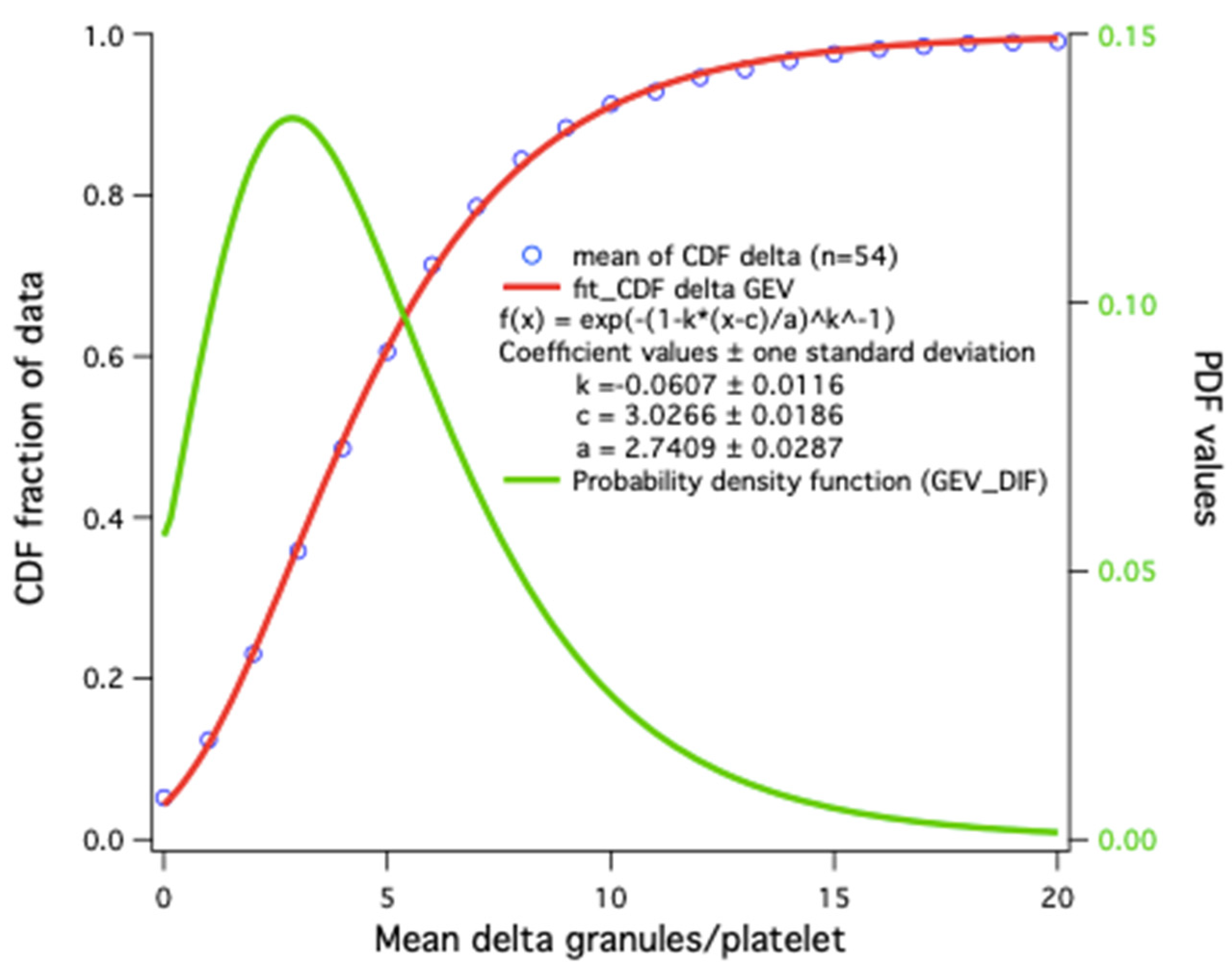
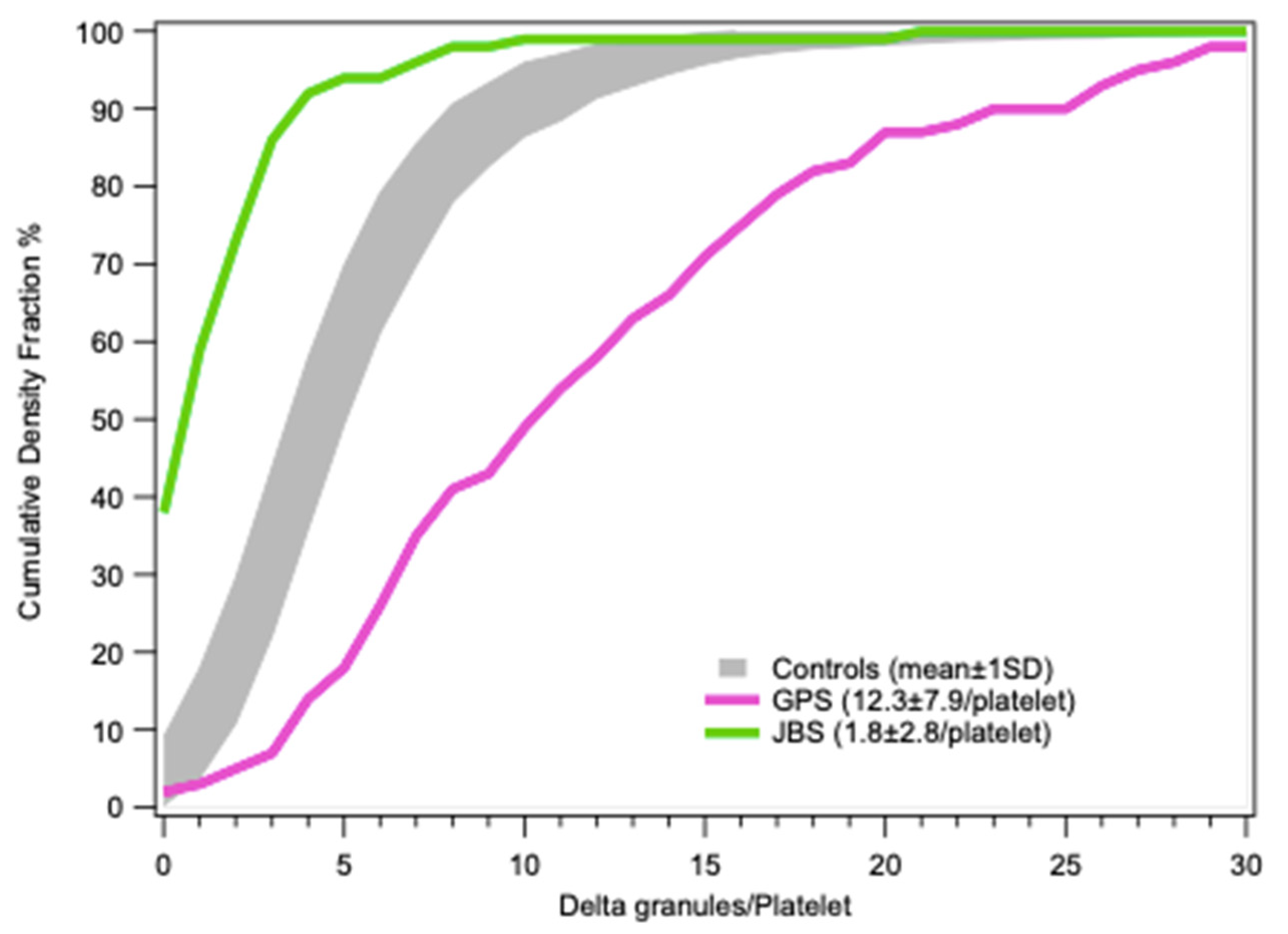
4.2.4. Distribution Statistics
4.3. Other Studies of Granular Content
4.3.1. ATP and ADP Contents
4.3.2. Serotonin Content
4.3.3. Fluorescence Microscopy
4.4. Secretion Tests
4.4.1. Uptake and Release of Radio-Labeled Serotonin
4.4.2. Chemical or Biochemical Measurement of Serotonin Release
4.4.3. Mepacrine Flow Cytometry
4.4.4. Polyphosphate Release
4.4.5. Flow Cytometry of CD63 Exposure
4.4.6. Lumi-Aggregometry
4.5. Genetic Studies
5. Diagnosis Strategy and Clinical Management of δ-SPD Patients
5.1. Diagnosis Strategy of δ-SPD Patients
5.2. Bleeding Risk and Clinical Management of δ-SPD Patients
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Versteeg, H.H.; Heemskerk, J.W.; Levi, M.; Reitsma, P.H. New fundamentals in hemostasis. Physiol. Rev. 2013, 93, 327–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharda, A.; Flaumenhaft, R. The life cycle of platelet granules. F1000Res 2018, 7, 236. [Google Scholar] [CrossRef] [PubMed]
- Gachet, C. Regulation of platelet functions by P2 receptors. Annu. Rev. Pharmacol. Toxicol. 2006, 46, 277–300. [Google Scholar] [CrossRef]
- Masliah-Planchon, J.; Darnige, L.; Bellucci, S. Molecular determinants of platelet delta storage pool deficiencies: An update. Br. J. Haematol. 2013, 160, 5–11. [Google Scholar] [CrossRef]
- Flaumenhaft, R. Platelet Secretion—Chapter 18. In Platelets, 3rd ed.; Michelson, A.D., Ed.; Academic Press: Cambridge, MA, USA, 2013; pp. 343–366. [Google Scholar] [CrossRef]
- Starcevic, M.; Dell’Angelica, E.C. Identification of snapin and three novel proteins (BLOS1, BLOS2, and BLOS3/reduced pigmentation) as subunits of biogenesis of lysosome-related organelles complex-1 (BLOC-1). J. Biol. Chem. 2004, 279, 28393–28401. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.; Kuo, Y.M.; Gitschier, J. The pallid gene encodes a novel, syntaxin 13-interacting protein involved in platelet storage pool deficiency. Nat. Genet. 1999, 23, 329–332. [Google Scholar] [CrossRef]
- Bultema, J.J.; Ambrosio, A.L.; Burek, C.L.; Di Pietro, S.M. BLOC-2, AP-3, and AP-1 proteins function in concert with Rab38 and Rab32 proteins to mediate protein trafficking to lysosome-related organelles. J. Biol. Chem. 2012, 287, 19550–19563. [Google Scholar] [CrossRef] [Green Version]
- Gerondopoulos, A.; Langemeyer, L.; Liang, J.R.; Linford, A.; Barr, F.A. BLOC-3 mutated in Hermansky-Pudlak syndrome is a Rab32/38 guanine nucleotide exchange factor. Curr. Biol. CB 2012, 22, 2135–2139. [Google Scholar] [CrossRef] [Green Version]
- Jedlitschky, G.; Greinacher, A.; Kroemer, H.K. Transporters in human platelets: Physiologic function and impact for pharmacotherapy. Blood 2012, 119, 3394–3402. [Google Scholar] [CrossRef] [Green Version]
- Ambrosio, A.L.; Boyle, J.A.; Di Pietro, S.M. Mechanism of platelet dense granule biogenesis: Study of cargo transport and function of Rab32 and Rab38 in a model system. Blood 2012, 120, 4072–4081. [Google Scholar] [CrossRef] [Green Version]
- Peden, A.A.; Oorschot, V.; Hesser, B.A.; Austin, C.D.; Scheller, R.H.; Klumperman, J. Localization of the AP-3 adaptor complex defines a novel endosomal exit site for lysosomal membrane proteins. J. Cell Biol. 2004, 164, 1065–1076. [Google Scholar] [CrossRef]
- Di Pietro, S.M.; Falcon-Perez, J.M.; Tenza, D.; Setty, S.R.; Marks, M.S.; Raposo, G.; Dell’Angelica, E.C. BLOC-1 interacts with BLOC-2 and the AP-3 complex to facilitate protein trafficking on endosomes. Mol. Biol. Cell 2006, 17, 4027–4038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carneiro, A.M.; Cook, E.H.; Murphy, D.L.; Blakely, R.D. Interactions between integrin alphaIIbbeta3 and the serotonin transporter regulate serotonin transport and platelet aggregation in mice and humans. J. Clin. Investig. 2008, 118, 1544–1552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Decouture, B.; Dreano, E.; Belleville-Rolland, T.; Kuci, O.; Dizier, B.; Bazaa, A.; Coqueran, B.; Lompre, A.M.; Denis, C.V.; Hulot, J.S.; et al. Impaired platelet activation and cAMP homeostasis in MRP4-deficient mice. Blood 2015, 126, 1823–1830. [Google Scholar] [CrossRef] [Green Version]
- Holmsen, H.; Weiss, H.J. Further evidence for a deficient storage pool of adenine nucleotides in platelets from some patients with thrombocytopathia—“Storage pool disease”. Blood 1972, 39, 197–209. [Google Scholar] [CrossRef] [Green Version]
- Holmsen, H.; Dangelmaier, C.A. Measurement of secretion of adenine nucleotides. Methods Enzym. 1989, 169, 195–205. [Google Scholar] [CrossRef]
- Morrissey, J.H. Polyphosphate: A link between platelets, coagulation and inflammation. Int. J. Hematol. 2012, 95, 346–352. [Google Scholar] [CrossRef] [Green Version]
- Smith, S.A.; Morrissey, J.H. Polyphosphate enhances fibrin clot structure. Blood 2008, 112, 2810–2816. [Google Scholar] [CrossRef] [Green Version]
- Golebiewska, E.M.; Harper, M.T.; Williams, C.M.; Savage, J.S.; Goggs, R.; Fischer von Mollard, G.; Poole, A.W. Syntaxin 8 regulates platelet dense granule secretion, aggregation, and thrombus stability. J. Biol. Chem. 2015, 290, 1536–1545. [Google Scholar] [CrossRef] [Green Version]
- Bolton-Maggs, P.H.; Chalmers, E.A.; Collins, P.W.; Harrison, P.; Kitchen, S.; Liesner, R.J.; Minford, A.; Mumford, A.D.; Parapia, L.A.; Perry, D.J.; et al. A review of inherited platelet disorders with guidelines for their management on behalf of the UKHCDO. Br. J. Haematol. 2006, 135, 603–633. [Google Scholar] [CrossRef]
- Barbosa, M.D.; Nguyen, Q.A.; Tchernev, V.T.; Ashley, J.A.; Detter, J.C.; Blaydes, S.M.; Brandt, S.J.; Chotai, D.; Hodgman, C.; Solari, R.C.; et al. Identification of the homologous beige and Chediak-Higashi syndrome genes. Nature 1996, 382, 262–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sepulveda, F.E.; Burgess, A.; Heiligenstein, X.; Goudin, N.; Menager, M.M.; Romao, M.; Cote, M.; Mahlaoui, N.; Fischer, A.; Raposo, G.; et al. LYST controls the biogenesis of the endosomal compartment required for secretory lysosome function. Traffic 2015, 16, 191–203. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Zhang, L.; Wang, S.; Chen, F.; Gu, Y.; Hong, E.; Yu, Y.; Ni, X.; Guo, Y.; Shi, T.; et al. Whole Genome Sequencing Identifies Novel Compound Heterozygous Lysosomal Trafficking Regulator Gene Mutations Associated with Autosomal Recessive Chediak-Higashi Syndrome. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martina, J.A.; Moriyama, K.; Bonifacino, J.S. BLOC-3, a protein complex containing the Hermansky-Pudlak syndrome gene products HPS1 and HPS4. J. Biol. Chem. 2003, 278, 29376–29384. [Google Scholar] [CrossRef] [Green Version]
- Feng, L.; Seymour, A.B.; Jiang, S.; To, A.; Peden, A.A.; Novak, E.K.; Zhen, L.; Rusiniak, M.E.; Eicher, E.M.; Robinson, M.S.; et al. The beta3A subunit gene (Ap3b1) of the AP-3 adaptor complex is altered in the mouse hypopigmentation mutant pearl, a model for Hermansky-Pudlak syndrome and night blindness. Hum. Mol. Genet. 1999, 8, 323–330. [Google Scholar] [CrossRef] [Green Version]
- Di Pietro, S.M.; Falcon-Perez, J.M.; Dell’Angelica, E.C. Characterization of BLOC-2, a complex containing the Hermansky-Pudlak syndrome proteins HPS3, HPS5 and HPS6. Traffic 2004, 5, 276–283. [Google Scholar] [CrossRef]
- Li, W.; Zhang, Q.; Oiso, N.; Novak, E.K.; Gautam, R.; O’Brien, E.P.; Tinsley, C.L.; Blake, D.J.; Spritz, R.A.; Copeland, N.G.; et al. Hermansky-Pudlak syndrome type 7 (HPS-7) results from mutant dysbindin, a member of the biogenesis of lysosome-related organelles complex 1 (BLOC-1). Nat. Genet. 2003, 35, 84–89. [Google Scholar] [CrossRef] [Green Version]
- Morgan, N.V.; Pasha, S.; Johnson, C.A.; Ainsworth, J.R.; Eady, R.A.; Dawood, B.; McKeown, C.; Trembath, R.C.; Wilde, J.; Watson, S.P.; et al. A germline mutation in BLOC1S3/reduced pigmentation causes a novel variant of Hermansky-Pudlak syndrome (HPS8). Am. J. Hum. Genet. 2006, 78, 160–166. [Google Scholar] [CrossRef] [Green Version]
- Pennamen, P.; Le, L.; Tingaud-Sequeira, A.; Fiore, M.; Bauters, A.; Van Duong Beatrice, N.; Coste, V.; Bordet, J.C.; Plaisant, C.; Diallo, M.; et al. BLOC1S5 pathogenic variants cause a new type of Hermansky-Pudlak syndrome. Genet. Med. Off. J. Am. Coll. Med. Genet. 2020, 1–10. [Google Scholar] [CrossRef]
- Cullinane, A.R.; Curry, J.A.; Carmona-Rivera, C.; Summers, C.G.; Ciccone, C.; Cardillo, N.D.; Dorward, H.; Hess, R.A.; White, J.G.; Adams, D.; et al. A BLOC-1 mutation screen reveals that PLDN is mutated in Hermansky-Pudlak Syndrome type 9. Am. J. Hum. Genet. 2011, 88, 778–787. [Google Scholar] [CrossRef] [Green Version]
- Ammann, S.; Schulz, A.; Krageloh-Mann, I.; Dieckmann, N.M.; Niethammer, K.; Fuchs, S.; Eckl, K.M.; Plank, R.; Werner, R.; Altmuller, J.; et al. Mutations in AP3D1 associated with immunodeficiency and seizures define a new type of Hermansky-Pudlak syndrome. Blood 2016, 127, 997–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohammed, M.; Al-Hashmi, N.; Al-Rashdi, S.; Al-Sukaiti, N.; Al-Adawi, K.; Al-Riyami, M.; Al-Maawali, A. Biallelic mutations in AP3D1 cause Hermansky-Pudlak syndrome type 10 associated with immunodeficiency and seizure disorder. Eur. J. Med. Genet. 2019, 62, 103583. [Google Scholar] [CrossRef] [PubMed]
- Huizing, M.; Malicdan, M.C.V.; Wang, J.A.; Pri-Chen, H.; Hess, R.A.; Fischer, R.; O’Brien, K.J.; Merideth, M.A.; Gahl, W.A.; Gochuico, B.R. Hermansky-Pudlak syndrome: Mutation update. Hum. Mutat. 2020, 41, 543–580. [Google Scholar] [CrossRef] [PubMed]
- Griscelli, C.; Durandy, A.; Guy-Grand, D.; Daguillard, F.; Herzog, C.; Prunieras, M. A syndrome associating partial albinism and immunodeficiency. Am. J. Med. 1978, 65, 691–702. [Google Scholar] [CrossRef]
- Westbroek, W.; Tuchman, M.; Tinloy, B.; De Wever, O.; Vilboux, T.; Hertz, J.M.; Hasle, H.; Heilmann, C.; Helip-Wooley, A.; Kleta, R.; et al. A novel missense mutation (G43S) in the switch I region of Rab27A causing Griscelli syndrome. Mol. Genet. Metab. 2008, 94, 248–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, J.G.; Keel, S.; Reyes, M.; Burris, S.M. Alpha-delta platelet storage pool deficiency in three generations. Platelets 2007, 18, 1–10. [Google Scholar] [CrossRef]
- Ferreira, C.R.; Chen, D.; Abraham, S.M.; Adams, D.R.; Simon, K.L.; Malicdan, M.C.; Markello, T.C.; Gunay-Aygun, M.; Gahl, W.A. Combined alpha-delta platelet storage pool deficiency is associated with mutations in GFI1B. Mol. Genet. Metab. 2017, 120, 288–294. [Google Scholar] [CrossRef] [Green Version]
- Jedlitschky, G.; Cattaneo, M.; Lubenow, L.E.; Rosskopf, D.; Lecchi, A.; Artoni, A.; Motta, G.; Niessen, J.; Kroemer, H.K.; Greinacher, A. Role of MRP4 (ABCC4) in platelet adenine nucleotide-storage: Evidence from patients with delta-storage pool deficiencies. Am. J. Pathol. 2010, 176, 1097–1103. [Google Scholar] [CrossRef]
- Mumford, A.D.; Frelinger, A.L.; Gachet, C.; Gresele, P.; Noris, P.; Harrison, P.; Mezzano, D. A review of platelet secretion assays for the diagnosis of inherited platelet secretion disorders. Thromb. Haemost. 2015, 114, 14–25. [Google Scholar] [CrossRef]
- Quiroga, T.; Goycoolea, M.; Panes, O.; Aranda, E.; Martinez, C.; Belmont, S.; Munoz, B.; Zuniga, P.; Pereira, J.; Mezzano, D. High prevalence of bleeders of unknown cause among patients with inherited mucocutaneous bleeding. A prospective study of 280 patients and 299 controls. Haematologica 2007, 92, 357–365. [Google Scholar] [CrossRef] [Green Version]
- Mezzano, D.; Quiroga, T.; Pereira, J. The level of laboratory testing required for diagnosis or exclusion of a platelet function disorder using platelet aggregation and secretion assays. Semin. Thromb. Hemost. 2009, 35, 242–254. [Google Scholar] [CrossRef] [PubMed]
- Fiore, M.; Garcia, C.; Sié, P.; Favier, R.; Lavenu-Bombled, C.; Hurtaud, M.F.; Gachet, C.; Alessi, M.C.; Dupuis, A. Déficit en granules denses plaquettaires: Une cause sous-estimée de saignements inexpliqués. Hématologie 2017, 23, 243–254. [Google Scholar] [CrossRef]
- Gresele, P.; Harrison, P.; Bury, L.; Falcinelli, E.; Gachet, C.; Hayward, C.P.; Kenny, D.; Mezzano, D.; Mumford, A.D.; Nugent, D.; et al. Diagnosis of suspected inherited platelet function disorders: Results of a worldwide survey. J. Thromb. Haemost. 2014, 12, 1562–1569. [Google Scholar] [CrossRef] [PubMed]
- Kerenyi, A.; Schlammadinger, A.; Ajzner, E.; Szegedi, I.; Kiss, C.; Pap, Z.; Boda, Z.; Muszbek, L. Comparison of PFA-100 closure time and template bleeding time of patients with inherited disorders causing defective platelet function. Thromb. Res. 1999, 96, 487–492. [Google Scholar] [CrossRef]
- Hechler, B.; Dupuis, A.; Mangin, P.H.; Gachet, C. Platelet preparation for function testing in the laboratory and clinic: Historical and practical aspects. Res. Pract. Thromb. Haemost. 2019, 3, 615–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieuwenhuis, H.K.; Akkerman, J.W.; Sixma, J.J. Patients with a prolonged bleeding time and normal aggregation tests may have storage pool deficiency: Studies on one hundred six patients. Blood 1987, 70, 620–623. [Google Scholar] [CrossRef] [Green Version]
- Ingerman, C.M.; Smith, J.B.; Shapiro, S.; Sedar, A.; Silver, M.J. Hereditary abnormality of platelet aggregation attributable to nucleotide storage pool deficiency. Blood 1978, 52, 332–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cattaneo, M.; Cerletti, C.; Harrison, P.; Hayward, C.P.; Kenny, D.; Nugent, D.; Nurden, P.; Rao, A.K.; Schmaier, A.H.; Watson, S.P.; et al. Recommendations for the Standardization of Light Transmission Aggregometry: A Consensus of the Working Party from the Platelet Physiology Subcommittee of SSC/ISTH. J. Thromb. Haemost. 2013, 11, 1183–1189. [Google Scholar] [CrossRef] [PubMed]
- Lanza, F.; Beretz, A.; Stierle, A.; Hanau, D.; Kubina, M.; Cazenave, J.P. Epinephrine potentiates human platelet activation but is not an aggregating agent. Am. J. Physiol. 1988, 255, H1276–H1288. [Google Scholar] [CrossRef]
- Kinlough-Rathbone, R.L.; Mustard, J.F.; Packham, M.A.; Perry, D.W.; Reimers, H.J.; Cazenave, J.P. Properties of washed human platelets. Thromb. Haemost. 1977, 37, 291–308. [Google Scholar] [CrossRef]
- Cazenave, J.P.; Ohlmann, P.; Cassel, D.; Eckly, A.; Hechler, B.; Gachet, C. Preparation of washed platelet suspensions from human and rodent blood. Methods Mol. Biol. 2004, 272, 13–28. [Google Scholar] [CrossRef] [PubMed]
- Bull, B.S. The ultrastructure of negatively stained platelets. Some physiologic implications. Blood 1966, 28, 901–912. [Google Scholar] [CrossRef] [PubMed]
- White, J.G. The dense bodies of human platelets: Inherent electron opacity of the serotonin storage particles. Blood 1969, 33, 598–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, J.H.; Carson, F.L.; Race, G.J. Calcium-containing platelet granules. J. Cell Biol. 1974, 60, 775–777. [Google Scholar] [CrossRef]
- Skaer, R.J.; Peters, P.D.; Emmines, J.P. The localization of calcium and phosphorus in human platelets. J. Cell Sci. 1974, 15, 679–682. [Google Scholar]
- Born, G.V.; Ingram, G.I.; Stacey, R.S. The relationship between 5-hydroxytryptamine and adenosine triphosphate in blood platelets. Br. J. Pharmacol. Chemother. 1958, 13, 62–64. [Google Scholar] [CrossRef] [Green Version]
- Fukami, M.H.; Dangelmaier, C.A.; Bauer, J.S.; Holmsen, H. Secretion, subcellular localization and metabolic status of inorganic pyrophosphate in human platelets. A major constituent of the amine-storing granules. Biochem. J. 1980, 192, 99–105. [Google Scholar] [CrossRef] [Green Version]
- Ruiz, F.A.; Lea, C.R.; Oldfield, E.; Docampo, R. Human platelet dense granules contain polyphosphate and are similar to acidocalcisomes of bacteria and unicellular eukaryotes. J. Biol. Chem. 2004, 279, 44250–44257. [Google Scholar] [CrossRef] [Green Version]
- Donovan, A.J.; Kalkowski, J.; Smith, S.A.; Morrissey, J.H.; Liu, Y. Size-controlled synthesis of granular polyphosphate nanoparticles at physiologic salt concentrations for blood clotting. Biomacromolecules 2014, 15, 3976–3984. [Google Scholar] [CrossRef]
- Kornberg, A. Inorganic polyphosphate: Toward making a forgotten polymer unforgettable. J. Bacteriol. 1995, 177, 491–496. [Google Scholar] [CrossRef] [Green Version]
- Mailer, R.K.W.; Hanel, L.; Allende, M.; Renne, T. Polyphosphate as a Target for Interference With Inflammation and Thrombosis. Front. Med. (Lausanne) 2019, 6, 76. [Google Scholar] [CrossRef] [Green Version]
- Baker, C.J.; Smith, S.A.; Morrissey, J.H. Polyphosphate in thrombosis, hemostasis, and inflammation. Res. Pract. Thromb. Haemost. 2019, 3, 18–25. [Google Scholar] [CrossRef]
- Verhoef, J.J.; Barendrecht, A.D.; Nickel, K.F.; Dijkxhoorn, K.; Kenne, E.; Labberton, L.; McCarty, O.J.; Schiffelers, R.; Heijnen, H.F.; Hendrickx, A.P.; et al. Polyphosphate nanoparticles on the platelet surface trigger contact system activation. Blood 2017, 129, 1707–1717. [Google Scholar] [CrossRef]
- White, J.G. Electron opaque structures in human platelets: Which are or are not dense bodies? Platelets 2008, 19, 455–466. [Google Scholar] [CrossRef] [PubMed]
- Brunet, J.G.; Iyer, J.K.; Badin, M.S.; Graf, L.; Moffat, K.A.; Timleck, M.; Spitzer, E.; Hayward, C.P.M. Electron microscopy examination of platelet whole mount preparations to quantitate platelet dense granule numbers: Implications for diagnosing suspected platelet function disorders due to dense granule deficiency. Int. J. Lab. Hematol. 2018, 40, 400–407. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Uhl, C.B.; Bryant, S.C.; Krumwiede, M.; Barness, R.L.; Olson, M.C.; Gossman, S.C.; Erdogan Damgard, S.; Gamb, S.I.; Cummins, L.A.; et al. Diagnostic laboratory standardization and validation of platelet transmission electron microscopy. Platelets 2018, 29, 574–582. [Google Scholar] [CrossRef] [PubMed]
- Weiss, H.J.; Lages, B.; Vicic, W.; Tsung, L.Y.; White, J.G. Heterogeneous abnormalities of platelet dense granule ultrastructure in 20 patients with congenital storage pool deficiency. Br. J. Haematol. 1993, 83, 282–295. [Google Scholar] [CrossRef]
- Gunning, W.T.; Raghavan, M.; Calomeni, E.P.; Turner, J.N.; Roysam, B.; Roysam, S.; Smith, M.R.; Kouides, P.A.; Lachant, N.A. A Morphometric Analysis of Platelet Dense Granules of Patients with Unexplained Bleeding: A New Entity of Delta-Microgranular Storage Pool Deficiency. J. Clin. Med. 2020, 9, 1734. [Google Scholar] [CrossRef] [PubMed]
- Markello, T.; Chen, D.; Kwan, J.Y.; Horkayne-Szakaly, I.; Morrison, A.; Simakova, O.; Maric, I.; Lozier, J.; Cullinane, A.R.; Kilo, T.; et al. York platelet syndrome is a CRAC channelopathy due to gain-of-function mutations in STIM1. Mol. Genet. Metab. 2015, 114, 474–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunning, W.T.; Calomeni, E.P. A brief review of transmission electron microscopy and applications in pathology. J. Histotechnol. 2000, 23, 237–246. [Google Scholar] [CrossRef]
- Sorokin, V.; Alkhoury, R.; Al-Rawabdeh, S.; Houston, R.H.; Thornton, D.; Kerlin, B.; O’Brien, S.; Baker, P.; Boesel, C.; Uddin, M.; et al. Reference Range of Platelet Delta Granules in the Pediatric Age Group: An Ultrastructural Study of Platelet Whole Mount Preparations from Healthy Volunteers. Pediatr. Dev. Pathol. 2016, 19, 498–501. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Stone, R.L.; Kaelber, J.T.; Rochat, R.H.; Nick, A.M.; Vijayan, K.V.; Afshar-Kharghan, V.; Schmid, M.F.; Dong, J.F.; Sood, A.K.; et al. Electron cryotomography reveals ultrastructure alterations in platelets from patients with ovarian cancer. Proc. Natl. Acad. Sci. USA 2015, 112, 14266–14271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eckly, A.; Rinckel, J.Y.; Proamer, F.; Ulas, N.; Joshi, S.; Whiteheart, S.W.; Gachet, C. Respective contributions of single and compound granule fusion to secretion by activated platelets. Blood 2016, 128, 2538–2549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pokrovskaya, I.D.; Yadav, S.; Rao, A.; McBride, E.; Kamykowski, J.A.; Zhang, G.; Aronova, M.A.; Leapman, R.D.; Storrie, B. 3D ultrastructural analysis of alpha-granule, dense granule, mitochondria, and canalicular system arrangement in resting human platelets. Res. Pract. Thromb. Haemost. 2020, 4, 72–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saultier, P.; Vidal, L.; Canault, M.; Bernot, D.; Falaise, C.; Pouymayou, C.; Bordet, J.C.; Saut, N.; Rostan, A.; Baccini, V.; et al. Macrothrombocytopenia and dense granule deficiency associated with FLI1 variants: Ultrastructural and pathogenic features. Haematologica 2017, 102, 1006–1016. [Google Scholar] [CrossRef]
- Asher, L.; Hata, J. Platelet Electron Microscopy: Utilizing LEAN Methodology to Optimize Laboratory Workflow. Pediatr. Dev. Pathol. 2020. [Google Scholar] [CrossRef]
- Marneth, A.E.; van Heerde, W.L.; Hebeda, K.M.; Laros-van Gorkom, B.A.; Barteling, W.; Willemsen, B.; de Graaf, A.O.; Simons, A.; Jansen, J.H.; Preijers, F.; et al. Platelet CD34 expression and alpha/delta-granule abnormalities in GFI1B- and RUNX1-related familial bleeding disorders. Blood 2017, 129, 1733–1736. [Google Scholar] [CrossRef] [Green Version]
- Urban, D.; Pluthero, F.G.; Christensen, H.; Baidya, S.; Rand, M.L.; Das, A.; Shah, P.S.; Chitayat, D.; Blanchette, V.S.; Kahr, W.H. Decreased numbers of dense granules in fetal and neonatal platelets. Haematologica 2017, 102, e36–e38. [Google Scholar] [CrossRef] [Green Version]
- Nessle, C.N.; Ghosal, S.; Mathews, C.; Taylor, D.; Myers, J.; Raj, A.; Panigrahi, A. Weak correlation of bleeding scores to platelet electron microscopy: A retrospective chart review of pediatric patients with delta-storage pool disorder. Pediatr. Blood Cancer 2019, 66, e27505. [Google Scholar] [CrossRef]
- Guicheney, P. Human platelet serotonin content: Methodological aspects and physiological variations. Methods Find. Exp. Clin. Pharmacol. 1988, 10, 253–258. [Google Scholar]
- Flachaire, E.; Beney, C.; Berthier, A.; Salandre, J.; Quincy, C.; Renaud, B. Determination of reference values for serotonin concentration in platelets of healthy newborns, children, adults, and elderly subjects by HPLC with electrochemical detection. Clin. Chem. 1990, 36, 2117–2120. [Google Scholar] [CrossRef]
- Gerrard, J.M.; Rao, G.H.; White, J.G. The influence of reserpine and ethylenediaminetetraacetic acid (EDTA) on serotonin storage organelles of blood platelets. Am. J. Pathol. 1977, 87, 633–646. [Google Scholar]
- Maurer-Spurej, E.; Pittendreigh, C.; Solomons, K. The influence of selective serotonin reuptake inhibitors on human platelet serotonin. Thromb. Haemost. 2004, 91, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Skaer, R.J.; Flemans, R.J.; McQuilkan, S. Mepacrine stains the dense bodies of human platelets and not platelet lysosomes. Br. J. Haematol. 1981, 49, 435–438. [Google Scholar] [CrossRef] [PubMed]
- Greinacher, A.; Pecci, A.; Kunishima, S.; Althaus, K.; Nurden, P.; Balduini, C.L.; Bakchoul, T. Diagnosis of inherited platelet disorders on a blood smear: A tool to facilitate worldwide diagnosis of platelet disorders. J. Thromb. Haemost. 2017, 15, 1511–1521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaninetti, C.; Greinacher, A. Diagnosis of Inherited Platelet Disorders on a Blood Smear. J. Clin. Med. 2020, 9, 539. [Google Scholar] [CrossRef] [Green Version]
- Westmoreland, D.; Shaw, M.; Grimes, W.; Metcalf, D.J.; Burden, J.J.; Gomez, K.; Knight, A.E.; Cutler, D.F. Super-resolution microscopy as a potential approach to diagnosis of platelet granule disorders. J. Thromb. Haemost. 2016, 14, 839–849. [Google Scholar] [CrossRef]
- Holmsen, H.; Ostvold, A.C.; Day, H.J. Behaviour of endogenous and newly absorbed serotonin in the platelet release reaction. Biochem. Pharmacol. 1973, 22, 2599–2608. [Google Scholar] [CrossRef]
- Anderson, G.M.; Hall, L.M.; Yang, J.X.; Cohen, D.J. Platelet dense granule release reaction monitored by high-performance liquid chromatography-fluorometric determination of endogenous serotonin. Anal. Biochem. 1992, 206, 64–67. [Google Scholar] [CrossRef]
- Bossant, M.J.; Ninio, E.; Delautier, D.; Bessou, G.; Trouvin, J.H.; Benveniste, J. Quantitation of paf-acether by release of endogenous platelet serotonin assessed by liquid chromatography with electrochemical detection. Anal. Biochem. 1989, 182, 419–423. [Google Scholar] [CrossRef]
- Ge, S.; Wittenberg, N.J.; Haynes, C.L. Quantitative and real-time detection of secretion of chemical messengers from individual platelets. Biochemistry 2008, 47, 7020–7024. [Google Scholar] [CrossRef] [PubMed]
- Ge, S.; Woo, E.; White, J.G.; Haynes, C.L. Electrochemical measurement of endogenous serotonin release from human blood platelets. Anal. Chem. 2011, 83, 2598–2604. [Google Scholar] [CrossRef] [Green Version]
- Gawaz, M.P.; Bogner, C.; Gurland, H.J. Flow-cytometric analysis of mepacrine-labelled platelets in patients with end-stage renal failure. Haemostasis 1993, 23, 284–292. [Google Scholar] [CrossRef]
- van Asten, I.; Blaauwgeers, M.; Granneman, L.; Heijnen, H.F.G.; Kruip, M.; Beckers, E.A.M.; Coppens, M.; Eikenboom, J.; Tamminga, R.Y.J.; Pasterkamp, G.; et al. Flow cytometric mepacrine fluorescence can be used for the exclusion of platelet dense granule deficiency. J. Thromb. Haemost. 2020, 18, 706–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aschar-Sobbi, R.; Abramov, A.Y.; Diao, C.; Kargacin, M.E.; Kargacin, G.J.; French, R.J.; Pavlov, E. High sensitivity, quantitative measurements of polyphosphate using a new DAPI-based approach. J. Fluoresc. 2008, 18, 859–866. [Google Scholar] [CrossRef] [PubMed]
- Schlagenhauf, A.; Pohl, S.; Haidl, H.; Leschnik, B.; Gallistl, S.; Muntean, W. Non-enzymatic quantification of polyphosphate levels in platelet lysates and releasates. J. Pharm. Biomed. Anal. 2016, 131, 1–5. [Google Scholar] [CrossRef]
- Smith, S.A.; Morrissey, J.H. Sensitive fluorescence detection of polyphosphate in polyacrylamide gels using 4′, 6-diamidino-2-phenylindol. Electrophoresis 2007, 28, 3461–3465. [Google Scholar] [CrossRef]
- Feinman, R.D.; Lubowsky, J.; Charo, I.; Zabinski, M.P. The lumi-aggregometer: A new instrument for simultaneous measurement of secretion and aggregation by platelets. J. Lab. Clin. Med. 1977, 90, 125–129. [Google Scholar]
- Cattaneo, M. Light transmission aggregometry and ATP release for the diagnostic assessment of platelet function. Semin. Thromb. Hemost. 2009, 35, 158–167. [Google Scholar] [CrossRef]
- Badin, M.S.; Graf, L.; Iyer, J.K.; Moffat, K.A.; Seecharan, J.L.; Hayward, C.P. Variability in platelet dense granule adenosine triphosphate release findings amongst patients tested multiple times as part of an assessment for a bleeding disorder. Int. J. Lab. Hematol. 2016, 38, 648–657. [Google Scholar] [CrossRef]
- Stockley, J.; Morgan, N.V.; Bem, D.; Lowe, G.C.; Lordkipanidze, M.; Dawood, B.; Simpson, M.A.; Macfarlane, K.; Horner, K.; Leo, V.C.; et al. Enrichment of FLI1 and RUNX1 mutations in families with excessive bleeding and platelet dense granule secretion defects. Blood 2013, 122, 4090–4093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latger-Cannard, V.; Philippe, C.; Bouquet, A.; Baccini, V.; Alessi, M.C.; Ankri, A.; Bauters, A.; Bayart, S.; Cornillet-Lefebvre, P.; Daliphard, S.; et al. Haematological spectrum and genotype-phenotype correlations in nine unrelated families with RUNX1 mutations from the French network on inherited platelet disorders. Orphanet J. Rare Dis. 2016, 11, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ringeisen, A.L.; Schimmenti, L.A.; White, J.G.; Schoonveld, C.; Summers, C.G. Hermansky-Pudlak syndrome (HPS5) in a nonagenarian. J. AAPOS 2013, 17, 334–336. [Google Scholar] [CrossRef] [PubMed]
- Novak, E.K.; Hui, S.W.; Swank, R.T. Platelet storage pool deficiency in mouse pigment mutations associated with seven distinct genetic loci. Blood 1984, 63, 536–544. [Google Scholar] [CrossRef] [Green Version]
- Cai, H.; Mullier, F.; Frotscher, B.; Briquel, M.E.; Toussaint, M.; Massin, F.; Lecompte, T.; Latger-Cannard, V. Usefulness of Flow Cytometric Mepacrine Uptake/Release Combined with CD63 Assay in Diagnosis of Patients with Suspected Platelet Dense Granule Disorder. Semin. Thromb. Hemost. 2016, 42, 282–291. [Google Scholar] [CrossRef]
- Rodeghiero, F.; Tosetto, A.; Abshire, T.; Arnold, D.M.; Coller, B.; James, P.; Neunert, C.; Lillicrap, D. ISTH/SSC bleeding assessment tool: A standardized questionnaire and a proposal for a new bleeding score for inherited bleeding disorders. J. Thromb. Haemost. 2010, 8, 2063–2065. [Google Scholar] [CrossRef]
- Selle, F.; James, C.; Tuffigo, M.; Pillois, X.; Viallard, J.F.; Alessi, M.C.; Fiore, M. Clinical and Laboratory Findings in Patients with delta-Storage Pool Disease: A Case Series. Semin. Thromb. Hemost. 2017, 43, 48–58. [Google Scholar] [CrossRef] [Green Version]
- Orsini, S.; Noris, P.; Bury, L.; Heller, P.G.; Santoro, C.; Kadir, R.A.; Butta, N.C.; Falcinelli, E.; Cid, A.R.; Fabris, F.; et al. Bleeding risk of surgery and its prevention in patients with inherited platelet disorders. Haematologica 2017, 102, 1192–1203. [Google Scholar] [CrossRef] [Green Version]
- Civaschi, E.; Klersy, C.; Melazzini, F.; Pujol-Moix, N.; Santoro, C.; Cattaneo, M.; Lavenu-Bombled, C.; Bury, L.; Minuz, P.; Nurden, P.; et al. Analysis of 65 pregnancies in 34 women with five different forms of inherited platelet function disorders. Br. J. Haematol. 2015, 170, 559–563. [Google Scholar] [CrossRef] [Green Version]
- Dupuis, A.; Gachet, C. Inherited platelet disorders: Management of the bleeding risk. Transfus. Clin. Biol. J. Soc. Fr. Transfus. Sang. 2018, 25, 228–235. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | DG/plt | Number of Subjects | Number of plts/Subject | Age Range | Anticoagulant | PRP Preparation | Reference |
|---|---|---|---|---|---|---|---|
| WP-TEM | >3.68 (m − 3SD) | NI | NI | NI | ACD | 100 g 15 min <48 h | Gunning 2000-20 [68,71] |
| WP-TEM | 1.95–4.37 | n = 40 (19M + 21F) | 100 | 2 m–21 year | ACD | 57 g 20 min | Sorokin 2016 [72] |
| WP-TEM | 1.2–4.0 | n = 113 (65M + 48F) | 100–200 | 18–70 year | ACD-A or -B | 200 g 20 min | Chen 2018 [67] |
| WT-TEM | >3 | n = 300 | 20(>3)–100(<3) | 6 w–21 year | EDTA | 100 g * 6 min <72 h | Asher 2020 [77] |
| WP-TEM | 4.9–8.2 | n = 60 M | 30–50 | 15–64 year | Na citrate | 115 g 15 min <4 h | Brunet 2018 [66] |
| WP-TEM | 4.9–8.8 | n = 66 F | 30–50 | 15–64 year | Na citrate | 115 g 15 min <4 h | Brunet 2018 [66] |
| WP-TEM | 4.14–7.74 | n = 54 | 100 | 18–70 year | CPDA1 | 150 g 10 min <4 h | Pennamen 2020 [30] |
| CT-TEM | 5.5 ± 1.9 | n = 6 F | 5 | NI | Na citrate | 150 g 20 min <1 h | Wang 2015 [73] |
| FIB-SEM | 8.0 ± 4.2 | 49 plts | NI | NI | ACD | 250 g 15 min ** | Eckly 2016 [74] |
| SBF-SEM | 5.5 ± 2.5 | n = 3 | 10 | NI | Na citrate | NI (after fixation) | Pokrovskaya 2020 [75] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dupuis, A.; Bordet, J.-C.; Eckly, A.; Gachet, C. Platelet δ-Storage Pool Disease: An Update. J. Clin. Med. 2020, 9, 2508. https://doi.org/10.3390/jcm9082508
Dupuis A, Bordet J-C, Eckly A, Gachet C. Platelet δ-Storage Pool Disease: An Update. Journal of Clinical Medicine. 2020; 9(8):2508. https://doi.org/10.3390/jcm9082508
Chicago/Turabian StyleDupuis, Arnaud, Jean-Claude Bordet, Anita Eckly, and Christian Gachet. 2020. "Platelet δ-Storage Pool Disease: An Update" Journal of Clinical Medicine 9, no. 8: 2508. https://doi.org/10.3390/jcm9082508
APA StyleDupuis, A., Bordet, J. -C., Eckly, A., & Gachet, C. (2020). Platelet δ-Storage Pool Disease: An Update. Journal of Clinical Medicine, 9(8), 2508. https://doi.org/10.3390/jcm9082508