

Detection of Xylella fastidiosa in Host Plants and Insect Vectors by Droplet Digital PCR

 , ,
, ,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strain
2.2. Plant Sources and Insects
2.3. Sample Preparation and DNA Extraction
2.4. Quantitative PCR Assay
2.5. Droplet Digital PCR Assay
2.6. Evaluation of ddPCR Performance in Naturally Infected Plant and Insect Samples
2.7. Data Analysis
3. Results
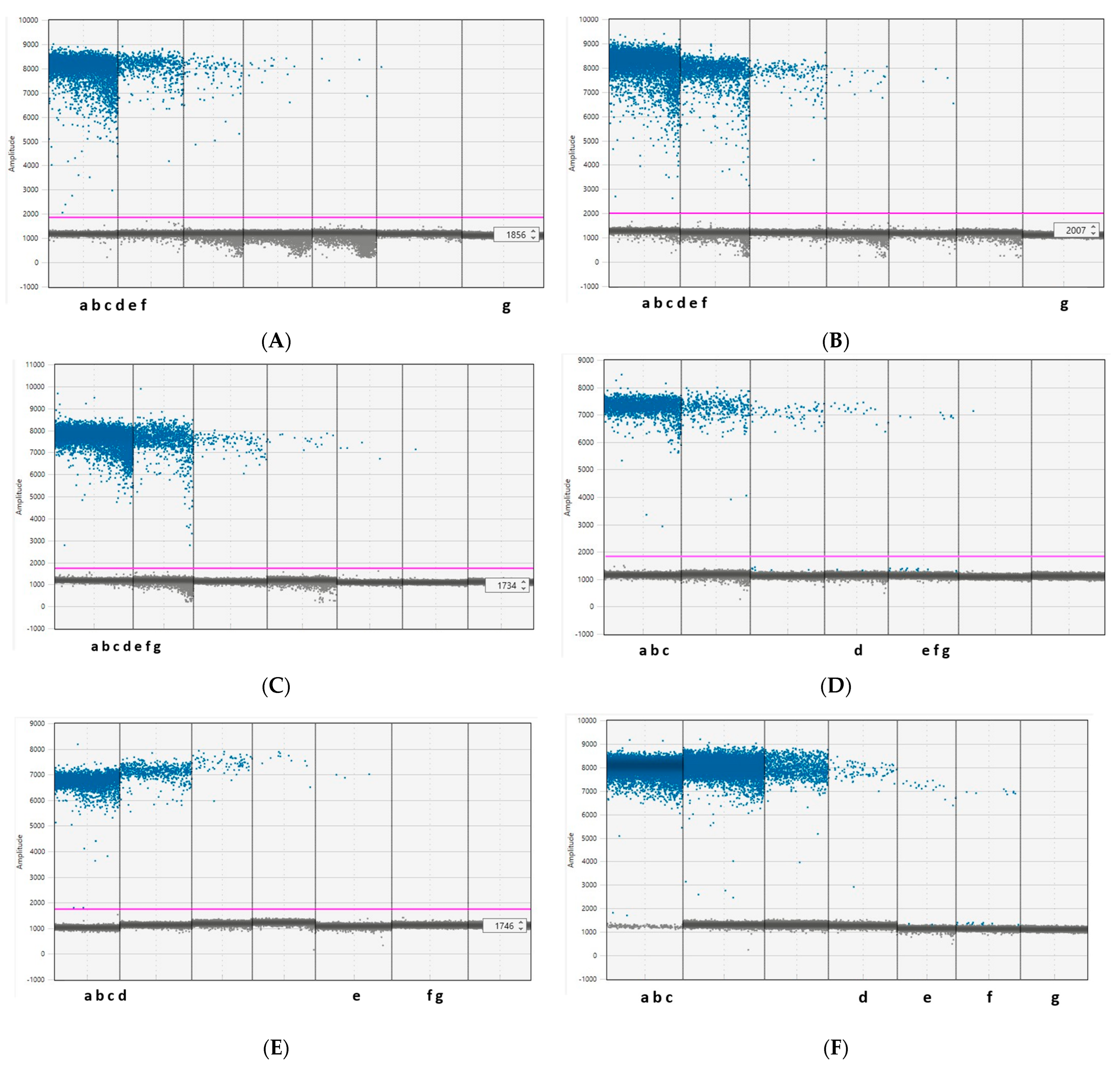
3.1. Optimization of the ddPCR Assay
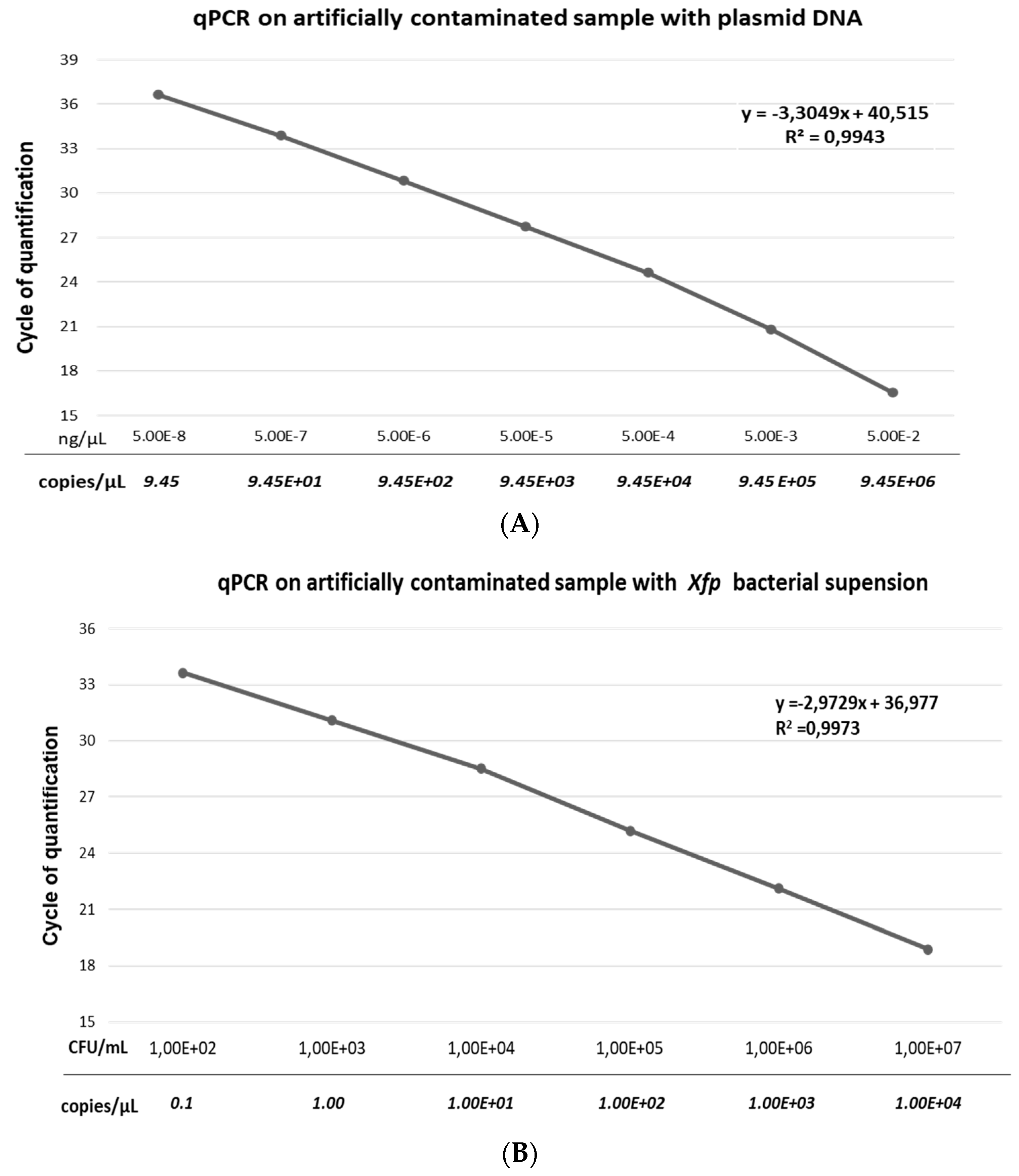
3.2. Linearity and Analytical Sensitivity of qPCR Assay
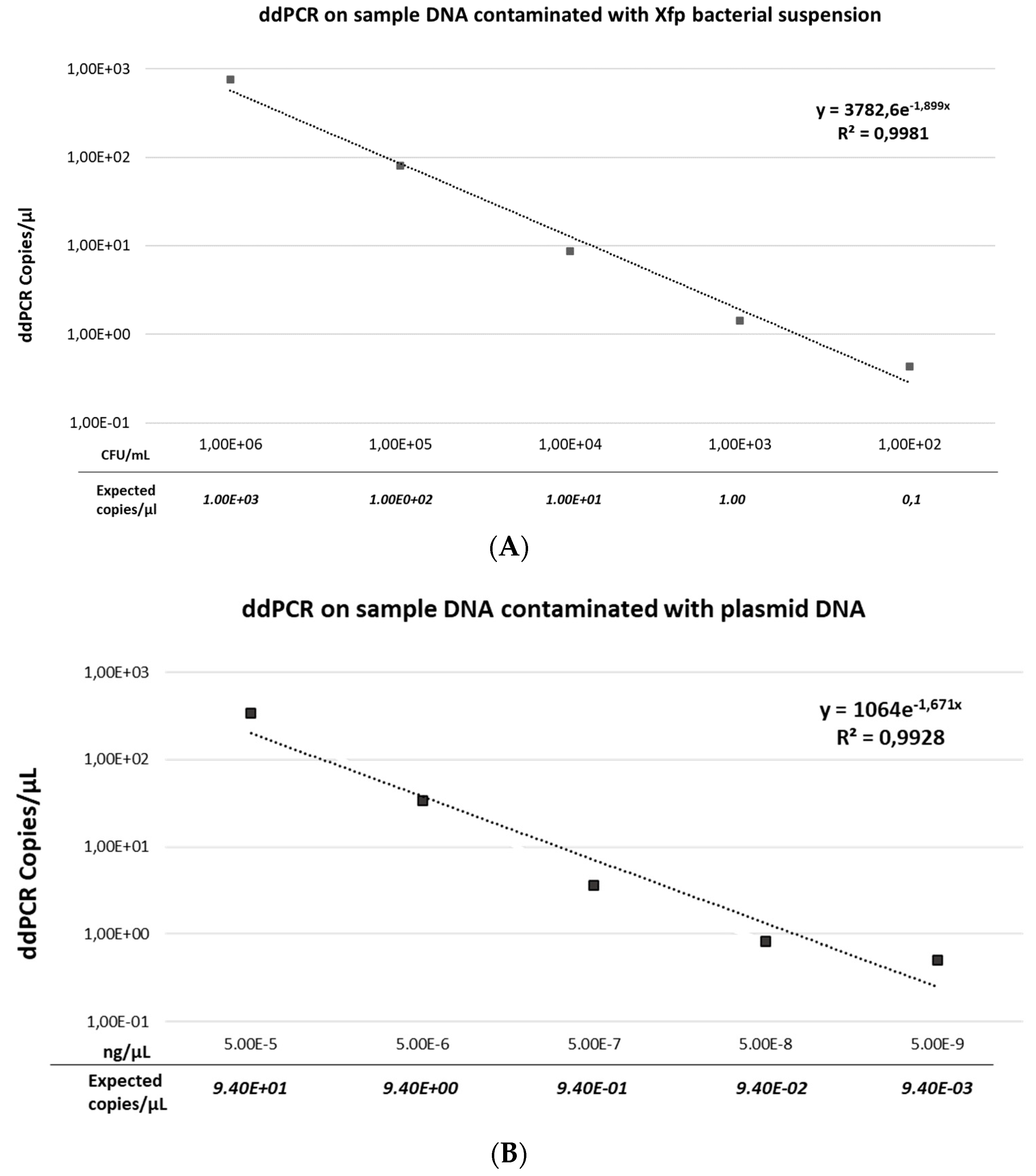
3.3. Linearity and Analytical Sensitivity of the ddPCR Assay
3.4. Comparison between ddPCR and qPCR Assays in Artificially Contaminated Samples
3.5. Evaluation of the ddPCR Assay in Field Samples
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wells, J.M.; Raju, B.C.; Hung, H.-Y.; Weisburg, W.G.; Mandelco-Paul, L.; Brenner, D.J. Xylella fastidiosa Gen. Nov., Sp. Nov: Gram-Negative, Xylem-Limited, Fastidious Plant Bacteria Related to Xanthomonas pp. Int. J. Syst. Bacteriol. 1987, 37, 136–143. [Google Scholar] [CrossRef]
- Delbianco, A.; Gibin, D.; Pasinato, L.; Morelli, M. Update of the Xylella Spp. Host Plant Database—Systematic Literature Search up to 30 June 2021. EFSA J. 2022, 20, 1–67. [Google Scholar] [CrossRef]
- Saponari, M.; Boscia, D.; Altamura, G.; Loconsole, G.; Zicca, S.; D’Attoma, G.; Morelli, M.; Palmisano, F.; Saponari, A.; Tavano, D.; et al. Isolation and Pathogenicity of Xylella fastidiosa Associated to the Olive Quick Decline Syndrome in Southern Italy. Sci. Rep. 2017, 7, 17723. [Google Scholar] [CrossRef] [Green Version]
- Coletta-Filho, H.; Francisco, C.; Lopes, J.; Oliveira, A.D.; Silva, L.F.D. First Report of Olive Leaf Scorch in Brazil, Associated with Xylella fastidiosa Subsp. pauca. Phytopathol. Mediterr. 2016, 55, 130–135. [Google Scholar] [CrossRef]
- Haelterman, R.M.; Tolocka, P.A.; Roca, M.E.; Guzmán, F.A.; Fernández, F.D.; Otero, M.L. First Presumptive Diagnosis of Xylella fastidiosa Causing Olive Scorch In Argentina. J. Plant Pathol. 2015, 97, 393. [Google Scholar] [CrossRef]
- Olmo, D.; Nieto, A.; Borràs, D.; Montesinos, M.; Adrover, F.; Pascual, A.; Gost, P.A.; Quetglas, B.; Urbano, A.; de Dios García, J.; et al. Landscape Epidemiology of Xylella fastidiosa in the Balearic Islands. Agronomy 2021, 11, 473. [Google Scholar] [CrossRef]
- Saponari, M.; Boscia, D.; Nigro, F.; Martelli, G.P. Identification of Dna Sequences Related to Xylella fastidiosa In Oleander, Almond And Olive Trees Exhibiting Leaf Scorch Symptoms In Apulia (Southern Italy). J. Plant Pathol. 2013, 95, 668. [Google Scholar] [CrossRef]
- Cornara, D.; Cavalieri, V.; Dongiovanni, C.; Altamura, G.; Palmisano, F.; Bosco, D.; Porcelli, F.; Almeida, R.P.P.; Saponari, M. Transmission of Xylella fastidiosa by Naturally Infected Philaenus spumarius (Hemiptera, Aphrophoridae) to Different Host Plants. J. Appl. Entomol. 2017, 141, 80–87. [Google Scholar] [CrossRef]
- Luvisi, A.; Nicolì, F.; De Bellis, L. Sustainable Management of Plant Quarantine Pests: The Case of Olive Quick Decline Syndrome. Sustainability 2017, 9, 659. [Google Scholar] [CrossRef] [Green Version]
- Morelli, M.; García-Madero, J.M.; Jos, Á.; Saldarelli, P.; Dongiovanni, C.; Kovacova, M.; Saponari, M.; Baños Arjona, A.; Hackl, E.; Webb, S.; et al. Xylella fastidiosa in Olive: A Review of Control Attempts and Current Management. Microorganisms 2021, 9, 1771. [Google Scholar] [CrossRef]
- Harper, S.J.; Ward, L.I.; Clover, G.R.G. Development of LAMP and Real-Time PCR Methods for the Rapid Detection of Xylella fastidiosa for Quarantine and Field Applications. Phytopathology 2010, 100, 1282–1288. [Google Scholar] [CrossRef]
- Cariddi, C.; Saponari, M.; Boscia, D.; Stradis, A.D.; Loconsole, G.; Nigro, F.; Porcelli, F.; Potere, O.; Martelli, G.P. Isolation of a Xylella fastidiosa Strain Infecting Olive and Oleander in Apulia, Italy. J. Plant Pathol. 2014, 96, 1–5. [Google Scholar] [CrossRef]
- Loconsole, G.; Potere, O.; Boscia, D.; Altamura, G.; Djelouah, K.; Elbeaino, T.; Frasheri, D.; Lorusso, D.; Palmisano, F.; Pollastro, P.; et al. Detection of Xylella fastidiosa in Olive Trees by Molecular and Serological Methods. J. Plant Pathol. 2014, 96, 1–8. [Google Scholar] [CrossRef]
- Waliullah, S.; Hudson, O.; Oliver, J.E.; Brannen, P.M.; Ji, P.; Ali, M.E. Comparative Analysis of Different Molecular and Serological Methods for Detection of Xylella fastidiosa in Blueberry. PLoS ONE 2019, 14, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horn, N. PM 7/24 (4) Xylella Fastidiosa. EPPO Bull. 2019, 49, 175–227. [Google Scholar] [CrossRef] [Green Version]
- Sykes, P.J.; Neoh, S.H.; Brisco, M.J.; Hughes, E.; Condon, J.; Morley, A.A. Quantitation of Targets for PCR by Use of Limiting Dilution. BioTechniques 1992, 13, 444–449. [Google Scholar]
- Vogelstein, B.; Kinzler, K.W. Digital PCR. Proc. Natl. Acad. Sci. USA 1999, 96, 9236–9241. [Google Scholar] [CrossRef] [Green Version]
- Hindson, B.J.; Ness, K.D.; Masquelier, D.A.; Belgrader, P.; Heredia, N.J.; Makarewicz, A.J.; Bright, I.J.; Lucero, M.Y.; Hiddessen, A.L.; Legler, T.C.; et al. High-Throughput Droplet Digital PCR System for Absolute Quantitation of DNA Copy Number. Anal. Chem. 2011, 83, 8604–8610. [Google Scholar] [CrossRef]
- Pinheiro, L.B.; Coleman, V.A.; Hindson, C.M.; Herrmann, J.; Hindson, B.J.; Bhat, S.; Emslie, K.R. Evaluation of a Droplet Digital Polymerase Chain Reaction Format for DNA Copy Number Quantification. Anal. Chem. 2012, 84, 1003–1011. [Google Scholar] [CrossRef]
- Huggett, J.F.; Whale, A. Digital PCR as a Novel Technology and Its Potential Implications for Molecular Diagnostics. Clin. Chem. 2013, 59, 1691–1693. [Google Scholar] [CrossRef] [Green Version]
- Whale, A.S.; De Spiegelaere, W.; Trypsteen, W.; Nour, A.A.; Bae, Y.-K.; Benes, V.; Burke, D.; Cleveland, M.; Corbisier, P.; Devonshire, A.S.; et al. The Digital MIQE Guidelines Update: Minimum Information for Publication of Quantitative Digital PCR Experiments for 2020. Clin. Chem. 2020, 66, 1012–1029. [Google Scholar] [CrossRef]
- Giampetruzzi, A.; Saponari, M.; Almeida, R.P.P.; Essakhi, S.; Boscia, D.; Loconsole, G.; Saldarelli, P. Complete Genome Sequence of the Olive-Infecting Strain Xylella fastidiosa Subsp. Pauca De Donno. Genome Announc. 2017, 5, e00569-17. [Google Scholar] [CrossRef] [Green Version]
- Davis, M.J. Isolation Media for the Pierce’s Disease Bacterium. Phytopathology 1980, 70, 425. [Google Scholar] [CrossRef]
- Dupas, E.; Legendre, B.; Olivier, V.; Poliakoff, F.; Manceau, C.; Cunty, A. Comparison of Real-Time PCR and Droplet Digital PCR for the Detection of Xylella fastidiosa in Plants. J. Microbiol. Methods 2019, 162, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Raguseo, C.; Gerin, D.; Pollastro, S.; Rotolo, C.; Rotondo, P.R.; Faretra, F.; Angelini, R.M.D.M. A Duplex-Droplet Digital PCR Assay for Simultaneous Quantitative Detection of Monilinia fructicola and Monilinia laxa on Stone Fruits. Front. Microbiol. 2021, 12, 747560. [Google Scholar] [CrossRef] [PubMed]
- Bogožalec Košir, A.; Demšar, T.; Štebih, D.; Žel, J.; Milavec, M. Digital PCR as an Effective Tool for GMO Quantification in Complex Matrices. Food Chem. 2019, 294, 73–78. [Google Scholar] [CrossRef]
- Taylor, S.C.; Laperriere, G.; Germain, H. Droplet Digital PCR versus QPCR for Gene Expression Analysis with Low Abundant Targets: From Variable Nonsense to Publication Quality Data. Sci. Rep. 2017, 7, 2409. [Google Scholar] [CrossRef] [Green Version]
- Sedlak, R.H.; Kuypers, J.; Jerome, K.R. A Multiplexed Droplet Digital PCR Assay Performs Better than QPCR on Inhibition Prone Samples. Diagn. Microbiol. Infect. Dis. 2014, 80, 285–286. [Google Scholar] [CrossRef]
- Koepfli, C.; Nguitragool, W.; Hofmann, N.E.; Robinson, L.J.; Ome-Kaius, M.; Sattabongkot, J.; Felger, I.; Mueller, I. Sensitive and Accurate Quantification of Human Malaria Parasites Using Droplet Digital PCR (DdPCR). Sci. Rep. 2016, 6, 39183. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Bai, R.; Zhao, Z.; Tao, L.; Ma, M.; Ji, Z.; Jian, M.; Ding, Z.; Dai, X.; Bao, F.; et al. Application of Droplet Digital PCR to Detect the Pathogens of Infectious Diseases. Biosci. Rep. 2018, 38, BSR20181170. [Google Scholar] [CrossRef] [Green Version]
- Baume, M.; Cariou, A.; Leveau, A.; Fessy, N.; Pastori, F.; Jarraud, S.; Pierre, S. Quantification of Legionella DNA Certified Reference Material by Digital Droplet PCR. J. Microbiol. Methods 2019, 157, 50–53. [Google Scholar] [CrossRef] [PubMed]
- Mahendran, P.; Liew, J.W.K.; Amir, A.; Ching, X.T.; Lau, Y.L. Droplet Digital Polymerase Chain Reaction (DdPCR) for the Detection of Plasmodium knowlesi and Plasmodium vivax. Malar. J. 2020, 19, 241. [Google Scholar] [CrossRef] [PubMed]
- Vasudevan, H.N.; Xu, P.; Servellita, V.; Miller, S.; Liu, L.; Gopez, A.; Chiu, C.Y.; Abate, A.R. Digital Droplet PCR Accurately Quantifies SARS-CoV-2 Viral Load from Crude Lysate without Nucleic Acid Purification. Sci. Rep. 2021, 11, 780. [Google Scholar] [CrossRef]
- Hepokoski, M.L.; Odish, M.; Lam, M.T.; Coufal, N.G.; Rolfsen, M.L.; Shadel, G.S.; Moyzis, A.G.; Sainz, A.G.; Takiar, P.G.; Patel, S.; et al. Absolute Quantification of Plasma Mitochondrial DNA by Droplet Digital PCR Marks COVID-19 Severity over Time during Intensive Care Unit Admissions. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2022, 323, L84–L92. [Google Scholar] [CrossRef]
- Milosevic, D.; Moyer, A.M.; Majumdar, R.; Kipp, B.R.; Yao, J.D. A Reverse-Transcription Droplet Digital PCR Assay to Detect and Quantify SARS-CoV-2 RNA in Upper Respiratory Tract Specimens. J. Clin. Virol. 2022, 153, 105216. [Google Scholar] [CrossRef]
- Morcia, C.; Ghizzoni, R.; Delogu, C.; Andreani, L.; Carnevali, P.; Terzi, V. Digital Pcr: What Relevance to Plant Studies? Biology 2020, 9, 433. [Google Scholar] [CrossRef]
- Martínez-Diz, M.d.P.; Andrés-Sodupe, M.; Berbegal, M.; Bujanda, R.; Díaz-Losada, E.; Gramaje, D. Droplet Digital PCR Technology for Detection of Ilyonectria liriodendri from Grapevine Environmental Samples. Plant Dis. 2020, 104, 1144–1150. [Google Scholar] [CrossRef] [PubMed]
- Maldonado-González, M.M.; del Pilar Martínez-Diz, M.; Andrés-Sodupe, M.; Bujanda, R.; Díaz-Losada, E.; Gramaje, D. Quantification of Cadophora luteo-olivacea from Grapevine Nursery Stock and Vineyard Soil Using Droplet Digital PCR. Plant Dis. 2020, 104, 2269–2274. [Google Scholar] [CrossRef]
- Wen, R.; Lee, J.; Chu, M.; Tonu, N.; Dumonceaux, T.; Gossen, B.D.; Yu, F.; Peng, G. Quantification of Plasmodiophora brassicae Resting Spores in Soils Using Droplet Digital PCR (DdPCR). Plant Dis. 2020, 104, 1188–1194. [Google Scholar] [CrossRef]
- Dreo, T.; Pirc, M.; Ramšak, Ž.; Pavšič, J.; Milavec, M.; Žel, J.; Gruden, K. Optimising Droplet Digital PCR Analysis Approaches for Detection and Quantification of Bacteria: A Case Study of Fire Blight and Potato Brown Rot. Anal. Bioanal. Chem. 2014, 406, 6513–6528. [Google Scholar] [CrossRef]
- Gutiérrez-Aguirre, I.; Rački, N.; Dreo, T.; Ravnikar, M. Droplet Digital PCR for Absolute Quantification of Pathogens. Methods Mol. Biol. 2015, 1302, 331–347. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Xia, Q.; Yin, Y.; Wang, Z. Comparison of Droplet Digital PCR and Quantitative PCR Assays for Quantitative Detection of Xanthomonas citri Subsp. Citri. PLoS ONE 2016, 11, e0159004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selvaraj, V.; Maheshwari, Y.; Hajeri, S.; Chen, J.; McCollum, T.G.; Yokomi, R. Development of a Duplex Droplet Digital PCR Assay for Absolute Quantitative Detection of “Candidatus liberibacter asiaticus”. PLoS ONE 2018, 13, e0197184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, X.; Liu, X.L.; Lou, B.H.; Zhou, C.Y.; Wang, X. Feng Development of a Sensitive and Reliable Droplet Digital PCR Assay for the Detection of ‘Candidatus liberibacter asiaticus’. J. Integr. Agric. 2018, 17, 483–487. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Zhang, H.J.; Zhao, Z.J.; Wen, C.L.; Wu, P.; Song, S.H.; YU, S.C.; LUO, L.X.; XU, X. lan Application of Droplet Digital PCR in Detection of Seed-Transmitted Pathogen Acidovorax citrulli. J. Integr. Agric. 2020, 19, 561–569. [Google Scholar] [CrossRef]
- Bahar, M.H.; Wist, T.J.; Bekkaoui, D.R.; Hegedus, D.D.; Olivier, C.Y. Aster Leafhopper Survival and Reproduction, and Aster Yellows Transmission under Static and Fluctuating Temperatures, Using DdPCR for Phytoplasma Quantification. Sci. Rep. 2018, 8, 227. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Wang, Y.; Wang, Q.; Zhang, Y.; Shen, W.; Li, R.; Cao, M.; Chen, L.; Li, X.; Zhou, C.; et al. Development of a Sensitive and Reliable Reverse Transcription Droplet Digital PCR Assay for the Detection of Citrus Yellow Vein Clearing Virus. Arch. Virol. 2019, 164, 691–697. [Google Scholar] [CrossRef]
- Vargas-Hernández, B.Y.; Ramírez-Pool, J.A.; Núñez-Muñoz, L.A.; Calderón-Pérez, B.; De La Torre-Almaráz, R.; Hinojosa-Moya, J.; Xoconostle-Cázares, B.; Ruiz-Medrano, R. Development of a Droplet Digital Polymerase Chain Reaction (DdPCR) Assay for the Detection of Tomato Brown Rugose Fruit Virus (ToBRFV) in Tomato and Pepper Seeds. J. Virol. Methods 2022, 302, 114466. [Google Scholar] [CrossRef]
- Lee, H.-J.; Cho, I.-S.; Ju, H.-J.; Jeong, R.-D. Development of a Reverse Transcription Droplet Digital PCR Assay for Sensitive Detection of Peach Latent Mosaic Viroid. Mol. Cell. Probes 2021, 58, 101746. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| ddPCR | Curve Equation | R2 |
|---|---|---|
| Olea europea | y = 1635.4 × 10−1.278x | R² = 0.9867 |
| Vitis vinifera | y = 6607.7 × 10−1.922x | R² = 0.9885 |
| Citrus sinensis | y = 2220.3 × 10−1.86x | R² = 0.9949 |
| Prunus dulcis | y = 4432.9 × 10−1.854x | R² = 0.9985 |
| Nerium oleander | y = 653.8 × 10−1.392x | R² = 0.9949 |
| Philaenus spumarius | y = 25,107 × 10−1.814x | R² = 0.9743 |
| qPCR | Curve Equation | R2 | Efficiency |
|---|---|---|---|
| Olea europea | y = −3.1993x + 38.963 | 0.9962 | 105.38% |
| Vitis vinifera | y = −3.1095x + 38.341 | 0.9987 | 109.70% |
| Citrus sinensis | y = −3.1376x + 38.847 | 0.9993 | 108.31% |
| Prunus dulcis | y = −3.0538x + 38.553 | 0.9905 | 112.55% |
| Nerium oleander | y = −2.94x + 37.563 | 0.9933 | 118.84% |
| Philaenus spumarius | y = −3.0427x + 38.005 | 0.9995 | 113.14% |
| Dilution Range | Olea europea | Vitis vinifera | Citrus sinensis | Prunus dulcis | Nerium oleander | Philaenus spumarius | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CFU/mL | copies/µL | Replicates * | copies/µL | Replicates * | copies/µL | Replicates * | copies/µL | Replicates * | copies/µL | Replicates * | copies/µL | Replicates * |
| 1.00 × 106 | 6.09 × 102 | 2/2 | 7.97 × 102 | 2/2 | 3.74 × 102 | 2/2 | 6.62 × 102 | 2/2 | 2.21 × 102 | 2/2 | 4.99 × 103 | 2/2 |
| 1.00 × 105 | 1.08 × 102 | 5/5 | 2.00 × 102 | 2/2 | 5.43 × 101 | 2/2 | 1.28 × 102 | 2/2 | 3.90 × 101 | 2/2 | 7.73 × 102 | 2/2 |
| 1.00 × 104 | 4.36 × 101 | 5/5 | 1.98 × 101 | 2/2 | 7.79 × 100 | 2/2 | 1.33 × 101 | 2/2 | 6.37 × 100 | 2/2 | 1.20 × 102 | 2/2 |
| 1.00 × 103 | 5.10 × 100 | 11/11 | 2.55 × 100 | 12/12 | 1.08 × 100 | 10/10 | 3.24 × 100 | 10/10 | 2.02 × 100 | 8/8 | 8.33 × 100 | 8/8 |
| 1.00 × 102 | 2.40 × 100 | 14/14 | 4.74 × 10−1 | 12/12 | 2.43 × 10−1 | 9/10 | 3.92 × 10−1 | 6/10 | 9.22 × 10−1 | 7/8 | 2.02 × 100 | 8/8 |
| 1.00 × 101 | 1.20 × 100 | 4/7 | n.d. ** | n.d. ** | n.d. ** | n.d. ** | 9.22 × 10−1 | 4/8 | ||||
| Dilution Range CFU/mL | Olea europea | Vitis vinifera | Citrus sinensis | Prunus dulcis | Nerium oleander | Philaenus spumarius |
|---|---|---|---|---|---|---|
| 1.00 × 106 | 22.67 | 22.97 | 26.20 | 23.02 | 26.02 | 22.92 |
| 1.00 × 105 | 26.24 | 25.61 | 29.53 | 26.19 | 28.60 | 25.69 |
| 1.00 × 104 | 29.74 | 29.04 | 32.67 | 30.23 | 31.31 | 28.84 |
| 1.00 × 103 | 32.76 | 32.23 | 35.61 | 32.25 | 34.92 | 31.90 |
| 1.00 × 102 | 35.41 | 35.21 | n.d | 35.26 | n.d | 35.03 |
| 1.00 × 101 | n.d. | n.d | n.d | n.d | n.d | n.d |
| Olive Samples Grouped Based on the Cq Values Obtained by qPCR | ddPCR | |||||
|---|---|---|---|---|---|---|
| Group | Cq Values | N. of Samples | qPCR Result | Positive | Negative | Total |
| (i) | 27–29 | N. 10 | Positive | 10 | 0 | 10 |
| (ii) | 30–32 | N. 9 | Positive | 9 | 0 | 9 |
| (iii) | 33–34 | N. 15 | Undetermined | 13 | 2 | 15 |
| Total | 32 | 2 | 34 | |||
| Insect Samples Grouped Based on the Cq Values Obtained by qPCR | ddPCR | ||||||
|---|---|---|---|---|---|---|---|
| Group | Cq Values | N. of Samples | qPCR Result | Positive | Negative | Undetermined | Total |
| (i) | 23–32 | N. 11 | Positive. | 11 | 0 | 0 | 11 |
| (ii) | 33–36 | N. 13 | Undetermined | 8 | 3 | 2 | 13 |
| (iii) | >36 | N. 3 | Negative | 0 | 3 | 0 | 3 |
| Total | 19 | 6 | 2 | 27 | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amoia, S.S.; Minafra, A.; Ligorio, A.; Cavalieri, V.; Boscia, D.; Saponari, M.; Loconsole, G. Detection of Xylella fastidiosa in Host Plants and Insect Vectors by Droplet Digital PCR. Agriculture 2023, 13, 716. https://doi.org/10.3390/agriculture13030716
Amoia SS, Minafra A, Ligorio A, Cavalieri V, Boscia D, Saponari M, Loconsole G. Detection of Xylella fastidiosa in Host Plants and Insect Vectors by Droplet Digital PCR. Agriculture. 2023; 13(3):716. https://doi.org/10.3390/agriculture13030716
Chicago/Turabian StyleAmoia, Serafina Serena, Angelantonio Minafra, Angela Ligorio, Vincenzo Cavalieri, Donato Boscia, Maria Saponari, and Giuliana Loconsole. 2023. "Detection of Xylella fastidiosa in Host Plants and Insect Vectors by Droplet Digital PCR" Agriculture 13, no. 3: 716. https://doi.org/10.3390/agriculture13030716
APA StyleAmoia, S. S., Minafra, A., Ligorio, A., Cavalieri, V., Boscia, D., Saponari, M., & Loconsole, G. (2023). Detection of Xylella fastidiosa in Host Plants and Insect Vectors by Droplet Digital PCR. Agriculture, 13(3), 716. https://doi.org/10.3390/agriculture13030716