


Why Do Bio-Carbonates Exist?



{kind=link}
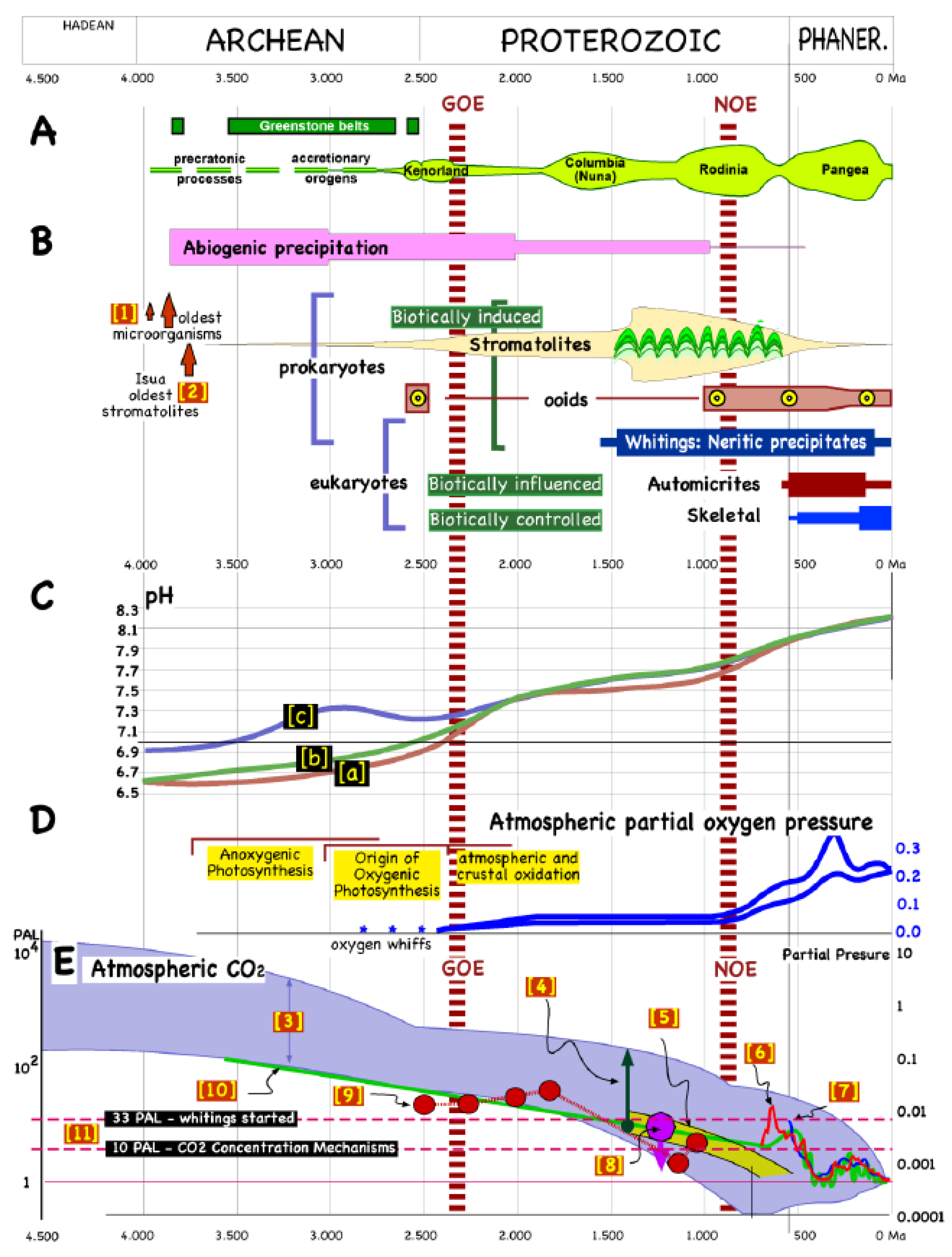{kind=link}
{kind=link}
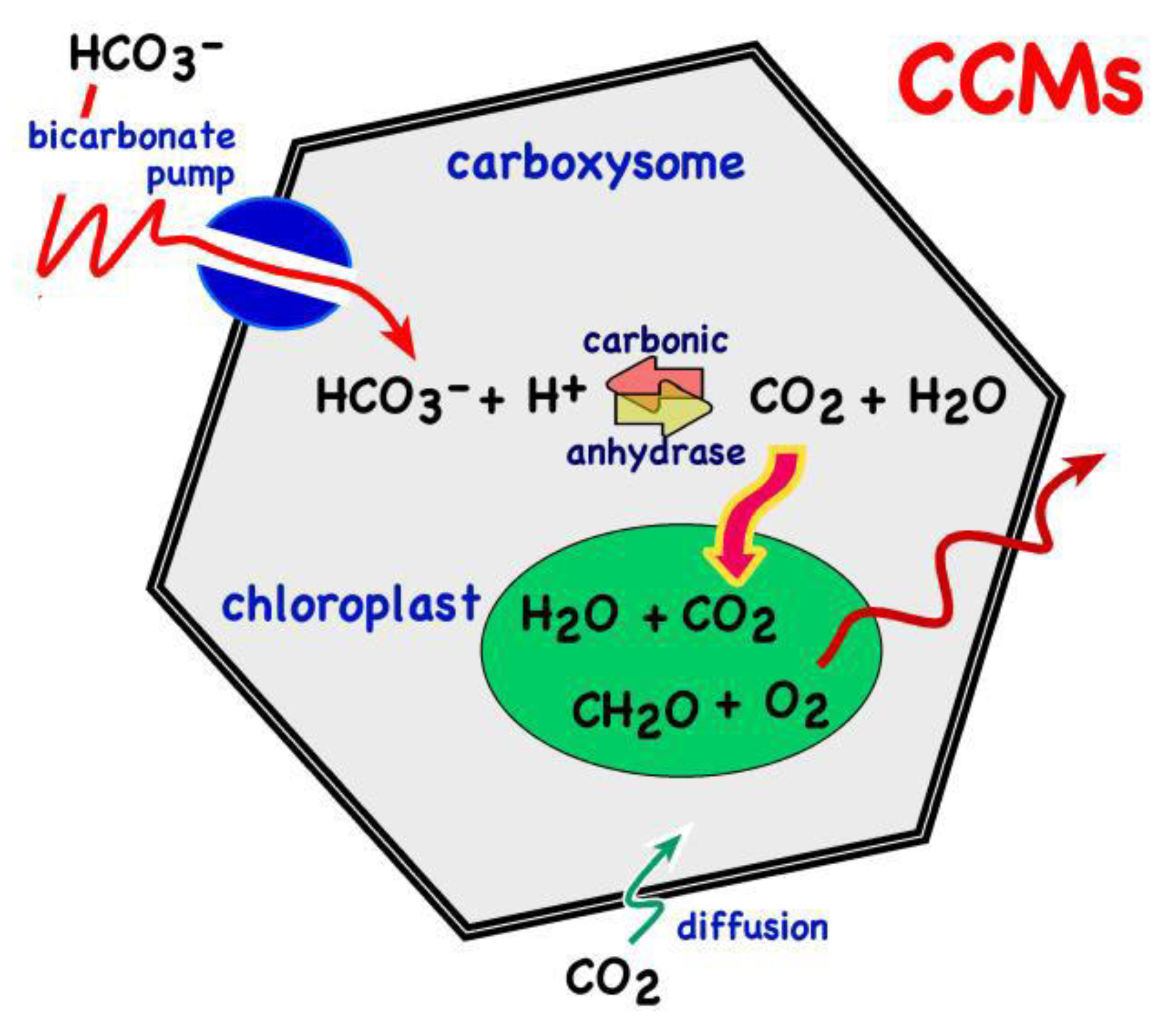{kind=link}
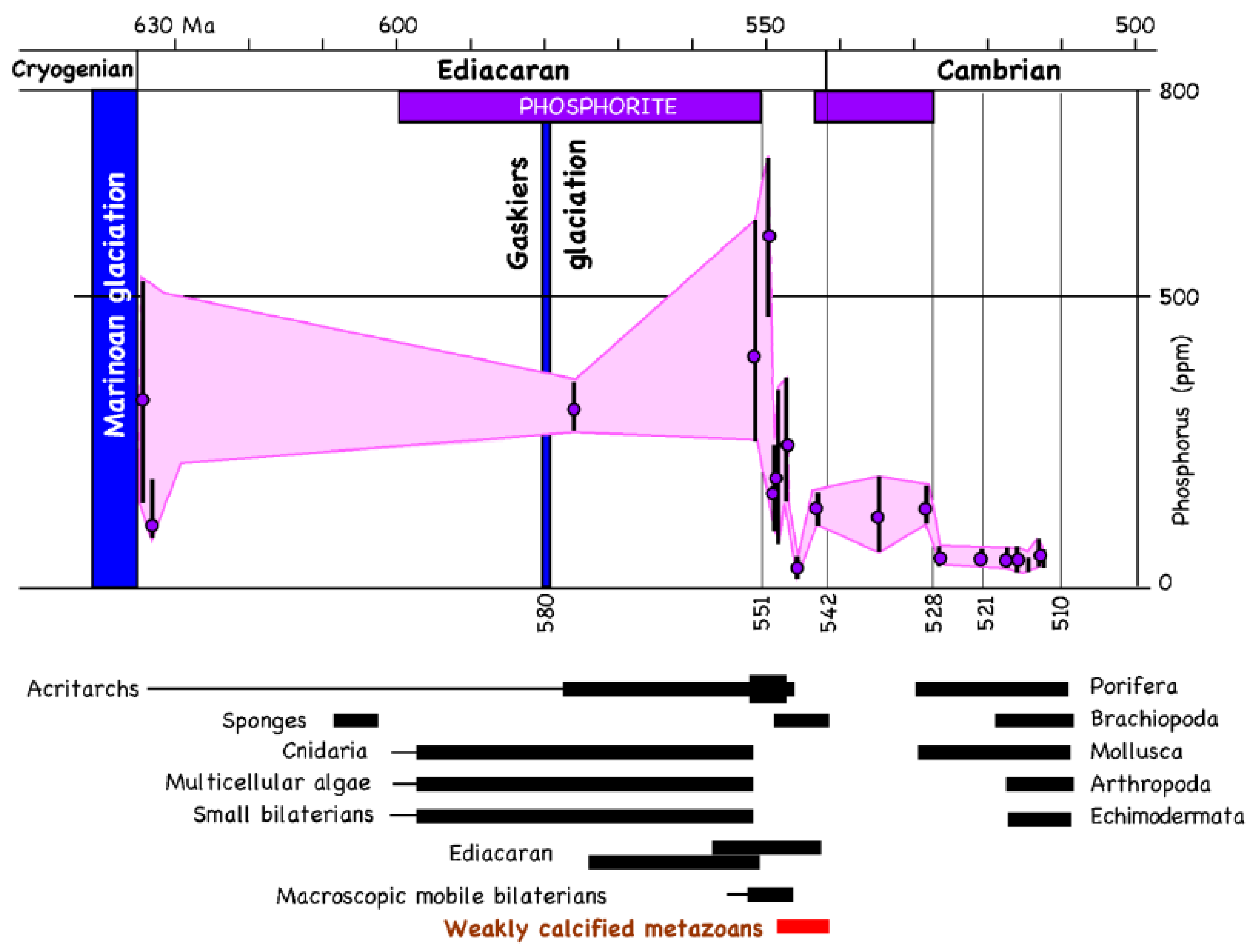{kind=link}
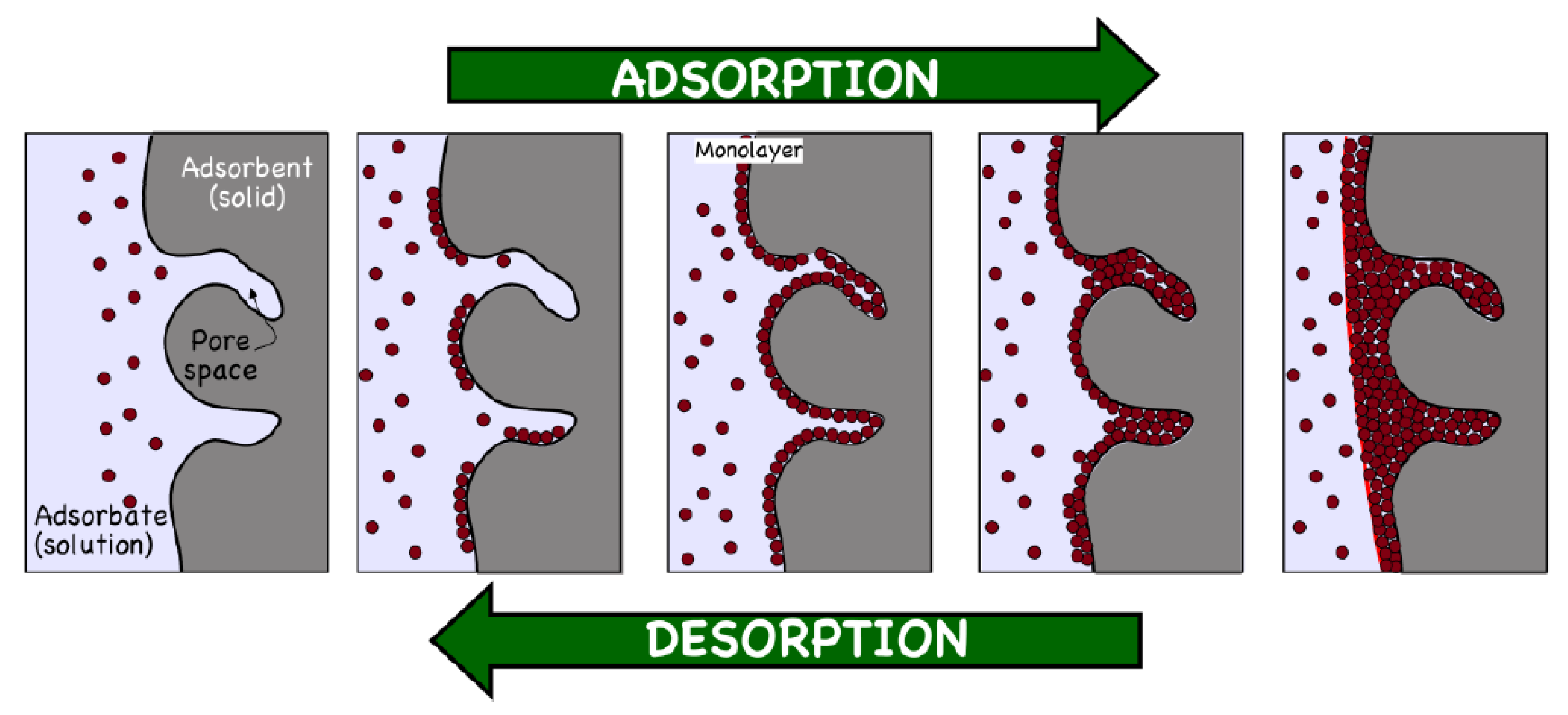{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Hypothesis
3. The Hadean–Archaean
4. Pertinent Chemical Processes
5. Pertinent Biological Processes
6. CCMs: CO2 Concentrating Mechanisms
7. Phosphorous
8. Calcification as a Sink and a Source of Both CO2 and PO43−
9. Prokaryotic Organo-Sedimentary Systems
10. Biocalcification in Photosynthetic Eukaryotes
11. CO2 and Carbonate Depositional History
12. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Selly, R.C.; Sonnenberg, S.A. Elements of Petroleum Geology, 3rd ed.; Academic Press: Boston, USA, 2015; pp. 1–507. [Google Scholar]
- Kharecha, P.; Kasting, J.; Siefert, J. A coupled atmosphere–ecosystem model of the early Archean Earth. Geobiology 2005, 3, 53–76. [Google Scholar] [CrossRef]
- Morse, J.W.; Arvidson, R.S.; Lüttge, A. Calcium carbonate formation and dissolution. Chem. Rev. 2007, 107, 342–381. [Google Scholar] [CrossRef] [PubMed]
- Burton, M.R.; Sawyer, G.M.; Granieri, D. Deep carbon emissions from volcanoes. Rev. Mineral. Geochem. 2013, 75, 323–354. [Google Scholar] [CrossRef] [Green Version]
- Global Carbon Budget 2021. Earth Syst. Sci. Data 2022, 14, 1917–2005. [CrossRef]
- Keeling, C.D. The concentration and isotopic abundances of carbon dioxide in the atmosphere. Tellus 1960, 12, 200–203. [Google Scholar] [CrossRef] [Green Version]
- Scripps CO2 Program. Global Stations CO2 Concentration Trends. 2022. Available online: https://scrippsco2.ucsd.edu/graphics_gallery/other_stations/global_stations_CO2_concentration_trends.html (accessed on 19 September 2022).
- Harris, D.C. Charles David Keeling and the story of atmospheric CO2 measurements. Anal. Chem. 2010, 82, 7865–7870. [Google Scholar] [CrossRef]
- IPCC (Intergovernment Panel on Climate Change). Available online: https://www.ipcc.ch/about/ (accessed on 22 August 2022).
- Kleypas, J.A.; Buddemeier, R.W.; Archer, D.; Gattuso, J.-P.; Langdon, C.; Opdyke, B.N. Geochemical consequences of increased atmospheric CO2 on coral reefs. Science 1999, 284, 118–120. [Google Scholar] [CrossRef]
- Caldeira, K.; Wickett, M.E. Anthropogenic carbon and ocean pH. Nature 2003, 425, 365. [Google Scholar] [CrossRef] [Green Version]
- Wigley, T.M. A combined mitigation/geoengineering approach to climate stabilization. Science 2006, 314, 452–454. [Google Scholar] [CrossRef] [Green Version]
- Wolf-Gladrow, D.; Riebesell, U.; Burkhardt, S.; Bijma, J. Direct effects of CO2 concentration on growth and isotopic composition of marine plankton. Tellus 1999, 51B, 461–476. [Google Scholar] [CrossRef]
- Orr, J.C.; Fabry, V.J.; Aumont, O.; Bopp, L.; Doney, S.C.; Feely, R.A.; Gnanadesikan, A.; Gruber, N.; Ishida, A.; Joos, F.; et al. Anthropogenic ocean acidification over the twenty-first century and its impact on calcifying organisms. Nature 2005, 437, 681–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veron, J.E.N. Mass extinctions and ocean acidification: Biological constraints on geological dilemmas. Coral Reefs 2008, 27, 459–472. [Google Scholar] [CrossRef]
- Kiessling, W.; Simpson, C. On the potential for ocean acidification to be a general cause of ancient reef crises. Glob. Chang. Biol. 2011, 17, 56–67. [Google Scholar] [CrossRef]
- Clarkson, M.O.; Kasemann, S.A.; Wood, R.A.; Lenton, T.M.; Dains, S.J.; Richoz, S.; Ohnemüller, F.; Meixner, A.; Poulton, S.W.; Tipper, E. Ocean acidification and the Permo-Triassic Mass Extinction. Science 2015, 348, 229–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodd, M.; Papineau, D.; Grenne, T.; Slack, J.F.; Rittner, M.; Pirajno, F.; O’Neil, J.; Little CT, S. Evidence for early life in Earth’s oldest hydrothermal vent precipitates. Nature 2017, 543, 60–64. [Google Scholar] [CrossRef]
- Nutman, A.P.; Bennett, V.C.; Friend CR, L.; Van Kranendonk, M.J.; Chivas, A.R. Rapid emergence of life shown by discovery of 3700-million-year-old microbial structures. Nature 2016, 537, 535–538. [Google Scholar] [CrossRef] [Green Version]
- Krissansen-Totton, J.; Arney, G.N.; Catling, D.C. Constraining the climate and ocean pH of the early Earth with a geological carbon cycle model. Proc. Natl. Acad. Sci. USA 2018, 115, 4105–4110. [Google Scholar] [CrossRef] [Green Version]
- Holland, H.D. The oxygenation of the atmosphere and oceans. Phil. Trans. R. Soc. B-Biol. Sci. 2006, 362, 903–915. [Google Scholar] [CrossRef] [Green Version]
- Kasting, J.F. Earth’s early atmosphere. Science 1993, 259, 920–926. [Google Scholar] [CrossRef]
- Kaufman, A.J.; Xiao, S. High CO2 levels in the Proterozoic atmosphere estimated from analyses of individual microfossils. Nature 2003, 425, 279–282. [Google Scholar] [CrossRef]
- Kah, L.C.; Bartley, J.K. Effect of marine carbon reservoir size on the duration of carbon isotope excursions. Interpreting the Mesoproterozoic carbon isotope record. Geol. Soc. Am. Abstr. Programs 2004, 36, 78. [Google Scholar]
- Berner, R.A.; Kothavala, Z. GEOCARB III: A revised model of atmospheric CO2 over Phanerozoic time. Am. J. Sci. 2001, 301, 182–204. [Google Scholar] [CrossRef]
- Royer, D.L.; Berner, R.A.; Montañez, I.P.; Tabor, N.J.; Beerling, D.J. CO2 as a primary driver of Phanerozoic climate. GSA Today 2014, 14, 4–10. [Google Scholar] [CrossRef]
- Kah, L.C.; Riding, R. Mesoproterozoic carbon dioxide levels inferred from calcified cyanobacteria. Geology 2007, 35, 799–802. [Google Scholar] [CrossRef]
- Sheldon, N.D. Precambrian paleosols and atmospheric CO2 levels. Precambrian Res. 2006, 147, 148–155. [Google Scholar] [CrossRef]
- Sleep, N.H.; Zahnle, K. Carbon dioxide cycling and implications for climate on ancient Earth. J. Geophys. Res. 2001, 106, 1373–1399. [Google Scholar] [CrossRef]
- Riding, R. Cyanobacterial calcification, carbon dioxide concentrating mechanisms, and Proterozoic–Cambrian changes in atmospheric composition. Geobiology 2006, 4, 299–316. [Google Scholar] [CrossRef]
- Frost, S.H. Cenozoic reef systems of Caribbean. Prospects for paleoecologic synthesis. Am. Assoc. Petrol. Geol. Stud. Geol. 1977, 4, 93–110. [Google Scholar]
- Kempe, S.; Degens, E.T. An early soda ocean. Chem. Geol. 1985, 53, 95–108. [Google Scholar] [CrossRef]
- Crockford, P.; Halevy, I. Questioning the paradigm of a phosphate-limited Archaean biosphere. Geophys. Res. Let. 2022, 49, e2022GL099818. [Google Scholar] [CrossRef]
- Ingalls, M.; Grotzinger, J.P.; Present, T.; Rasmussen, B.; Fischer, W.W. Carbonate-associated phosphate (CAP) indicates elevated phosphate availability in Neoarchean shallow marine environments. Geophys. Res. Let. 2022, 49, e2022GL098100. [Google Scholar] [CrossRef]
- Zahnle, K.; Arndt, N.; Cockell, C.; Halliday, A.; Nisbet, E.; Selsis, F.; Sleep, N.H. Emergence of a habitable planet. Space Sci. Rev. 2007, 129, 35–78. [Google Scholar] [CrossRef]
- Sleep, N.H. The Hadean-Archaean environment. Cold Spring Harb. Perspect. Biol. 2010, 2, a002527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Neil, J.; Carlson, R.W.; Francis, D.; Stevenson, R.K. Neodymium-142 evidence for Hadean mafic crust. Science 2009, 321, 1828–1831. [Google Scholar] [CrossRef] [Green Version]
- Sleep, N.H.; Zahnle, K.; Neuhoff, P.S. Initiation of clement surface conditions on the early Earth. Proc. Natl. Acad. Sci. USA 2001, 98, 3666–3672. [Google Scholar] [CrossRef] [Green Version]
- Lowe, D.R. Abiological origin of described stromatolites older than 3.2 Ga. Geology 1994, 22, 387–390. [Google Scholar] [CrossRef]
- Goldblatt, C.; Zahnle, K.J. Clouds and the faint young Sun paradox. Clim. Past 2011, 7, 203–220. [Google Scholar] [CrossRef] [Green Version]
- Hallock, P. Changing influences between life and limestones in Earth history. In Coral Reefs in the Anthropocene; Birkeland, C., Ed.; Springer: Dordrecht, The Netherlands, 2015; pp. 17–42. [Google Scholar]
- Mentel, M.; Martin, W. Energy metabolism among eukaryotic anaerobes in light of Proterozoic ocean chemistry. Phil. Trans. R. Soc. B-Biol. Sci. 2008, 363, 2717–2729. [Google Scholar] [CrossRef] [Green Version]
- Ward, L.M.; Rasmussen, B.; Fischer, W.W. Primary productivity was limited by electron donors prior to the advent of oxygenic photosynthesis. J. Geophys. Res. Biogeosci. 2019, 124, 211–226. [Google Scholar] [CrossRef]
- Fischer, W.W.; Hemp, J.; Johnson, J.E. Evolution of oxygenic photosynthesis. Ann. Rev. Earth Planet. Sci. 2016, 44, 647–683. [Google Scholar] [CrossRef]
- Erb, T.J.; Zarzycki, J. A short history of RubisCO: The rise and fall (?) of Nature’s predominant CO2 fixing enzyme. Curr. Opin. Biotech. 2018, 49, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Badger, M.R.; Price, G.D. CO2 concentrating mechanisms in cyanobacteria: Molecular components, their diversity and evolution. J. Exper. Bot. 2003, 54, 609–622. [Google Scholar] [CrossRef] [PubMed]
- Raven, J.A.; Beardall, J.; Sánchez-Baracaldo, P. The possible evolution and future of CO2-concentrating mechanisms. J. Exper. Bot. 2017, 68, 3701–3716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papineau, D. Global biogeochemical changes at both ends of the Proterozoic: Insights from phosphorites. Astrobiology 2010, 10, 165–181. [Google Scholar] [CrossRef]
- Large, R.R.; Halpin, J.A.; Lounejeva, E.; Danyushevsky, L.V.; Maslennikov, V.V.; Gregory, D.; Sack, P.J.; Haines, P.W.; Long, J.A.; Makoundi, C.; et al. Cycles of nutrient trace elements in the Phanerozoic ocean. Gondwana Res. 2015, 28, 1282–1293. [Google Scholar] [CrossRef]
- Pufahl, P.K.; Groat, L.A. Sedimentary and igneous phosphate deposits: Formation and exploration: An invited paper. Econ. Geol. 2017, 112, 483–516. [Google Scholar] [CrossRef]
- Paytan, A.; McLaughlin, K. The oceanic phosphorus cycle. Chem. Rev. 2007, 107, 563–576. [Google Scholar] [CrossRef]
- Poulton, S.W.; Canfield, D.E. Ferruginous conditions: A dominant feature of the ocean through Earth’s history. Elements 2011, 7, 107–112. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.; Webb, G.A.; Jell, J.S. Platform margins, reef facies and microbial carbonates; a comparison of Devonian reef complexes in the Canning Basin, Western Australia and the Guilin region, South China. Earth-Sci. Rev. 2008, 88, 33–59. [Google Scholar] [CrossRef]
- Baturin, G.N. Phosphorus cycle in the ocean. Lithol. Min. Res. 2003, 38, 101–119. [Google Scholar] [CrossRef]
- Chernoff, C.B.; Orris, G.J. Data set of world phosphate mines, deposits, and occurrences: Part A. geologic data; Part B. location and mineral economic data. U.S. Geol. Surv. Open-File Rep. 2002, C6, 680. [Google Scholar]
- Vescei, A.; Berger, W.H. Increase of atmospheric CO2 during deglaciation: Constraints on the coral reef hypothesis from patterns of deposition. Glob. Biogeochem. Cycles 2004, 18, GB1035. [Google Scholar]
- Danen-Louwerse, H.J.; Lijklema, L.; Coenraats, M. Coprecipitation of phosphate with calcium carbonate in Lake Veluwe. Water Resour. 1995, 29, 1781–1785. [Google Scholar] [CrossRef]
- Gulbrandsen, R.A.; Cremer, M. Coprecipitation of carbonate and phosphate from seawater. U.S. Geol. Surv. Prof. Pap. 1970, 700-C, C125–C126. [Google Scholar]
- Kitano, Y.; Okumura, M.; Idogaki, M. Uptake of phosphate ions by calcium carbonate. Geochem. J. 1978, 12, 29–37. [Google Scholar] [CrossRef]
- Millero, F.; Huang, F.; Zhu, X.; Liu, X.; Zhang, J.-Z. Adsorption and desorption of phosphate on calcite and aragonite in seawater. Aquat. Geochem. 2001, 7, 33–56. [Google Scholar] [CrossRef]
- Dupraz, C.; Reid, R.P.; Braissant, O.; Decho, A.W.; Norman, R.S.; Visscher, P.T. Processes of carbonate precipitation in modern microbial mats. Earth-Sci. Rev. 2009, 96, 141–162. [Google Scholar] [CrossRef]
- Stal, L.J. Coastal microbial mats: The physiology of a small-scale ecosystem. S. Afr. J. Bot. 2001, 67, 399–410. [Google Scholar] [CrossRef] [Green Version]
- Van Mooy, B.A.S.; Rocap, G.; Fredricks, H.F.; Evans, C.T.; Devol, A.H. Sulfolipids dramatically decrease phosphorus demand by picocyanobacteria in oligotrophic marine environments. Proc. Natl. Acad. Sci. USA 2006, 103, 8607–8612. [Google Scholar] [CrossRef] [Green Version]
- McConnaughey, T.A.; Whelan, J.F. Calcification generates protons for nutrient and bicarbonate uptake. Earth-Sci. Rev. 2006, 42, 95–117. [Google Scholar] [CrossRef]
- Visscher, P.T.; Hoeft, S.E.; Surgeon, T.-M.L.; Rogers, D.R.; Bebout, B.M.; Thompson, J.S.; Reid, R.P. Microelectrode measurements in stromatolites. Unraveling the Earth’s past? In Environmental Electrochemistry: Analyses of Trace Element Biogeochemistry; ACS Symposium Series 811; Taillefert, M., Rozan, T., Eds.; Oxford University Press: New York, NY, 2002; pp. 265–282. [Google Scholar]
- Wright, D.T.; Altermann, W. Microfacies development in Late Archaean stromatolites and oolites of the Ghaap Group of South Africa. Geol. Soc. Lond. Spec. Pub. 2000, 178, 51–70. [Google Scholar] [CrossRef]
- Batchelor, M.T.; Burne, R.V.; Henry, B.I.; Li, F.; Paul, P. A biofilm and organomineralisation model for the growth and limiting size of ooids. Sci. Rep. 2018, 8, 559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grotzinger, J.P.; James, N.P. Precambrian carbonates: Evolution of understanding. In Carbonate Sedimentation and Diagenesis in the Evolving Precambrian World; Grotzinger, J.P., James, N.P., Eds.; SEPM Society for Sedimentary Geology Special Publications: Tulsa, OK, USA, 2000; Volume 67, pp. 3–20. [Google Scholar]
- Brehm, U.; Krumbein, W.E.; Palinska, K.A. Biomicrospheres generate ooids in the laboratory. Geomicrobiol. J. 2006, 23, 545–550. [Google Scholar] [CrossRef]
- Diaz, M.R.; Eberli, G.P.; Blackwelder, P.; Phillips, B.; Swart, P.K. Microbially mediated organomineralization in the formation of ooids. Geology 2017, 45, 771–774. [Google Scholar] [CrossRef]
- Diaz, M.R.; Eberli, G.P. Decoding the mechanism of formation in marine ooids: A review. Earth-Sci. Rev. 2019, 190, 536–556. [Google Scholar] [CrossRef]
- Yates, K.K.; Robbins, L.L. Microbial lime-mud production and its relation to climate change. In Geological Perspectives of Global Climate Change; Gerhard, L.C., Harrison, W.H., Hanson, B.M., Eds.; American Association of Petroleum Geologists: Tulsa, OK, USA, 2001; Volume 47, pp. 267–283. [Google Scholar]
- Robbins, L.L.; Tao, Y.; Evans, C.A. Temporal and spatial distribution of whitings on Great Bahama Bank and a new lime mud budget. Geology 1997, 25, 947–950. [Google Scholar] [CrossRef]
- Robbins, L.L.; Blackwelder, P.L. Biochemical and ultrastructural evidence for the origin of whitings: A biologically induced calcium carbonate precipitation mechanism. Geology 1992, 20, 464–468. [Google Scholar] [CrossRef]
- Bhattacharya, D.; Medlin, L. The phylogeny of plastids: A review based on comparisons of small-subunit ribosomal RNA coding regions. J. Phycol. 1995, 31, 489–498. [Google Scholar] [CrossRef] [Green Version]
- Stiller, J.W.; Hall, B.D. The origin of the red algae: Implications for plastid evolution. P. Natl. Acad. Sci. USA 1997, 94, 4520–4525. [Google Scholar] [CrossRef] [Green Version]
- Demes, K.W.; Littler, M.M.; Littler, D.S. Comparative phosphate acquisition in giant-celled rhizophytic algae (Bryopsidales, Chlorophyta): Fleshy vs. calcified forms. Aquat. Bot. 2010, 92, 157–160. [Google Scholar] [CrossRef]
- Wizemann, A.; Meyer, F.W.; Westphal, H. A new model for the calcification of the green macro-alga Halimeda opuntia (Lamouroux). Coral Reefs 2015, 33, 951–964. [Google Scholar] [CrossRef]
- Wizemann, A.; Meyer, F.W.; Hofmann, L.C.; Wild, C.; Westphal, H. Ocean acidification alters the calcareous microstructure of the green macro-alga Halimeda opuntia. Coral Reefs 2015, 34, 941–954. [Google Scholar] [CrossRef]
- Nash, M.C.; Diaz-Pulido, G.; Harvey, A.S.; Adey, W. Coralline algal calcification: A morphological and process-based understanding. PLoS ONE 2019, 14, e0221396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comeau, S.; Edmunds, P.J.; Spindel, N.B.; Carpenter, R.C. The responses of eight coral reef calcifiers to increasing partial pressure of CO2 do not exhibit a tipping point. Limnol. Oceanogr. 2003, 58, 388–398. [Google Scholar] [CrossRef] [Green Version]
- Hurd, C.L.; Cornwall, C.E.; Currie, K.; Hepburn, C.D.; McGraw, C.M.; Hunter, K.A.; Boyd, P.W. Metabolically induced pH fluctuations by some coastal calcifiers exceed projected 22nd century ocean acidification: A mechanism for differential susceptibility? Glob. Chang. Biol. 2011, 17, 3254–3262. [Google Scholar] [CrossRef]
- Hay, W.W. Carbonate fluxes and calcareous nannoplankton. In Coccolithophores–From Molecular Processes to Global Impact; Thierstein, H., Young, J.R., Eds.; Springer: New York, NY, USA, 2004; pp. 509–527. [Google Scholar]
- Monteiro, F.M.; Bach, L.T.; Brownlee, C.; Bown, P.; Rickaby RE, M.; Poulton, A.J.; Tyrrell, T.; Beaufort, L.; Dutkiewicz, S.; Gibbs, S.; et al. Why marine phytoplankton calcify. Sci. Adv. 2016, 2, e1501822. [Google Scholar] [CrossRef] [Green Version]
- Young, J.R. Possible functional interpretations of coccolith morphology. Abh. Geolog. Bundesanst.-A 1987, 39, 305–313. [Google Scholar]
- Paasche, E. Roles of nitrogen and phosphorus in coccolith formation in Emiliania huxleyi (Prymnesiophyceae). Eur. J. Phycol. 1998, 33, 33–42. [Google Scholar] [CrossRef]
- Sheward, R.M.; Poulton, A.J.; Gibbs, S.J.; Daniels, C.J.; Bown, P.R. Physiology regulates the relationship between coccosphere geometry and growth phase in coccolithophores. Biogeosciences 2017, 14, 1493–1509. [Google Scholar] [CrossRef] [Green Version]
- Brownlee, C.; Wheeler, G.L.; Taylor, A.R. Coccolithophore biomineralization: New questions, new answers. Sem. Cell Devel. Biol. 2015, 46, 11–16. [Google Scholar] [CrossRef] [Green Version]
- Walker, C.E.; Heath, S.; Salmon, D.L.; Smirnoff, N.; Langer, G.; Taylor, A.R.; Brownlee, C.; Wheeler, G.L. An extracellular polysaccharide-rich organic layer contributes to organization of the coccosphere in coccolithophores. Front. Mar. Sci. 2018, 5, 306. [Google Scholar] [CrossRef]
- Sviben, S.; Gal, A.; Hood, M.A.; Bertinetti, L.; Politi, Y.; Bennet, M.; Krishnamoorthy, P.; Schertel, A.; Wirth, R.; Sorrentino, A.; et al. A vacuole-like compartment concentrates a disordered calcium phase in a key coccolithophorid alga. Nat. Com. 2016, 7, 11228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gal, A.; Sviben, S.; Wirth, R.; Schreiber, A.; Lassalle-Kaiser, B.; Faivre, D.; Scheffel, A. Trace-element incorporation into intracellular pools uncovers calcium-pathways in a coccolithophore. Adv. Sci. 2017, 4, 1700088. [Google Scholar] [CrossRef] [PubMed]
- Pomar, L.; Hallock, P. Carbonate factories: A conundrum in sedimentary geology. Earth-Sci. Rev. 2008, 87, 134–169. [Google Scholar] [CrossRef]
- Newell, N.D. Mass extinctions at the end of the Cretaceous Period. Science 1965, 149, 922–924. [Google Scholar] [CrossRef]
- Keller, G.; Mateo, P.; Punekar, J.; Khozyem, H.; Gertsch, B.; Spangenberg, J.; Bitchong, A.M.; Adatte, T. Environmental Changes during the Cretaceous-Paleogene Mass Extinction and Paleocene-Eocene Thermal Maximum: Implications for the Anthropocene. Gondwana Res. 2018, 56, 69–89. [Google Scholar] [CrossRef]
- Hallock, P. Fluctuations in the trophic resource continuum: A factor in global diversity cycles? Paleoceanography 1987, 2, 457–471. [Google Scholar] [CrossRef]
- d’Halloy, J.-J. Observations sur un essai de carte géologique de la France, des Pays-Bas, et des contrées voisines [Observations on a trial geological map of France, the Low Countries, and neighboring countries]. Ann. Des Mines 1822, 7, 353–376. [Google Scholar]
- Lear, C.H.; Rosenthal, Y.; Wright, J.D. The closing of a seaway: Ocean water masses and global climate change. Earth Planet. Sci. Lett. 2003, 210, 425–436. [Google Scholar] [CrossRef]
- Adams, C.G.; Lee, D.E.; Rosen, B.R. Conflicting isotopic and biotic evidence for tropical sea-surface temperatures during the Tertiary. Palaeogeog. Palaeoecol. 1990, 77, 289–313. [Google Scholar] [CrossRef]
- Stanley, S.M.; Hardie, L.A. Secular oscillations in the carbonate mineralogy of reef-building and sediment-producing organisms driven by tectonically forced shifts in seawater chemistry. Palaeogeog. Palaeoecol. 1998, 144, 3–19. [Google Scholar] [CrossRef]
- Pearson, P.N.; Palmer, M.R. Atmospheric carbon dioxide concentrations over the past 60 million years. Nature 2000, 406, 695–699. [Google Scholar] [CrossRef] [PubMed]
- Pochon, X.; Montoya-Burgos, J.I.; Stadelmann, B.; Pawlowski, J. Molecular phylogeny, evolutionary rates, and divergence timing of the symbiotic dinoflagellate genus Symbiodinium. Mol. Phylogenet. Evol. 2006, 38, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Wooldridge, S.A. Breakdown of the coral-algae symbiosis: Towards formalising a linkage between warm-water bleaching thresholds and the growth rate of the intracellular zooxanthellae. Biogeosciences 2013, 10, 1647–1658. [Google Scholar] [CrossRef]












Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pomar, L.; Hallock, P.; Mateu-Vicens, G.; Baceta, J.I. Why Do Bio-Carbonates Exist? J. Mar. Sci. Eng. 2022, 10, 1648. https://doi.org/10.3390/jmse10111648
Pomar L, Hallock P, Mateu-Vicens G, Baceta JI. Why Do Bio-Carbonates Exist? Journal of Marine Science and Engineering. 2022; 10(11):1648. https://doi.org/10.3390/jmse10111648
Chicago/Turabian StylePomar, Luis, Pamela Hallock, Guillem Mateu-Vicens, and Juan I. Baceta. 2022. "Why Do Bio-Carbonates Exist?" Journal of Marine Science and Engineering 10, no. 11: 1648. https://doi.org/10.3390/jmse10111648
APA StylePomar, L., Hallock, P., Mateu-Vicens, G., & Baceta, J. I. (2022). Why Do Bio-Carbonates Exist? Journal of Marine Science and Engineering, 10(11), 1648. https://doi.org/10.3390/jmse10111648