Computational Models of the NF-KB Signalling Pathway



Abstract
:1. Introduction
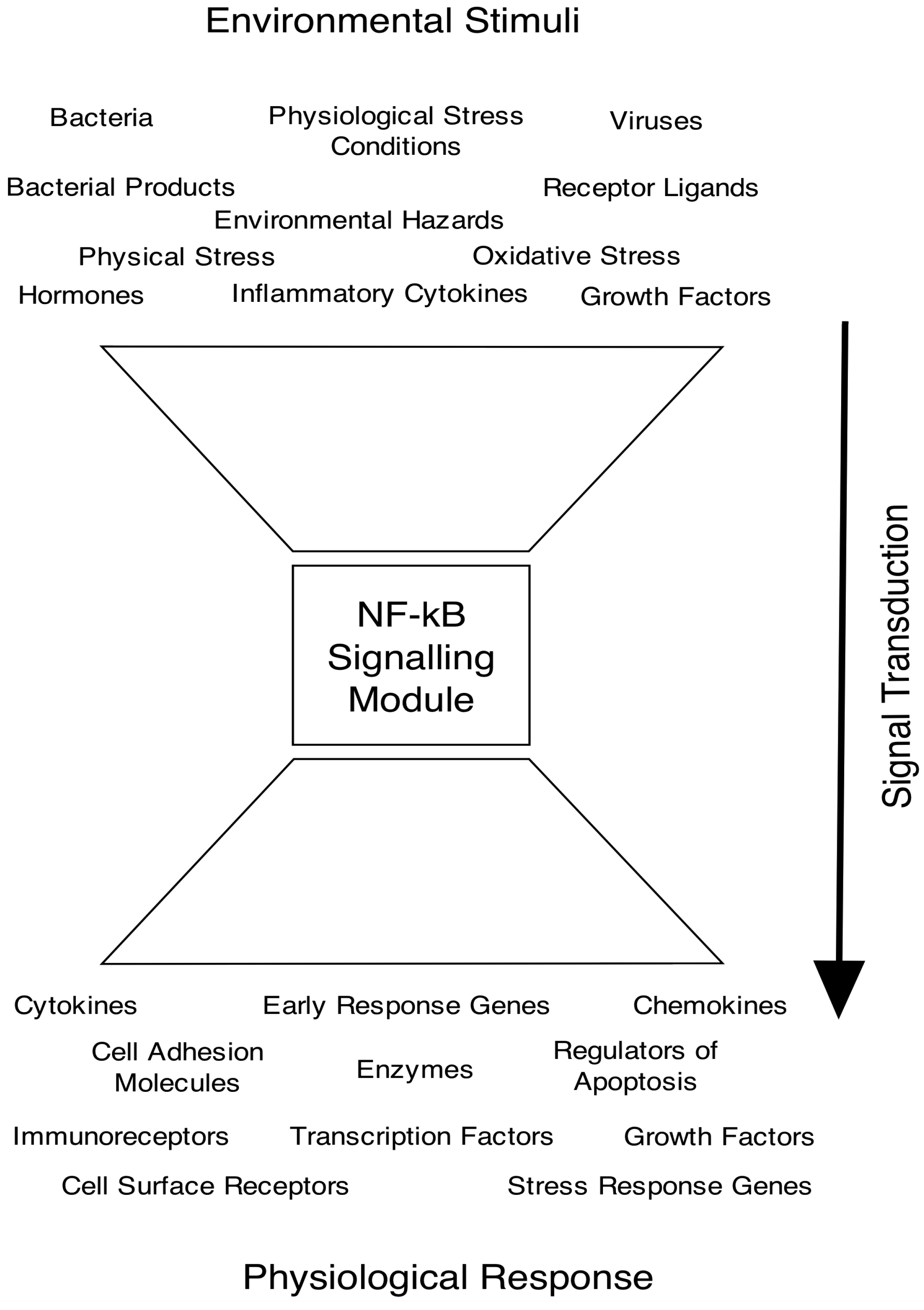
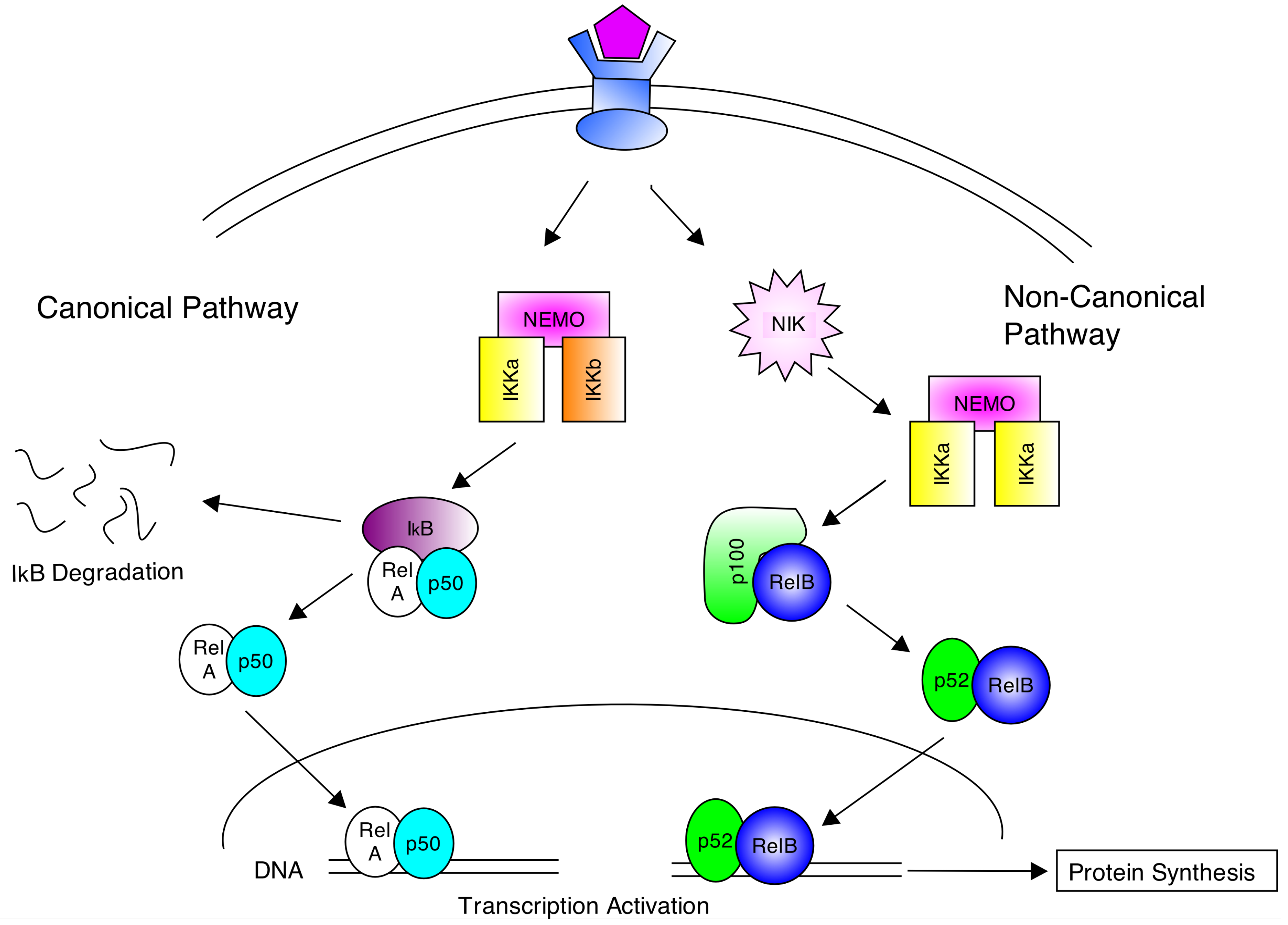
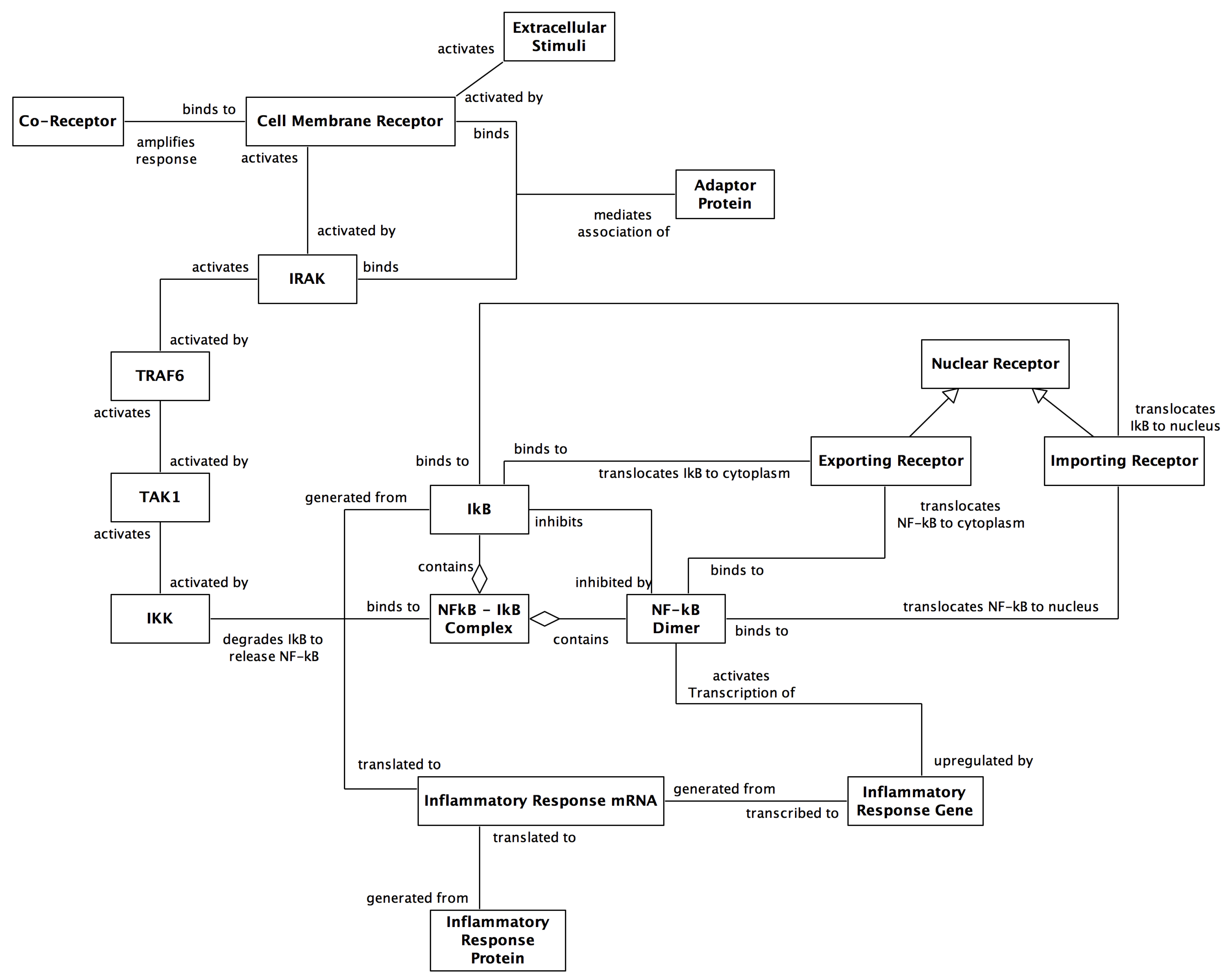
2. The NF-κB Signalling Pathway



3. Existing Computational Models of NF-κB
3.1. Deterministic Differential Equation Models
3.2. Semi-Stochastic (Hybrid) Differential Equation Models
3.3. Agent-Based Models
3.4. Peer-Validation of These Computational Models
3.5. Minimal Models
4. Discussion and Perspectives

{kind=link}
{kind=link}
{kind=link}
| Year | Authors | Modelling Paradigm | Stimuli | Cell Type | Level | Pathway Components | Key Advances |
|---|---|---|---|---|---|---|---|
| 2000 | Carlotti et al. [34] | Deterministic | IL-1 | Monkey Smooth Muscle | Single-Cell | NF-κB, IκBα | First known model of NF-κB and IκBα dissociation and translocation dynamics. |
| 2002 | Hoffmann et al. [35] | Deterministic | TNFα | Mouse Fibroblast | Population | IKK, NF-κB, IκBα, IκBβ, IκBϵ | First model of TNFα induced activation of the signalling pathway. |
| 2004 | Lipniacki et al. [47] | Deterministic | - | - | Population | IKK, NF-κB, IκBα, A20 | Incorporated 2-feedback loop (IκBα and A20) to Hoffmann model and parameter refit. |
| 2004 | Nelson et al. [45] | Deterministic | TNFα | Human HeLa and SK-N-AS | Single-Cell | IKK, NF-κB, IκBα | First model to show NF-κB oscillations at single-cell level. Augmented Hoffmann model using single-cell data for calibration and validation. |
| 2006 | Pogson et al. [69] | Agent-Based | IL-1 | - | Single-Cell | TIR, IKK, NF-κB, Nuclear Membrane Transporters | First known agent-based model of signalling pathway. |
| 2006 | Lipniacki et al. [59] | Semi-Stochastic | TNFα | - | Single-Cell | IKK, NF-κB, IκBα, A20, mRNA, genes | Stochastic transcription to generate cellular heterogeneity. |
| 2006 | Cheong et al. [54] | Deterministic | TNFα | Mouse Embryonic Fibroblast | Population | IKK, NF-κB, IκBα | Incorporated temporal profiles of IKK activation. |
| 2006 | Kearns et al. [40] | Deterministic | TNFα | Mouse Embryonic Fibroblast | Population | IKK, NF-κB, IκBα, IκBβ, IκBϵ | Reimplementation of Hoffmann model in Matlab. Showed that IκBϵ provides negative feedback control of NF-κB oscillations. |
| 2007 | Basak et al. [42] | Deterministic | LTβR | Mouse Embryonic Fibroblast | Population | IKK1, IKK2, NF-κB, IκBδ | Incorporated fourth IκB inhibtor (nfkb2 p100, or IκBδ). Models cross-talk of canonical NF-κB/RelA activity in response to non-canonical IKK1-induction. |
| 2007 | O’Dea et al. [38] | Deterministic | TNFα | Mouse Embryonic Fibroblast | Population | IKK, NF-κB, IκBα, IκBβ, IκBϵ | Distinguished between NF-κB bound and free IκB pools. Investigated steady-state regulation of NF-κB signalling module. |
| 2007 | Lipniacki et al. [60] | Semi-Stochastic | TNFα | - | Single-Cell | TNFR1, IKKK, IKK, NF-κB, IκBα, A20, mRNA, genes | Incorporated stochastic switches for cell membrane receptor activation by TNFα ligand, and transcription of IκBα and A20 genes. |
| 2008 | Pogson et al. [50] | Agent-Based | IL-1 | Human HeLa | Single-Cell | TIR, IKK, NF-κB, IκBα, genes, cytoskeleton, Nuclear Membrane Transporters | Updated earlier ABM with transcription and translation of IκBα to provide negative feedback. Also incorporated sequestration of excess IκBα to cytoskeleton. |
| 2009 | Shih et al. [43] | Deterministic | TNFα, IL-1, LPS | Mouse Embryonic Fibroblast | Population | IKK, NF-κB, IκBα, IκBβ, IκBδ, IκBϵ | Modelled the 4 distinct IκB variants and utilised TNFα, IL-1 and LPS stimulation to determine signal specificity for the negative feedback loops. |
| 2009 | Ashall et al. [63] | Semi-Stochastic | TNFα | SK-N-AS and Mouse Embryonic Fibroblast | Single-Cell | IKK, NF-κB, IκBα, IκBϵ, A20, genes | Incorporated delayed stochastic transcription of IκBϵ, and stochastic transcription of IκBα and A20. |
| 2010 | Tay et al. [56] | Semi-Stochastic | TNFα | Mouse Fibroblast | Single-Cell | TNFR1, IKKK, IKK, NF-κB, IκBα, A20, mRNA, genes | Updated to reflect the heterogeneous, digital response of single cells, and the analogue dynamics of peak NF-κB intensity, response time and oscillation number, to modulate the overall population response. |
| 2010 | Paszek et al. [41] | Semi-Stochastic | TNFα | Mouse Embryonic Fibroblast | Population | IKKK, IKK, NF-κB, IκBα, IκBϵ, A20, mRNA, genes | Aggregated large-scale single-cell dynamics to show that cellular heterogeneity (for timings of NF-κB oscillations) is important for population-level robustness. |
| 2010 | Turner et al. [57] | Semi-Stochastic | TNFα | SK-N-AS | Single-Cell | TNFR1, IKK, NF-κB, IκBα, A20, mRNA, genes | Updated Ashall model to incorporate stochastic processes for IKK activation. |
| 2012 | Fallahi-Sichani et al. [62] | Semi-Stochastic | TNFα | Macrophage | Population | TNFR1, IKKK, IKK, NF-κB, IκBα, A20, mRNA, genes | Merged their previous ABM of granuloma formation (did not model NF-κB) with the ODE model of Tay, to develop a multi-scale hybrid model. |
| 2013 | Choudhary et al. [58] | Deterministic | TNFα | Human Epithelial | Population | TRAF1, NIK, TRAF2, NF-κB, IκBα, IκBδ, A20, mRNA, genes | Integrated canonical and non-canonical pathways, using TRAF1-NIK as a feed-forward complex. |
| 2013 | Pekalski et al. [61] | Semi-Stochastic | TNFα | 3T3 | Single-Cell | TNFR1, IKKK, IKK, NF-κB, IκBα, A20, mRNA, genes, TNFα | Built on Tay model to integrate negative and positive feedback loops due to IκBα and A20, and TNFα respectively. |
Acknowledgments
Conflicts of Interest
References
- Bhalla, U.S.; Iyengar, R. Emergent properties of networks of biological signaling pathways. Science 1999, 283, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Barkai, N.; Leibler, S. Robustness in simple biochemical networks. Nature 1997, 387, 913–917. [Google Scholar] [PubMed]
- Kitano, H. Systems biology: A brief overview. Science 2002, 295, 1662–1664. [Google Scholar] [CrossRef] [PubMed]
- Oltvai, Z.N.; Barabasi, A.L. Life’s complexity pyramid. Science 2002, 298, 763–764. [Google Scholar] [CrossRef] [PubMed]
- Wolkenhauer, O. Systems biology: The reincarnation of systems theory applied in biology? Brief. Bioinform. 2001, 2, 258–270. [Google Scholar] [CrossRef] [PubMed]
- Kitano, H. Computational systems biology. Nature 2002, 420, 206–210. [Google Scholar] [CrossRef] [PubMed]
- Mardinoglu, A.; Nielsen, J. Systems medicine and metabolic modelling. J. Intern. Med. 2012, 271, 142–154. [Google Scholar] [CrossRef] [PubMed]
- Perelson, A.S.; Weisbuch, G. Immunology for physicists. Rev. Mod. Phys. 1997, 69, 1219–1267. [Google Scholar] [CrossRef]
- Alberts, B.; Bray, D.; Lewis, J.; Raff, M.; Roberts, K.; Watson, J.D. Molecular Biology of the Cell; Garland Publishing Inc.: New York, NY, USA, 1994. [Google Scholar]
- Tian, B.; Brasier, A.R. Identification of a nuclear factor kappa b-dependent gene network. Recent Prog. Horm. Res. 2003, 58, 95–130. [Google Scholar] [CrossRef] [PubMed]
- Pahl, H.L. Activators and target genes of Rel/NF-κB transcription factors. Oncogene 1999, 18, 6853–6866. [Google Scholar] [CrossRef] [PubMed]
- Sen, R.; Baltimore, D. Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell 1986, 46, 705–716. [Google Scholar] [CrossRef]
- Oeckinghaus, A.; Ghosh, S. The NF-κB family of transcription factors and its regulation. In NF-κB A Network Hub Controlling Immunity, Inflammation, and Cancer; Karin, M., Staudt, L.M., Eds.; Cold Spring Harbour Perspectives in Biology, Cold Spring Harbour Press: New York, NY, USA, 2009; pp. 5–18. [Google Scholar]
- Karin, M.; Ben-Neriah, Y. Phosphorylation meets ubiquitination: The control of NF-κB activity. Annu. Rev. Immunol. 2000, 18, 621–663. [Google Scholar] [CrossRef] [PubMed]
- Stylianou, E.; O’Neill, L.A.J.; Rawlinson, L.; Edbrooke, M.R.; Woo, P.; Saklatvala, J. Interleukin 1 induces NF-κB through its type I but not its type II receptor in lymphocytes. J. Biol. Chem. 1992, 267, 15836–15841. [Google Scholar] [PubMed]
- Bubici, C.; Papa, S.; Dean, K.; Franzoso, G. Mutual cross-talk between reactice oxygen species and nuclear factor-kappa B: Molecular basis and biological basis. Oncogene 2006, 25, 6731–6748. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.J.; Liu, Z.G. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Hiscott, J.; Nguyen, T.L.; Arguello, M.; Nakhaei, P.; Paz, S. Manipulation of the nuclear factor-κB pathway and the innate immune response by viruses. Oncogene 2006, 25, 6844–6867. [Google Scholar] [CrossRef] [PubMed]
- Laflamme, N.; Rivest, S. Toll-Like receptor 4: The missing link of cerebral innate immune response triggered by circulating gram-negative bacterial cell wall components. FASEB J. 2001, 15, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Bender, K.; Gottlicher, M.; Whiteside, S.; Rahmsdorf, H.; Herrlich, P. Sequential DNA damage-independent and -dependent activation of NF-κB by UV. EMBO J. 1998, 17, 5170–5181. [Google Scholar] [CrossRef] [PubMed]
- Ganguli, A.; Persson, L.; Palmer, I.R.; Evans, I.; Yang, L.; Smallwood, R.; Black, R.; Qwarnstrom, E.E. Distinct NF-κB regulation by shear stress through Ras-dependent IκBα oscillations: Real time analysis of flow-mediated activation in live cells. Circ. Res. 2005, 96, 626–634. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, Z.H.; Deitch, E.A.; Davidson, M.T.; Szabo, C.; Vizi, E.S.; Hasko, G. Disruption of the actin cytoskeleton results in nuclear factor-κB activation and inflammatory mediator production in cultured human epithelial cells. J. Cell. Physiol. 2004, 200, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Brasier, A.R. The NF-κB regulatory network. Cardiovasc. Toxicol. 2006, 6, 111–130. [Google Scholar] [CrossRef] [PubMed]
- Ben-Neriah, Y.; Karin, M. Inflammation meets Cancer, with NF-kappaB as the Matchmaker. Nat. Immunol. 2011, 12, 715–723. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; May, M.J.; Kopp, E.B. NF-κB and Rel Proteins: Evolutionarily conserved mediators of immune response. Annu. Rev. Immunol. 1998, 16, 225–260. [Google Scholar] [CrossRef] [PubMed]
- Siebenlist, U.; Franzoso, G.; Brown, K. Structure, regulation and function of NF-κB. Annu. Rev. Cell Biol. 1994, 10, 405–455. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, A.S. The NF-κB and IκB Proteins: New discoveries and insights. Annu. Rev. Immunol. 1996, 14, 649–681. [Google Scholar] [CrossRef] [PubMed]
- Karin, M. The beginning of the end: IκB Kinase (IKK) and NF-κB Activation. J. Biol. Chem. 1999, 274, 27339–27342. [Google Scholar] [CrossRef] [PubMed]
- Karin, M. How NF-κB is activated: The role of the IκB Kinase (IKK) complex. Oncogene 1999, 18, 6867–6874. [Google Scholar] [CrossRef] [PubMed]
- Senftleben, U.; Cao, Y.; Xiao, G.; Greten, F.R.; Krahn, G.; Bonizzi, G.; Chen, Y.; Hu, Y.; Fong, A.; Sun, S.C.; et al. Activation by IKKα of a second, evolutionary conserved, NF-κB signaling pathway. Science 2001, 293, 1495–1499. [Google Scholar] [CrossRef] [PubMed]
- Murphy, K.; Travers, P.; Walport, M. Janeway’s Immunobiology, 7th ed.; Garland Science: New York, NY, USA, 2008. [Google Scholar]
- Babur, O.; Demir, E.; Gonen, M.; Sander, C.; Dogrusoz, U. Discovering modulators of gene expression. Nucleic Acids Res. 2010, 38, 5648–5656. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhao, Y.; Tian, B.; Jamaluddin, M.; Mitra, A.; Yang, J.; Rowicka, M.; Brasier, A.R.; Kudlicki, A. Modulation of gene expression regulated by the transcription factor NF-κB/RelA. J. Biol. Chem. 2014, 289, 11927–11944. [Google Scholar] [CrossRef] [PubMed]
- Carlotti, F.; Dower, S.K.; Qwarnstrom, E.E. Dynamic shuttling of nuclear factor κB between the nucleus and cytoplasm as a consequence of inhibitor dissociation. J. Biol. Chem. 2000, 275, 41028–41034. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, A.; Levchenko, A.; Scott, M.L.; Baltimore, D. The IκB - NF-κB signaling module: Temporal control and selective gene activation. Science 2002, 298, 1241–1245. [Google Scholar] [CrossRef] [PubMed]
- Mendes, P. GEPASI: A software package for modelling the dynamics, steady staes and control of biochemical and other systems. Comput. Appl. Biosci. 1993, 9, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Mendes, P. Biochemistry by numbers: Simulation of biochemical pathways with Gepasi 3. Trends Biol. Sci. 1997, 22, 361–363. [Google Scholar] [CrossRef]
- O’Dea, E.L.; Barken, D.; Peralta, R.Q.; Tran, K.T.; Werner, S.L.; Kearns, J.D.; Levchenko, A.; Hoffmann, A. A homeostatic model of IκB metabolism to control constitutive NF-κB activity. Mol. Syst. Biol. 2007, 3, 111. [Google Scholar] [CrossRef] [PubMed]
- Cheong, R.; Hoffmann, A.; Levchenko, A. Understanding NF-κB signalling via mathematical modeling. Mol. Syst. Biol. 2008, 4, 192. [Google Scholar] [CrossRef] [PubMed]
- Kearns, J.D.; Basak, S.; Werner, S.L.; Huang, C.S.; Hoffmann, A. IκBϵ provides negative feedback to control NF-κB oscillations, signaling dynamics, and inflammatory gene expression. J. Cell Biol. 2006, 173, 659–664. [Google Scholar] [CrossRef] [PubMed]
- Paszek, P.; Ryan, S.; Ashall, L.; Sillitoe, K.; Harper, C.V.; Spiller, D.G.; Rand, D.A.; White, M.R.H. Population robustness arising from cellular heterogeneity. Proc. Natl. Acad. Sci. USA 2010, 107, 11644–11649. [Google Scholar] [CrossRef] [PubMed]
- Basak, S.; Kim, H.; Kearns, J.D.; Tergaonkar, V.; O’Dea, E.; Werner, S.L.; Benedict, C.A.; Ware, C.F.; Ghosh, G.; Verma, I.M.; et al. A fourth IkappaB protein within the NF-kappaB signaling module. Cell 2007, 128, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Shih, V.F.S.; Kearns, J.D.; Basak, S.; Savinova, O.V.; Ghosh, G.; Hoffmann, A. Kinetic control of negative feedback regulators of NF-κB/RelA determines their pathogen- and cytokine-receptor signaling specificity. Proc. Natl. Acad. Sci. USA 2009, 106, 9619–9624. [Google Scholar] [CrossRef] [PubMed]
- Shih, V.F.S.; Davis-Turak, J.; Macal, M.; Huang, J.Q.; Ponomarenko, J.; Kearns, J.D.; Yu, T.; Fagerlund, R.; Asagiri, M.; Zuniga, E.I.; et al. Control of RelB during dendritic cell activation integrates canonica and noncanonical NF-κB pathways. Nat. Immunol. 2012, 13, 1162–1172. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.E.; Ihekwaba, A.E.C.; Elliott, M.; Johnson, J.R.; Gibney, C.A.; Foreman, B.E.; Nelson, G.; See, V.; Horton, C.A.; Spiller, D.G.; et al. Oscillations in NF-κB signaling control the dynamics of gene expression. Science 2004, 306, 704–708. [Google Scholar] [CrossRef] [PubMed]
- Lucey, B.P.; Nelson-Rees, W.A.; Hutchins, G.M. Henrietta Lacks, HeLa cells, and Cell culture contamination. Arch. Pathol. Lab. Med. 2009, 133, 1463–1467. [Google Scholar] [PubMed]
- Lipniacki, T.; Paszek, P.; Brasier, A.R.; Luxon, B.; Kimmel, M. Mathematical model of NF-κB regulatory module. J. Theor. Biol. 2004, 228, 195–215. [Google Scholar] [CrossRef] [PubMed]
- Carlotti, F.; Chapman, R.; Dower, S.K.; Qwarnstrom, E.E. Activation of nuclear factor κB in single living cells. J. Biol. Chem. 1999, 274, 37941–37949. [Google Scholar] [CrossRef] [PubMed]
- Rice, N.R.; Ernst, M.K. In vivo control of NF-κB activation by IκBα. EMBO J. 1993, 12, 4685–4695. [Google Scholar] [PubMed]
- Pogson, M.; Holcombe, M.; Smallwood, R.; Qwarnstrom, E.E. Introducing spatial information into predictive NF-κB modelling — An agent-based approach. PLoS One 2008, 3, e2367. [Google Scholar] [CrossRef] [PubMed]
- Krikos, A.; Laherty, C.D.; Dixit, V.M. Transcriptional activation of the tumor necrosis factor alpha-inducible zinc finger protein, A20, is mediated by kappa B elements. J. Biol. Chem. 1992, 267, 17971–17976. [Google Scholar] [PubMed]
- Yang, L.; Chen, H.; Qwarnstrom, E. Degradation of IκBα is limited by a postphosphorylation/ubiquitination event. Biochem. Biophys. Res. Commun. 2001, 285, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.G.; Boone, D.L.; Chai, S.; Libby, S.L.; Chien, M.; Lodolce, J.P.; Ma, A. Failure to regulate TNF-induced NF-κB and cell death responses in A20-deficient mice. Science 2000, 289, 2350–2354. [Google Scholar] [CrossRef] [PubMed]
- Cheong, R.; Bergmann, A.; Werner, S.L.; Regal, J.; Hoffmann, A.; Levchenko, A. Transient IκB kinase activity mediates temporal NF-κB dynamics in response to wide range of tumour necrosis factor-α doses. J. Biol. Chem. 2006, 281, 2945–2950. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, B.E.; Levchenko, A.; Meyerowitz, E.M.; Wold, B.J.; Mjolsness, E.D. Cellerator: Extending a computer algebra system to include biochemical arrows for signal transduction simulations. Bioinformatics 2003, 19, 677–678. [Google Scholar] [CrossRef] [PubMed]
- Tay, S.; Hughey, J.J.; Lee, T.K.; Lipniacki, T.; Quake, S.R.; Covert, M.W. Single-cell NF-κB dynamics reveal digital activation and analogue information processing. Nature 2010, 466, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Turner, D.A.; Paszek, P.; Woodcock, D.J.; Nelson, D.E.; Horton, C.A.; Wang, Y.; Spiller, D.G.; Rand, D.A.; White, M.R.H.; Harper, C.V. Physiological levels of TNFα stimulation induce stochastic dynamics of NF-κB responses in single living cells. J. Cell Sci. 2010, 123, 2834–2843. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, S.; Kalita, M.; Fang, L.; Patel, K.; Tian, B.; Zhao, Y.; Edeh, C.B.; Brasier, A.R. Inducible TNF receptor associated factor-1 expression coules the canonical to the non-canonical NF-κB pathway in TNF stimulation. J. Biol. Chem. 2013, 288, 14612–14623. [Google Scholar] [CrossRef] [PubMed]
- Lipniacki, T.; Paszek, P.; Brasier, A.R.; Luxon, B.A.; Kimmel, M. Stochastic regulation in early immune response. Biophys. J. 2006, 90, 725–742. [Google Scholar] [CrossRef] [PubMed]
- Lipniacki, T.; Puszynski, K.; Paszek, P.; Brasier, A.R. Single TNFα trimers mediating NF-κB activation: Stochastic robustness of NF-κB signaling. BMC Bioinform. 2007, 8, 376. [Google Scholar] [CrossRef] [PubMed]
- Pekalski, J.; Zuk, P.J.; Kochanczyk, M.; Junkin, M.; Kellogg, R.; Tay, S.; Lipniacki, T. Spontaneous NF-κB activation by autocrine TNFα signaling: A computational analysis. PLoS One 2013, 8, e78887. [Google Scholar] [CrossRef] [PubMed]
- Fallahi-Sichani, M.; Kirschner, D.E.; Linderman, J.J. NF-κB Signaling dynamics play a key role in infection control in tuberculosis. Front. Physiol. 2012, 3, 170. [Google Scholar] [CrossRef] [PubMed]
- Ashall, L.; Horton, C.A.; Nelson, D.E.; Paszek, P.; Harper, C.V.; Sillitoe, K.; Ryan, S.; Spiller, D.G.; Unitt, J.F.; Broomhead, D.S.; et al. Pulsatile stimulation determines timing and specificity of NF-κB-dependent transcription. Science 2009, 324, 242–246. [Google Scholar] [CrossRef] [PubMed]
- Acerbi, E.; Decraene, J.; Gouaillard, A. Computational reconstruction of biochemical networks. In Proceedings of the 15th International Conference on Information Fusion, Singapore, Singapore, 9–12 July 2012; IEEE: New York, NY, USA, 2012; pp. 1134–1141. [Google Scholar]
- Cohn, M.; Mata, J. Quantitative modeling of immune responses. Immunol. Rev. 2007, 216, 5–8. [Google Scholar] [PubMed]
- Stark, J.; Chan, C.; George, A.J. Oscillations in the immune system. Immunol. Rev. 2007, 216, 213–231. [Google Scholar] [PubMed]
- Kam, N.; Cohen, I.R.; Harel, D. The immune system as a reactive system: Modeling T cell activation with statecharts. In Proceedings of the Symposium on Human Centric Computing Languages and Environments Conference, Stresa, Italy, 5–7 September 2001; IEEE: New York, NY, USA, 2001. [Google Scholar]
- Macal, C.M.; North, M.J. Tutorial on agent-based modeling and simulation. In Proceedings of the Winter Simulation Conference, New Orleans, LA, USA, 4–7 December 2005; Kuhl, M.E., Steiger, N.M., Armstrong, F.B., Jones, J.A., Eds.; ACM: New York, NY, USA, 2005; pp. 2–15. [Google Scholar]
- Pogson, M.; Smallwood, R.; Qwarnstrom, E.E.; Holcombe, M. Formal agent-based modelling of intracellular chemical interactions. BioSystems 2006, 85, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Barnard, J.; Whitworth, J.; Woodward, M. Communicating X-Machines. Inf. Softw. Technol. 1996, 38, 401–407. [Google Scholar] [CrossRef]
- Terry, A.J.; Chaplain, M.A.J. Spatio-temporal modelling of the NF-κB intracellular signalling pathway: The roles of diffusion, active transport, and cell geometry. J. Theor. Biol. 2011, 290, 7–26. [Google Scholar] [CrossRef] [PubMed]
- Ohshima, D.; Inoue, J.I.; Ichikawa, K. Roles of spatial parameters on the oscillation of nuclear NF-κB: Computer simulations of a 3D spherica cell. PLoS One 2012, 7, e46911. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Ross, K.; Qwarnstrom, E.E. RelA control of IκBα phosphorylation. J. Biol. Chem. 2003, 278, 30881–30888. [Google Scholar] [CrossRef] [PubMed]
- Barken, D.; Wang, C.J.; Kearns, J.; Cheong, R.; Hoffmann, A.; Levchenko, A. Comment of “oscillations in NF-κB signaling control the dynamics of gene expression”. Science 2005, 308, 52. [Google Scholar] [CrossRef] [PubMed]
- Sung, M.H.; Li, N.; Lao, Q.; Gottschalk, R.A.; Hager, G.L.; Fraser, I.D.C. Switching of the relative dominance between feedback mechanisms in lipopolysaccharide-Induced NF-κB signaling. Sci. Signal. 2014, 7, ra6. [Google Scholar] [CrossRef] [PubMed]
- Sung, M.H.; Salvatore, L.; de Lorenzi, R.; Indrawan, A.; Pasparakis, M.; Hager, G.L.; Bianchi, M.E.; Agresti, A. Sustained oscillations of NF-κB produce distinct genome scanning and gene expression profiles. PLoS One 2009, 4, e7163. [Google Scholar] [CrossRef] [PubMed]
- Zambrano, S.; Bianchi, M.E.; Agresti, A. High-throughput analysis of NF-κB dynamics in single cells reveal basal nuclear localization of NF-κB and spontaneous activation of oscillations. PLoS One 2014, 9, e90104. [Google Scholar] [CrossRef] [PubMed]
- Hayot, F.; Jayaprakash, C. NF-κB oscillations and cell-to-cell variability. J. Theor. Biol. 2006, 240, 583–591. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, D.T. Exact stochastic simulation of coupled chemical reactions. J. Phys. Chem. 1977, 81, 2340–2361. [Google Scholar] [CrossRef]
- Ihekwaba, A.E.C.; Broomhead, D.S.; Grimley, R.L.; Benson, N.; Kell, D.B. Sensitivity analysis of parameters controlling oscillatory signalling in the NF-κB pathway: The roles of IKK and IκBα. Syst. Biol. 2004, 1, 93–103. [Google Scholar] [CrossRef]
- Ihekwaba, A.E.C.; Broomhead, D.S.; Grimley, R.; Benson, N.; White, M.R.; Kell, D.B. Synergistic control of oscillations in the nf-kappab signalling pathway. IEEE Proc. Syst. Biol. 2005, 152, 153–160. [Google Scholar] [CrossRef]
- Mathes, E.; O’Dea, E.L.; Hoffmann, A.; Ghosh, G. NF-κB dictates the degradation pathway of IκBα. EMBO J. 2008, 27, 1357–1367. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Paszek, P.; Horton, C.A.; Kell, D.B.; White, M.R.H.; Broomhead, D.S.; Muldoon, M.R. Interactions among oscillatory pathways in NF-κB signalling. BMC Syst. Biol. 2011, 5, 23. [Google Scholar] [CrossRef] [PubMed]
- Yue, H.; Brown, M.; Knowles, J.; Wang, H.; Broomhead, D.S.; Kell, D.B. Insights into the behaviour of systems biology models from dynamic sensitivity and identifiability analysis: A case study of an NF-κB signalling pathway. Mol. BioSyst. 2006, 2, 640–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joo, J.; Plimpton, S.; Martin, S.; Swiler, L.; Faulon, J.L. Sensitivity analysis of a computational model of the IKK-NF-κB-IκBα-A20 signal transduction network. Wiley: New York, NY, USA, 2007; Volume 1115, pp. 221–239. [Google Scholar]
- Krishna, S.; Jensen, M.H.; Sneppen, K. Minimal model of spiky oscillations in NF-κB signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 10840–10845. [Google Scholar] [CrossRef] [PubMed]
- Yde, P.; Mengel, B.; Jensen, M.H.; Krishna, S.; Trusina, A. Modeling the NF-κB mediated inflammatory response predicts cytokine waves in tissue. BMC Syst. Biol. 2011, 5, 115. [Google Scholar] [CrossRef] [PubMed]
- Werner, S.L.; Barken, D.; Hoffmann, A. Stimulus specificity of gene expression programs determined by temporal control of IKK activity. Science 2005, 309, 1857–1861. [Google Scholar] [CrossRef] [PubMed]
- Longo, D.M.; Selimkhanov, J.; Kearns, J.D.; Hasty, J.; Hoffmann, A.; Tsimring, L.S. Dual delayed feedback provides sensitivity and robustness to the NF-κB signaling module. PLoS Comput. Biol. 2013, 9, e1003112. [Google Scholar] [CrossRef] [PubMed]
- Zambrano, S.; Bianchi, M.E.; Agresti, A. A simple model of NF-κB dynamics reproduces experimental observations. J. Theor. Biol. 2014, 347, 44–53. [Google Scholar] [CrossRef] [PubMed]
- West, S.; Bridge, L.J.; White, M.R.H.; Paszek, P.; Biktashev, V.N. A method of “speed coefficients” for biochemical model reduction applied to the NF-κB system. J. Math. Biol. 2014. [Google Scholar] [CrossRef] [Green Version]
- Gilmore, T.D.; Herscovitch, M. Inhibitors of NF-κB signalling: 785 and counting. Oncogene 2006, 25, 6887–6899. [Google Scholar] [CrossRef] [PubMed]
- Luke, S.; Cioffi-Revilla, C.; Panait, L.; Sullivan, K.; Balan, G. MASON: A multi-agent simulation environment. Simulation 2005, 81, 517–527. [Google Scholar] [CrossRef]
- Coakley, S.; Smallwood, R.; Holcombe, M. Using X-machines as a formal basis for describing agents in agent-based modelling. Simul. Ser. 2006, 38, 33–40. [Google Scholar]
- Coakley, S.; Gheorghe, M.; Holcombe, M.; Chin, S.; Worth, D.; Greenough, C. Exploitation of high-performance computing in the FLAME agent-based simulation framework. In Proceedings of the 14th IEEE International Conference on High-Performance Computing and Communications (HPCC), Liverpool, UK, 25–27 June 2012.
- Richmond, P.; Coakley, S.; Romano, D. Cellular level agent-based modelling on the graphics processing unit. In Proceedings of the International Workshop on High-Performance Computational Systems Biology (HiBi’09), Trento, Italy, 14–16 October 2009; IEEE: New York, NY, USA; pp. 43–50.
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Williams, R.A.; Timmis, J.; Qwarnstrom, E.E. Computational Models of the NF-KB Signalling Pathway. Computation 2014, 2, 131-158. https://doi.org/10.3390/computation2040131
Williams RA, Timmis J, Qwarnstrom EE. Computational Models of the NF-KB Signalling Pathway. Computation. 2014; 2(4):131-158. https://doi.org/10.3390/computation2040131
Chicago/Turabian StyleWilliams, Richard A., Jon Timmis, and Eva E. Qwarnstrom. 2014. "Computational Models of the NF-KB Signalling Pathway" Computation 2, no. 4: 131-158. https://doi.org/10.3390/computation2040131
APA StyleWilliams, R. A., Timmis, J., & Qwarnstrom, E. E. (2014). Computational Models of the NF-KB Signalling Pathway. Computation, 2(4), 131-158. https://doi.org/10.3390/computation2040131