
Binding and Action of Triphenylphosphonium Analog of Chloramphenicol upon the Bacterial Ribosome

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
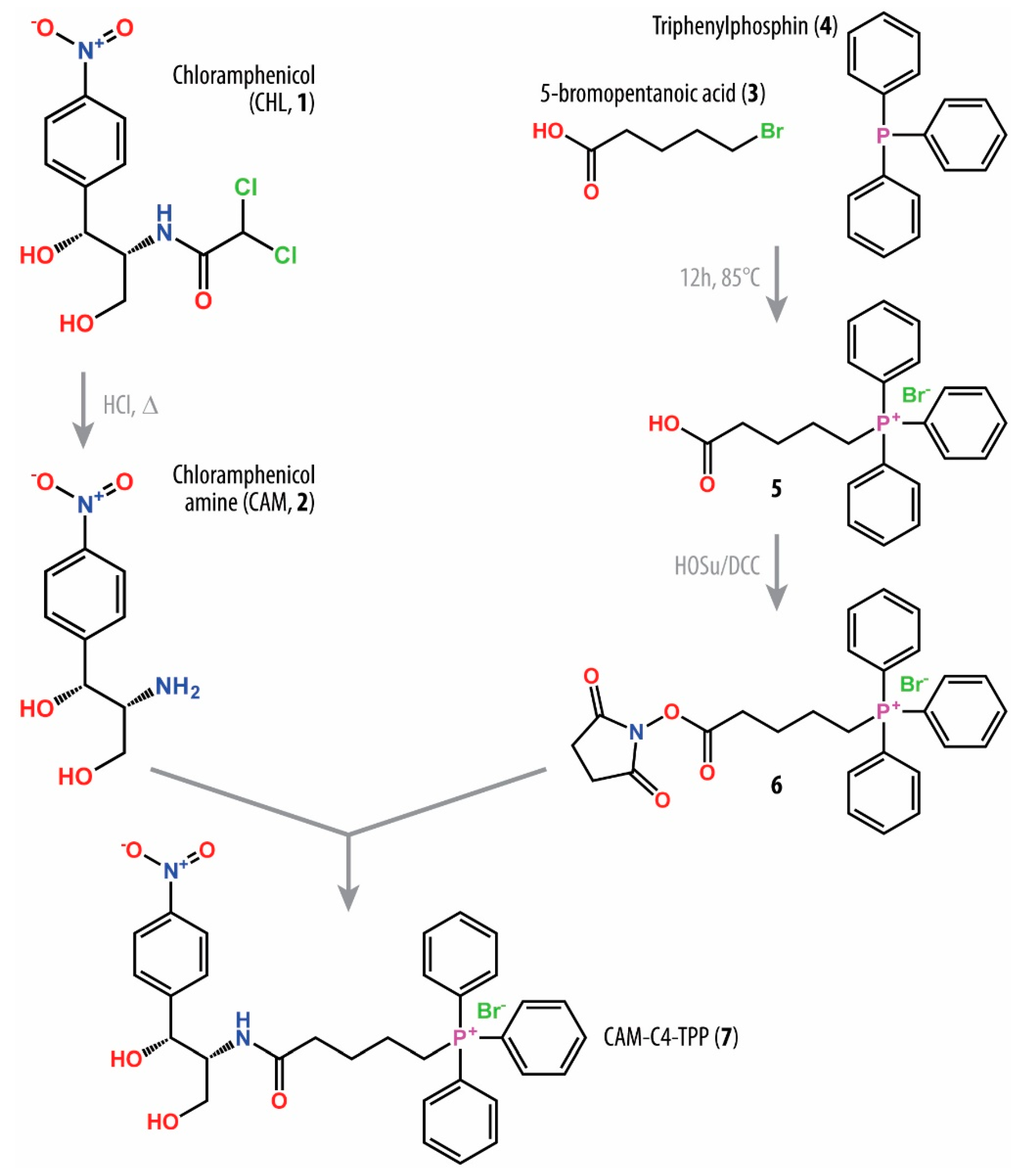
2.1. Synthesis of Triphenylphosphonium Chloramphenicol Analog
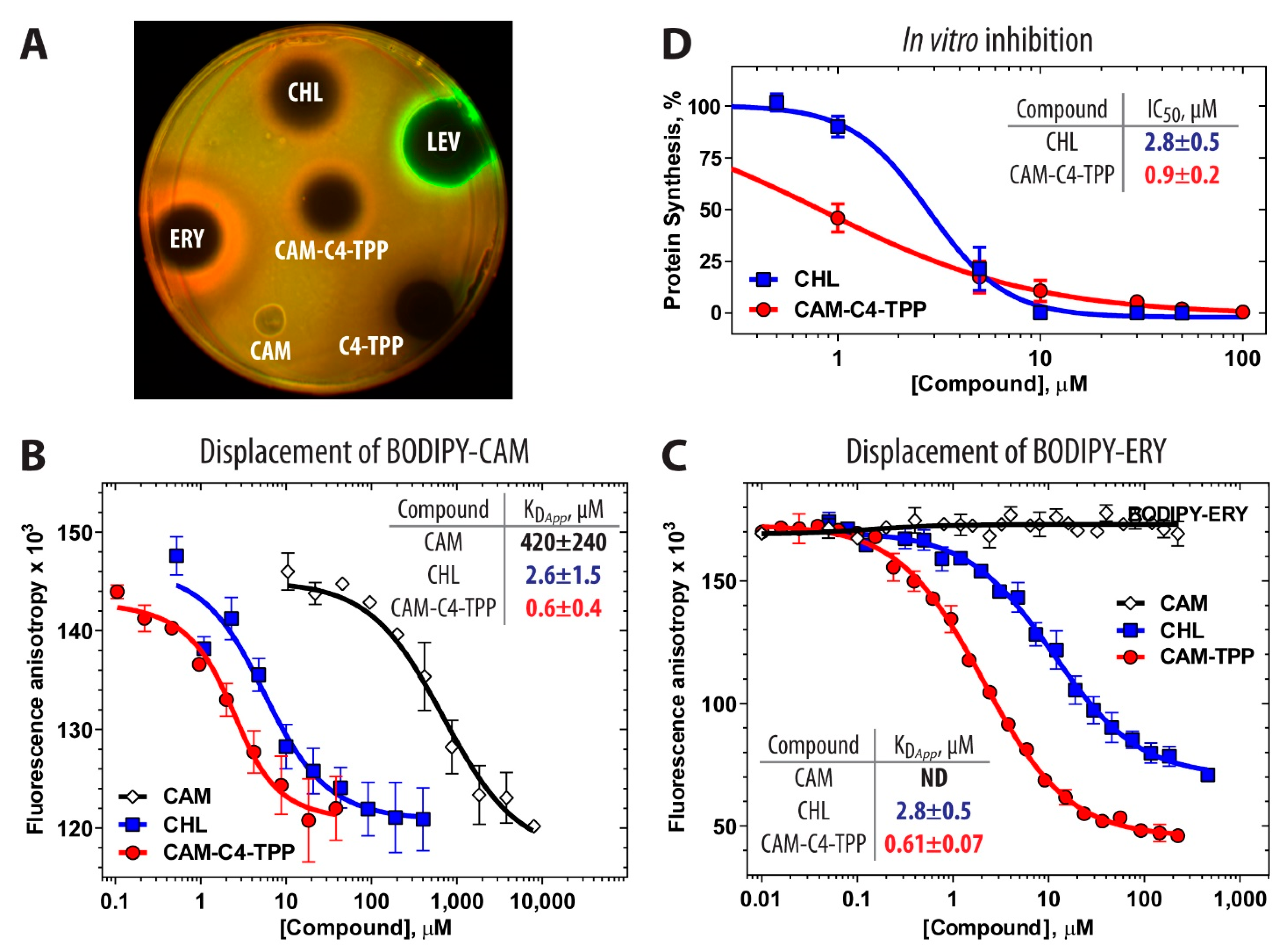
2.2. Protein Synthesis Is the Primary Target of CAM-C4-TPP Action In Vivo
2.3. CAM-C4-TPP Binds Tightly to the Bacterial Ribosome and Inhibits Protein Synthesis
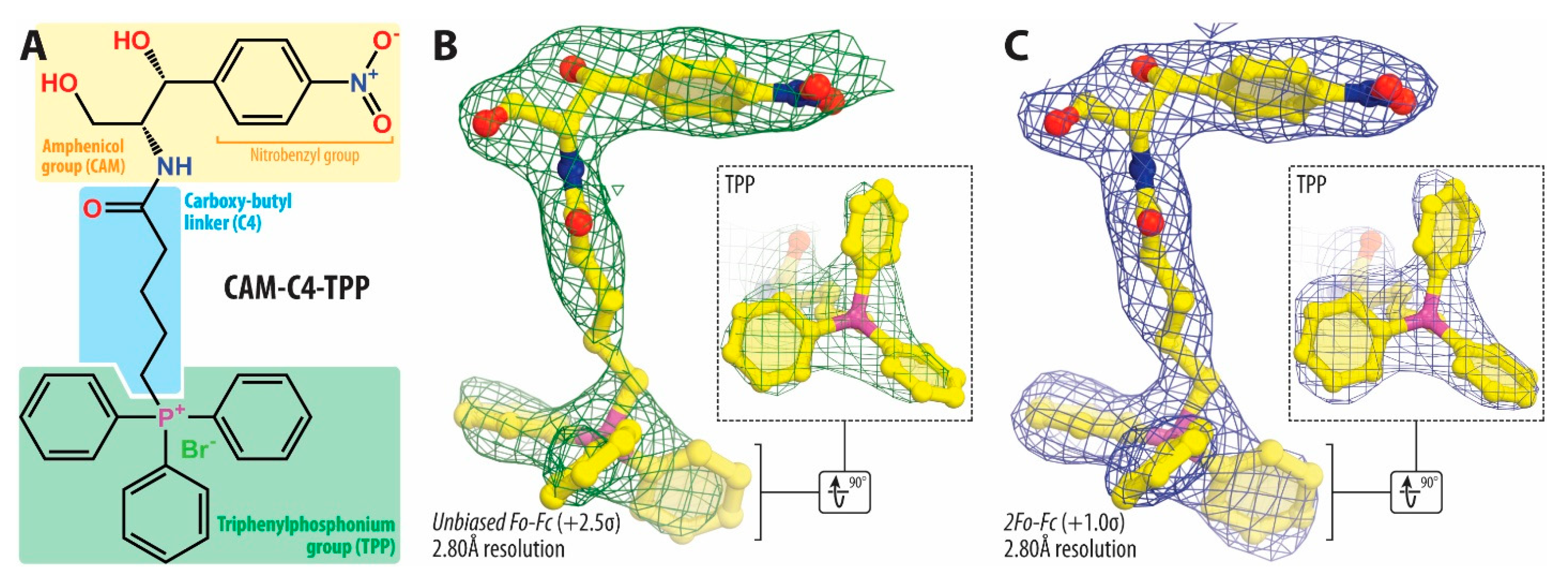
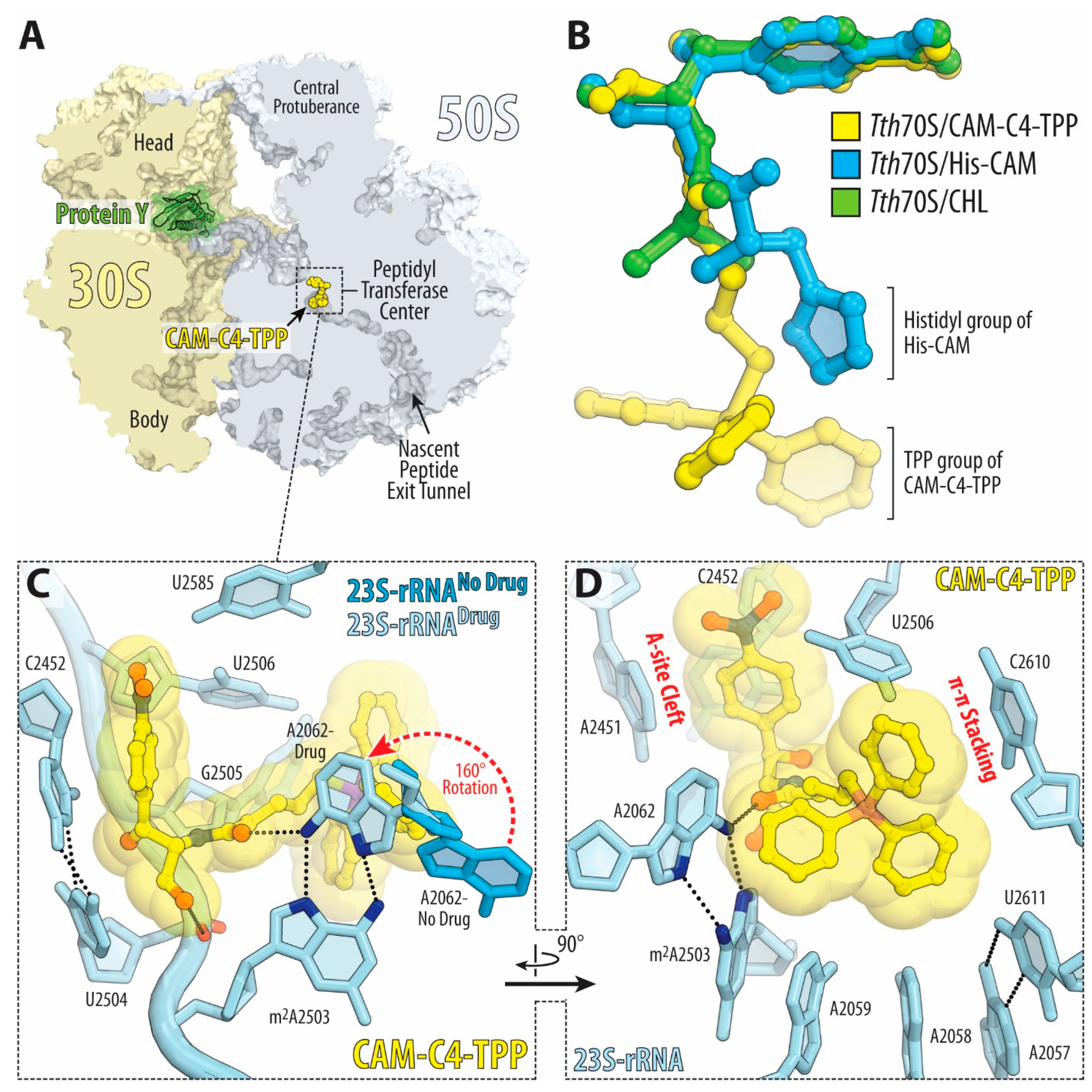
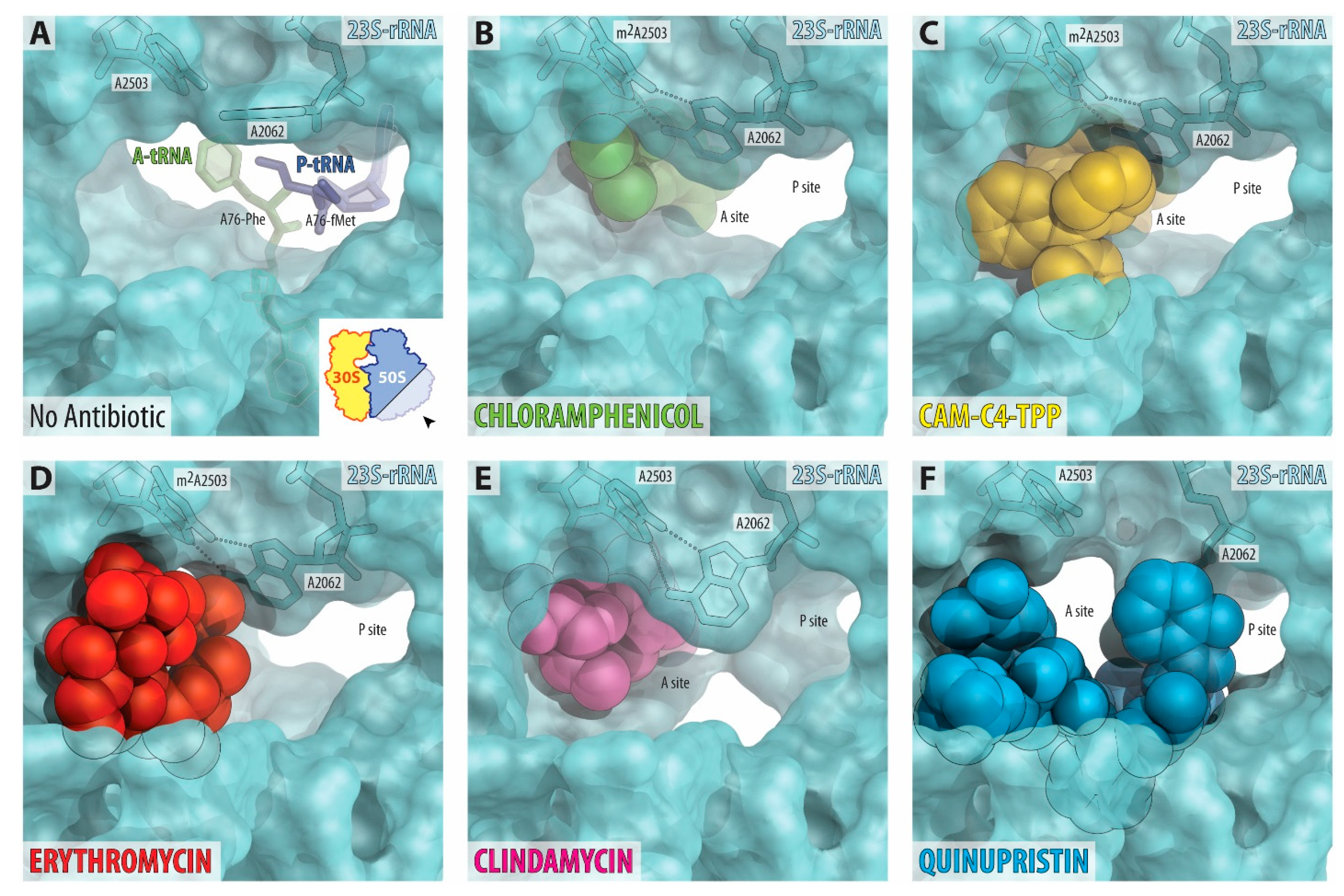
2.4. TPP Moiety of CAM-C4-TPP Establishes Unique Interactions with the Ribosome
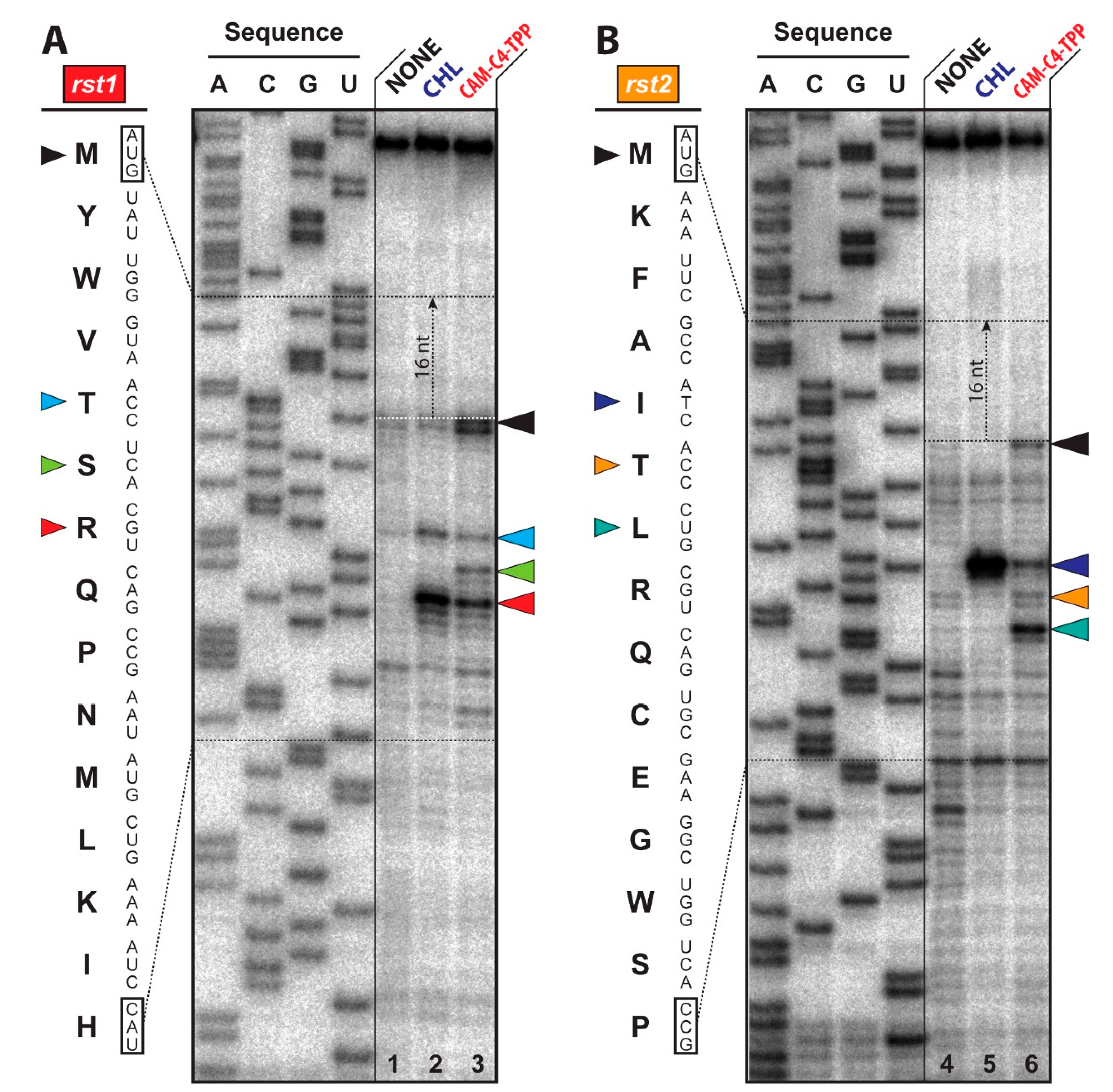
2.5. CAM-C4-TPP Shows Unique Context-Specificity of Action

3. Materials and Methods
3.1. Reagents
3.2. Chemical Synthesis of CAM-C4-TPP
3.3. In Vivo Detection of Translation Inhibitors Using Pdualrep2 Reporter Strain
3.4. In Vitro Binding Assay
3.5. In Vitro Translation Inhibition Assays
3.6. Toe-Printing Analysis
3.7. Crystallographic Structure Determination
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nissen, P.; Hansen, J.; Ban, N.; Moore, P.B.; Steitz, T.A. The structural basis of ribosome activity in peptide bond synthesis. Science 2000, 289, 920–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svetlov, M.S.; Plessa, E.; Chen, C.W.; Bougas, A.; Krokidis, M.G.; Dinos, G.P.; Polikanov, Y.S. High-resolution crystal structures of ribosome-bound chloramphenicol and erythromycin provide the ultimate basis for their competition. RNA 2019, 25, 600–606. [Google Scholar] [CrossRef] [PubMed]
- Bulkley, D.; Innis, C.A.; Blaha, G.; Steitz, T.A. Revisiting the structures of several antibiotics bound to the bacterial ribosome. Proc. Natl. Acad. Sci. USA 2010, 107, 17158–17163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunkle, J.A.; Xiong, L.; Mankin, A.S.; Cate, J.H. Structures of the Escherichia coli ribosome with antibiotics bound near the peptidyl transferase center explain spectra of drug action. Proc. Natl. Acad. Sci. USA 2010, 107, 17152–17157. [Google Scholar] [CrossRef] [Green Version]
- Marks, J.; Kannan, K.; Roncase, E.J.; Klepacki, D.; Kefi, A.; Orelle, C.; Vazquez-Laslop, N.; Mankin, A.S. Context-specific inhibition of translation by ribosomal antibiotics targeting the peptidyl transferase center. Proc. Natl. Acad. Sci. USA 2016, 113, 12150–12155. [Google Scholar] [CrossRef] [Green Version]
- Barnhill, A.E.; Brewer, M.T.; Carlson, S.A. Adverse effects of antimicrobials via predictable or idiosyncratic inhibition of host mitochondrial components. Antimicrob. Agents Chemother. 2012, 56, 4046–4051. [Google Scholar] [CrossRef] [Green Version]
- Jones, C.N.; Miller, C.; Tenenbaum, A.; Spremulli, L.L.; Saada, A. Antibiotic effects on mitochondrial translation and in patients with mitochondrial translational defects. Mitochondrion 2009, 9, 429–437. [Google Scholar] [CrossRef]
- Singh, R.; Sripada, L.; Singh, R. Side effects of antibiotics during bacterial infection: Mitochondria, the main target in host cell. Mitochondrion 2014, 16, 50–54. [Google Scholar] [CrossRef]
- Li, C.H.; Cheng, Y.W.; Liao, P.L.; Yang, Y.T.; Kang, J.J. Chloramphenicol causes mitochondrial stress, decreases atp biosynthesis, induces matrix metalloproteinase-13 expression, and solid-tumor cell invasion. Toxicol. Sci. 2010, 116, 140–150. [Google Scholar] [CrossRef] [Green Version]
- Cohen, B.H.; Saneto, R.P. Mitochondrial translational inhibitors in the pharmacopeia. Biochim. Biophys. Acta 2012, 1819, 1067–1074. [Google Scholar] [CrossRef]
- Prestinaci, F.; Pezzotti, P.; Pantosti, A. Antimicrobial resistance: A global multifaceted phenomenon. Pathog. Glob. Health 2015, 109, 309–318. [Google Scholar] [CrossRef] [Green Version]
- Dinos, G.P.; Athanassopoulos, C.M.; Missiri, D.A.; Giannopoulou, P.C.; Vlachogiannis, I.A.; Papadopoulos, G.E.; Papaioannou, D.; Kalpaxis, D.L. Chloramphenicol derivatives as antibacterial and anticancer agents: Historic problems and current solutions. Antibiotics 2016, 5, 20. [Google Scholar] [CrossRef]
- Tereshchenkov, A.G.; Dobosz-Bartoszek, M.; Osterman, I.A.; Marks, J.; Sergeeva, V.A.; Kasatsky, P.; Komarova, E.S.; Stavrianidi, A.N.; Rodin, I.A.; Konevega, A.L.; et al. Binding and action of amino acid analogs of chloramphenicol upon the bacterial ribosome. J. Mol. Biol. 2018, 430, 842–852. [Google Scholar] [CrossRef]
- Mamos, P.; Krokidis, M.G.; Papadas, A.; Karahalios, P.; Starosta, A.L.; Wilson, D.N.; Kalpaxis, D.L.; Dinos, G.P. On the use of the antibiotic chloramphenicol to target polypeptide chain mimics to the ribosomal exit tunnel. Biochimie 2013, 95, 1765–1772. [Google Scholar] [CrossRef]
- Kostopoulou, O.N.; Kouvela, E.C.; Magoulas, G.E.; Garnelis, T.; Panagoulias, I.; Rodi, M.; Papadopoulos, G.; Mouzaki, A.; Dinos, G.P.; Papaioannou, D.; et al. Conjugation with polyamines enhances the antibacterial and anticancer activity of chloramphenicol. Nucleic Acids Res. 2014, 42, 8621–8634. [Google Scholar] [CrossRef]
- Giannopoulou, P.C.; Missiri, D.A.; Kournoutou, G.G.; Sazakli, E.; Papadopoulos, G.E.; Papaioannou, D.; Dinos, G.P.; Athanassopoulos, C.M.; Kalpaxis, D.L. New chloramphenicol derivatives from the viewpoint of anticancer and antimicrobial activity. Antibiotics 2019, 8, 9. [Google Scholar] [CrossRef] [Green Version]
- Pavlova, J.A.; Khairullina, Z.Z.; Tereshchenkov, A.G.; Nazarov, P.A.; Lukianov, D.A.; Volynkina, I.A.; Skvortsov, D.A.; Makarov, G.I.; Abad, E.; Murayama, S.Y.; et al. Triphenilphosphonium analogs of chloramphenicol as dual-acting antimicrobial and antiproliferating agents. Antibiotics 2021. under revision. [Google Scholar]
- Long, K.S.; Poehlsgaard, J.; Kehrenberg, C.; Schwarz, S.; Vester, B. TheCfr rRNA methyltransferase confers resistance to phenicols, lincosamides, oxazolidinones, pleuromutilins, and streptogramin a antibiotics. Antimicrob. Agents Chemother. 2006, 50, 2500–2505. [Google Scholar] [CrossRef] [Green Version]
- Liberman, E.A.; Topaly, V.P.; Tsofina, L.M.; Jasaitis, A.A.; Skulachev, V.P. Mechanism of coupling of oxidative phosphorylation and the membrane potential of mitochondria. Nature 1969, 222, 1076–1078. [Google Scholar] [CrossRef]
- Khailova, L.S.; Nazarov, P.A.; Sumbatyan, N.V.; Korshunova, G.A.; Rokitskaya, T.I.; Dedukhova, V.I.; Antonenko, Y.N.; Skulachev, V.P. Uncoupling and toxic action of alkyltriphenylphosphonium cations on mitochondria and the bacterium Bacillus subtilis as a function of alkyl chain length. Biochemistry 2015, 80, 1589–1597. [Google Scholar] [CrossRef]
- Nazarov, P.A.; Kirsanov, R.S.; Denisov, S.S.; Khailova, L.S.; Karakozova, M.V.; Lyamzaev, K.G.; Korshunova, G.A.; Lukyanov, K.A.; Kotova, E.A.; Antonenko, Y.N. Fluorescein derivatives as antibacterial agents acting via membrane depolarization. Biomolecules 2020, 10, 309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iaubasarova, I.R.; Khailova, L.S.; Nazarov, P.A.; Rokitskaya, T.I.; Silachev, D.N.; Danilina, T.I.; Plotnikov, E.Y.; Denisov, S.S.; Kirsanov, R.S.; Korshunova, G.A.; et al. Linking 7-nitrobenzo-2-oxa-1,3-diazole (NBD) to triphenylphosphonium yields mitochondria-targeted protonophore and antibacterial agent. Biochemistry 2020, 85, 1578–1590. [Google Scholar] [PubMed]
- Rebstock, M.C.; Crooks, H.M.; Controulis, J.; Bartz, Q.R. Chloramphenicol (chloromycetin). 1 iv. 1a chemical studies. J. Am. Chem. Soc. 1949, 71, 2458–2462. [Google Scholar] [CrossRef]
- Osterman, I.A.; Komarova, E.S.; Shiryaev, D.I.; Korniltsev, I.A.; Khven, I.M.; Lukyanov, D.A.; Tashlitsky, V.N.; Serebryakova, M.V.; Efremenkova, O.V.; Ivanenkov, Y.A.; et al. Sorting out antibiotics’ mechanisms of action: A double fluorescent protein reporter for high-throughput screening of ribosome and DNA biosynthesis inhibitors. Antimicrob. Agents Chemother. 2016, 60, 7481–7489. [Google Scholar] [CrossRef] [Green Version]
- Yan, K.; Hunt, E.; Berge, J.; May, E.; Copeland, R.A.; Gontarek, R.R. Fluorescence polarization method to characterize macrolide-ribosome interactions. Antimicrob. Agents Chemother. 2005, 49, 3367–3372. [Google Scholar] [CrossRef] [Green Version]
- Tereshchenkov, A.G.; Shishkina, A.V.; Karpenko, V.V.; Chertkov, V.A.; Konevega, A.L.; Kasatsky, P.S.; Bogdanov, A.A.; Sumbatyan, N.V. New fluorescent macrolide derivatives for studying interactions of antibiotics and their analogs with the ribosomal exit tunnel. Biochemistry 2016, 81, 1163–1172. [Google Scholar] [CrossRef]
- Hansen, J.L.; Moore, P.B.; Steitz, T.A. Structures of five antibiotics bound at the peptidyl transferase center of the large ribosomal subunit. J. Mol. Biol. 2003, 330, 1061–1075. [Google Scholar] [CrossRef]
- Svetlov, M.S.; Syroegin, E.A.; Aleksandrova, E.V.; Atkinson, G.C.; Gregory, S.T.; Mankin, A.S.; Polikanov, Y.S. Structure of erm-modified 70s ribosome reveals the mechanism of macrolide resistance. Nat. Chem. Biol. 2021, 17, 412–420. [Google Scholar] [CrossRef]
- Vazquez, D. Binding of chloramphenicol to ribosomes the effect of a number of antibiotics. Biochim. Biophys. Acta Nucleic Acids Protein Synth. 1966, 114, 277–288. [Google Scholar] [CrossRef]
- Lessard, J.L.; Pestka, S. Studies on the formation of transfer ribonucleic acid-ribosome complexes. 23. Chloramphenicol, aminoacyl-oligonucleotides, and Escherichia coli ribosomes. J. Biol. Chem. 1972, 247, 6909–6912. [Google Scholar] [CrossRef]
- Polikanov, Y.S.; Blaha, G.M.; Steitz, T.A. How hibernation factors rmf, hpf, and yfia turn off protein synthesis. Science 2012, 336, 915–918. [Google Scholar] [CrossRef] [Green Version]
- Polikanov, Y.S.; Melnikov, S.V.; Soll, D.; Steitz, T.A. Structural insights into the role of rRNA modifications in protein synthesis and ribosome assembly. Nat. Struct. Mol. Biol. 2015, 22, 342–344. [Google Scholar] [CrossRef]
- Kannan, K.; Vazquez-Laslop, N.; Mankin, A.S. Selective protein synthesis by ribosomes with a drug-obstructed exit tunnel. Cell 2012, 151, 508–520. [Google Scholar] [CrossRef] [Green Version]
- Kannan, K.; Kanabar, P.; Schryer, D.; Florin, T.; Oh, E.; Bahroos, N.; Tenson, T.; Weissman, J.S.; Mankin, A.S. The general mode of translation inhibition by macrolide antibiotics. Proc. Natl. Acad. Sci. USA 2014, 111, 15958–15963. [Google Scholar] [CrossRef] [Green Version]
- Davis, A.R.; Gohara, D.W.; Yap, M.N. Sequence selectivity of macrolide-induced translational attenuation. Proc. Natl. Acad. Sci. USA 2014, 111, 15379–15384. [Google Scholar] [CrossRef] [Green Version]
- Hartz, D.; McPheeters, D.S.; Traut, R.; Gold, L. Extension inhibition analysis of translation initiation complexes. Methods Enzymol. 1988, 164, 419–425. [Google Scholar]
- Orelle, C.; Carlson, S.; Kaushal, B.; Almutairi, M.M.; Liu, H.; Ochabowicz, A.; Quan, S.; Pham, V.C.; Squires, C.L.; Murphy, B.T.; et al. Tools for characterizing bacterial protein synthesis inhibitors. Antimicrob. Agents Chemother. 2013, 57, 5994–6004. [Google Scholar] [CrossRef] [Green Version]
- Noeske, J.; Huang, J.; Olivier, N.B.; Giacobbe, R.A.; Zambrowski, M.; Cate, J.H. Synergy of streptogramin antibiotics occurs independently of their effects on translation. Antimicrob. Agents Chemother. 2014, 58, 5269–5279. [Google Scholar] [CrossRef] [Green Version]
- Haughland, R.P.; Kang, H.C.; Young, S.L.; Melner, M.H. Fluorescent chloramphenicol derivatives for determination of chloramphenicol acetyltransferase activity. US005364764A, 15 November 1994. [Google Scholar]
- Zakalyukina, Y.V.; Birykov, M.V.; Lukianov, D.A.; Shiriaev, D.I.; Komarova, E.S.; Skvortsov, D.A.; Kostyukevich, Y.; Tashlitsky, V.N.; Polshakov, V.I.; Nikolaev, E.; et al. Nybomycin-producing Streptomyces isolated from carpenter ant Camponotus vagus. Biochimie 2019, 160, 93–99. [Google Scholar] [CrossRef]
- Shishkina, A.; Makarov, G.; Tereshchenkov, A.; Korshunova, G.; Sumbatyan, N.; Golovin, A.; Svetlov, M.; Bogdanov, A. Conjugates of amino acids and peptides with 5-O-mycaminosyltylonolide and their interaction with the ribosomal exit tunnel. Bioconjug. Chem. 2013, 24, 1861–1869. [Google Scholar] [CrossRef]
- Wang, Z.X. An exact mathematical expression for describing competitive binding of two different ligands to a protein molecule. FEBS Lett. 1995, 360, 111–114. [Google Scholar] [CrossRef] [Green Version]
- Polikanov, Y.S.; Steitz, T.A.; Innis, C.A. A proton wire to couple aminoacyl-tRNA accommodation and peptide-bond formation on the ribosome. Nat. Struct. Mol. Biol. 2014, 21, 787–793. [Google Scholar] [CrossRef]
- Schuttelkopf, A.W.; van Aalten, D.M. Prodrg: A tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 1355–1363. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, C.-W.; Pavlova, J.A.; Lukianov, D.A.; Tereshchenkov, A.G.; Makarov, G.I.; Khairullina, Z.Z.; Tashlitsky, V.N.; Paleskava, A.; Konevega, A.L.; Bogdanov, A.A.; et al. Binding and Action of Triphenylphosphonium Analog of Chloramphenicol upon the Bacterial Ribosome. Antibiotics 2021, 10, 390. https://doi.org/10.3390/antibiotics10040390
Chen C-W, Pavlova JA, Lukianov DA, Tereshchenkov AG, Makarov GI, Khairullina ZZ, Tashlitsky VN, Paleskava A, Konevega AL, Bogdanov AA, et al. Binding and Action of Triphenylphosphonium Analog of Chloramphenicol upon the Bacterial Ribosome. Antibiotics. 2021; 10(4):390. https://doi.org/10.3390/antibiotics10040390
Chicago/Turabian StyleChen, Chih-Wei, Julia A. Pavlova, Dmitrii A. Lukianov, Andrey G. Tereshchenkov, Gennady I. Makarov, Zimfira Z. Khairullina, Vadim N. Tashlitsky, Alena Paleskava, Andrey L. Konevega, Alexey A. Bogdanov, and et al. 2021. "Binding and Action of Triphenylphosphonium Analog of Chloramphenicol upon the Bacterial Ribosome" Antibiotics 10, no. 4: 390. https://doi.org/10.3390/antibiotics10040390
APA StyleChen, C. -W., Pavlova, J. A., Lukianov, D. A., Tereshchenkov, A. G., Makarov, G. I., Khairullina, Z. Z., Tashlitsky, V. N., Paleskava, A., Konevega, A. L., Bogdanov, A. A., Osterman, I. A., Sumbatyan, N. V., & Polikanov, Y. S. (2021). Binding and Action of Triphenylphosphonium Analog of Chloramphenicol upon the Bacterial Ribosome. Antibiotics, 10(4), 390. https://doi.org/10.3390/antibiotics10040390