
Actinomycetes: A Never-Ending Source of Bioactive Compounds—An Overview on Antibiotics Production



Abstract
:1. Introduction
2. Actinomycetes: Biology and Bioactive Compounds
3. Mechanisms of Resistance and Antibiotics from Actinomycetes: An Overview
- Permeability alteration;
- Target modification/amplification;
- Drug inactivation.
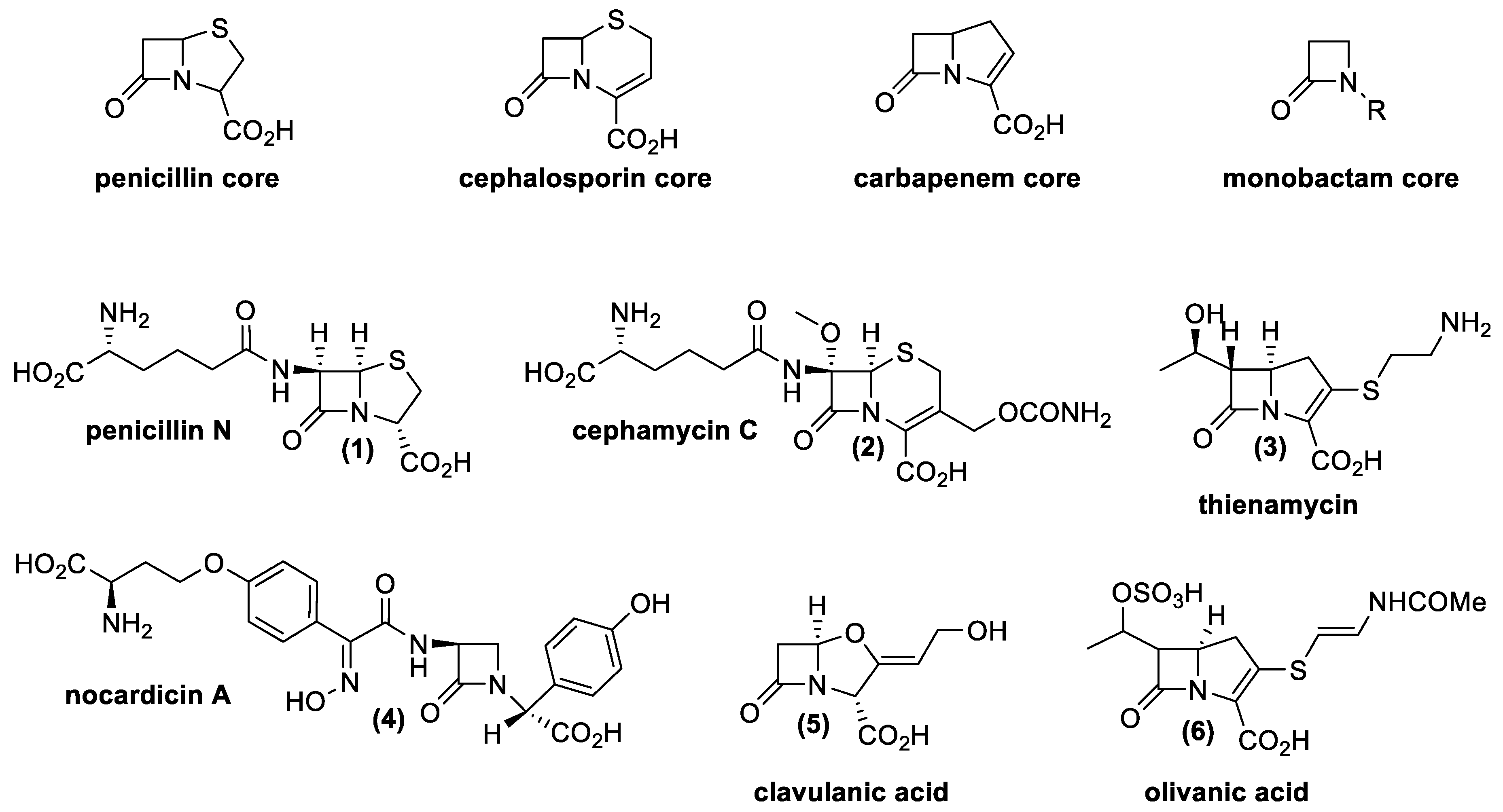
3.1. β-Lactam Antibiotics
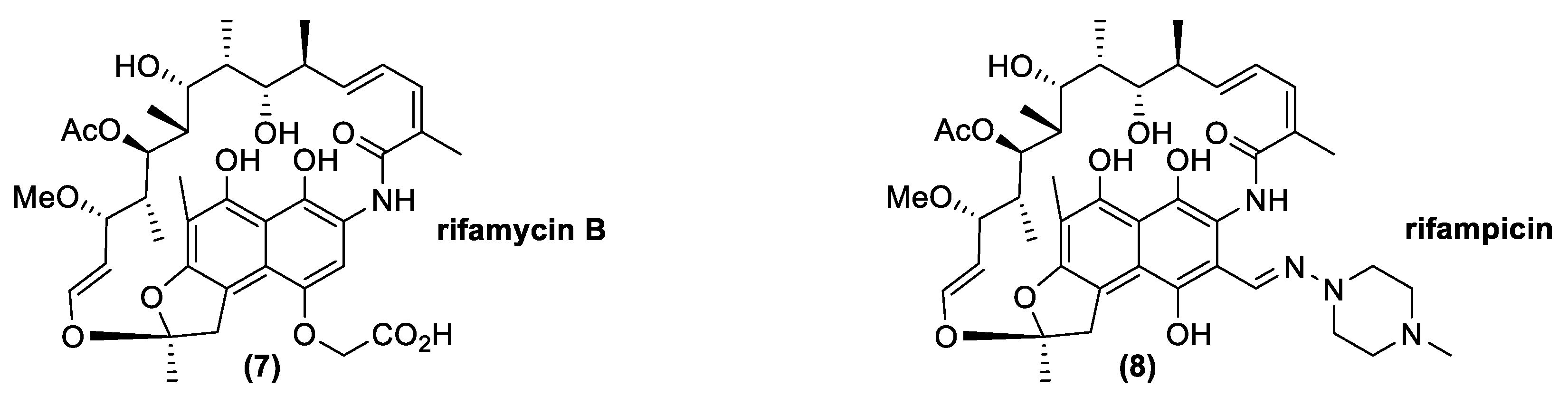
3.2. Ansamycines
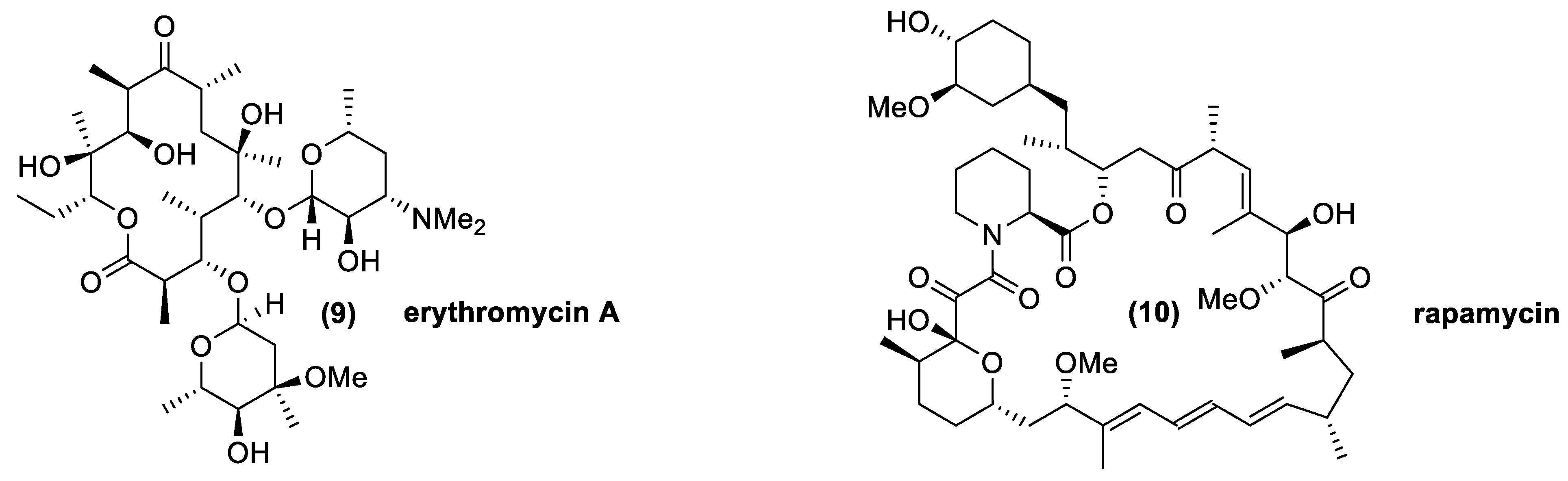
3.3. Macrolides
3.4. Lincosamides
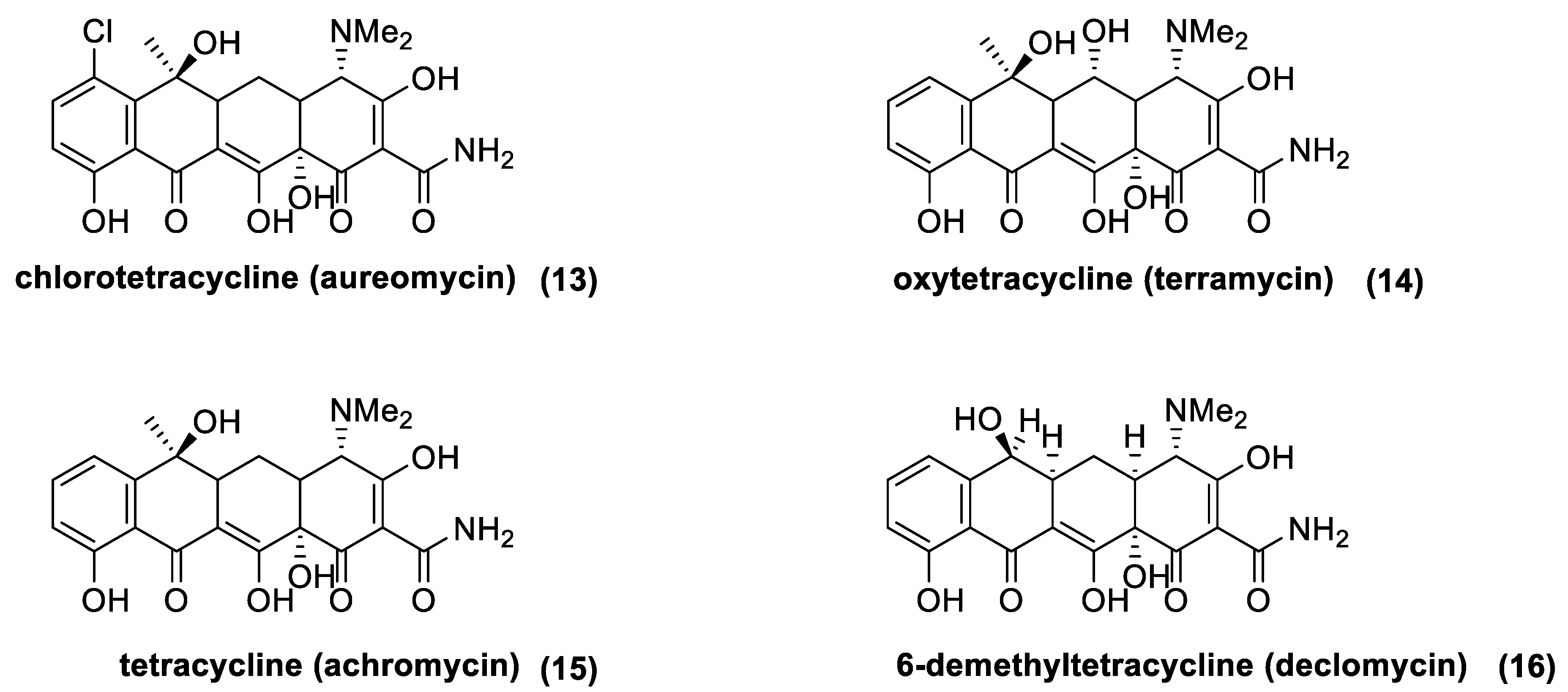
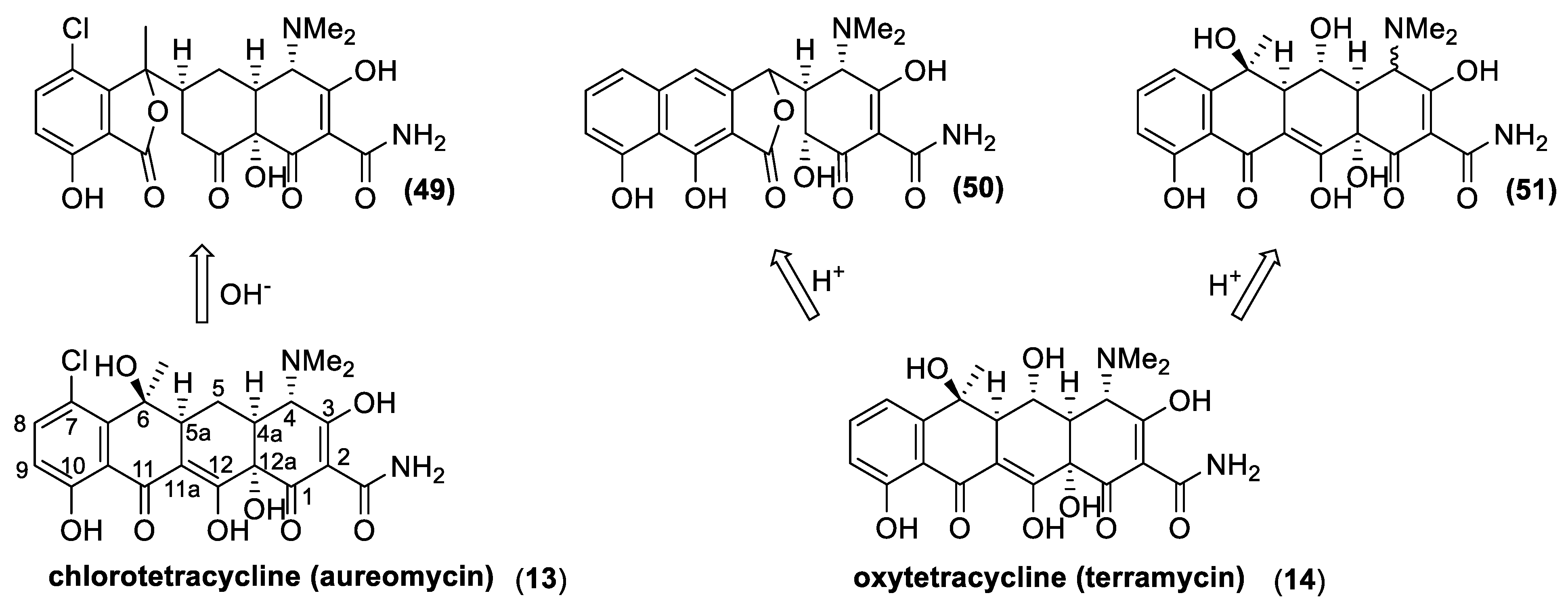
3.5. Tetracyclines
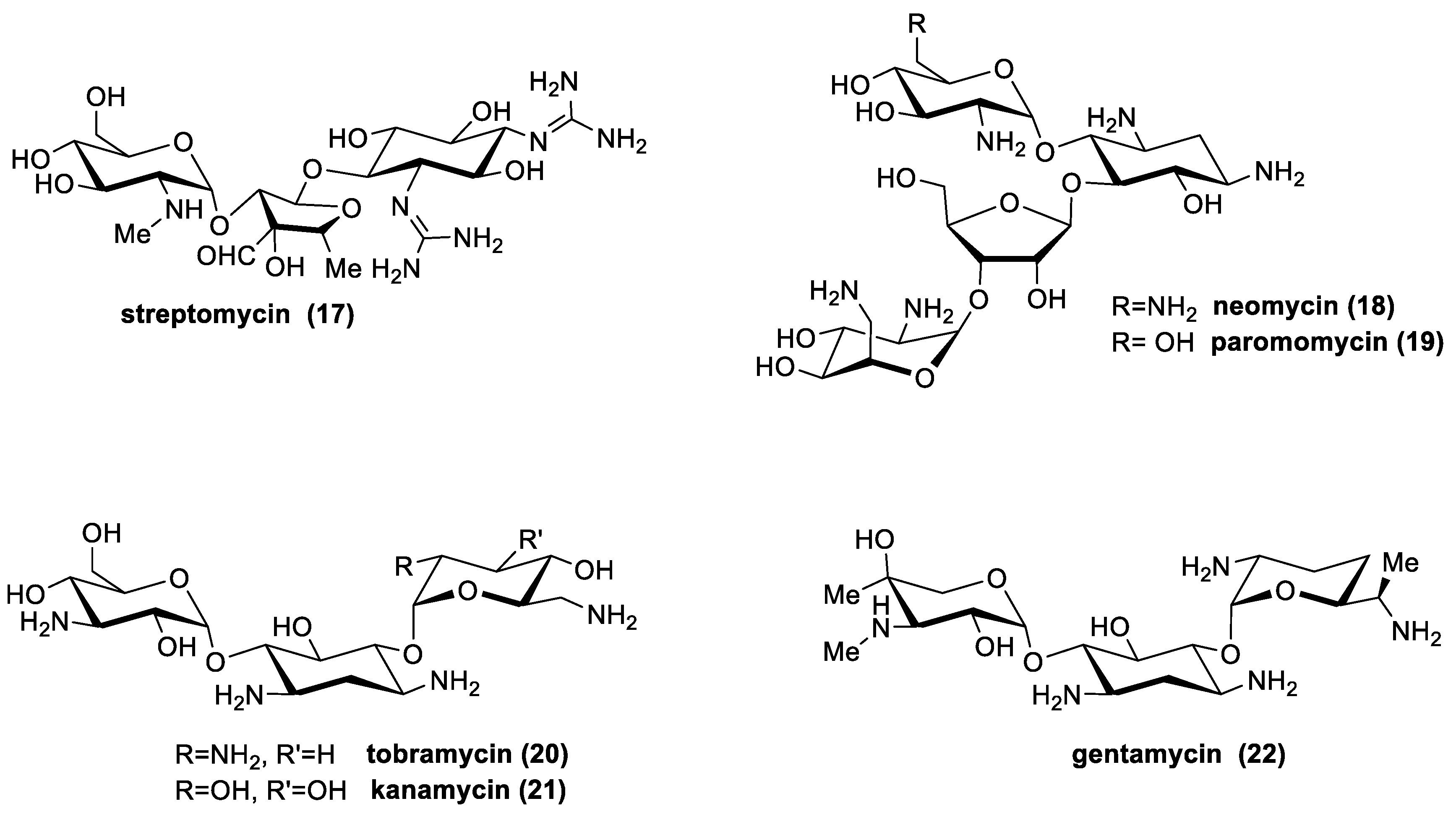
3.6. Aminoglycosides
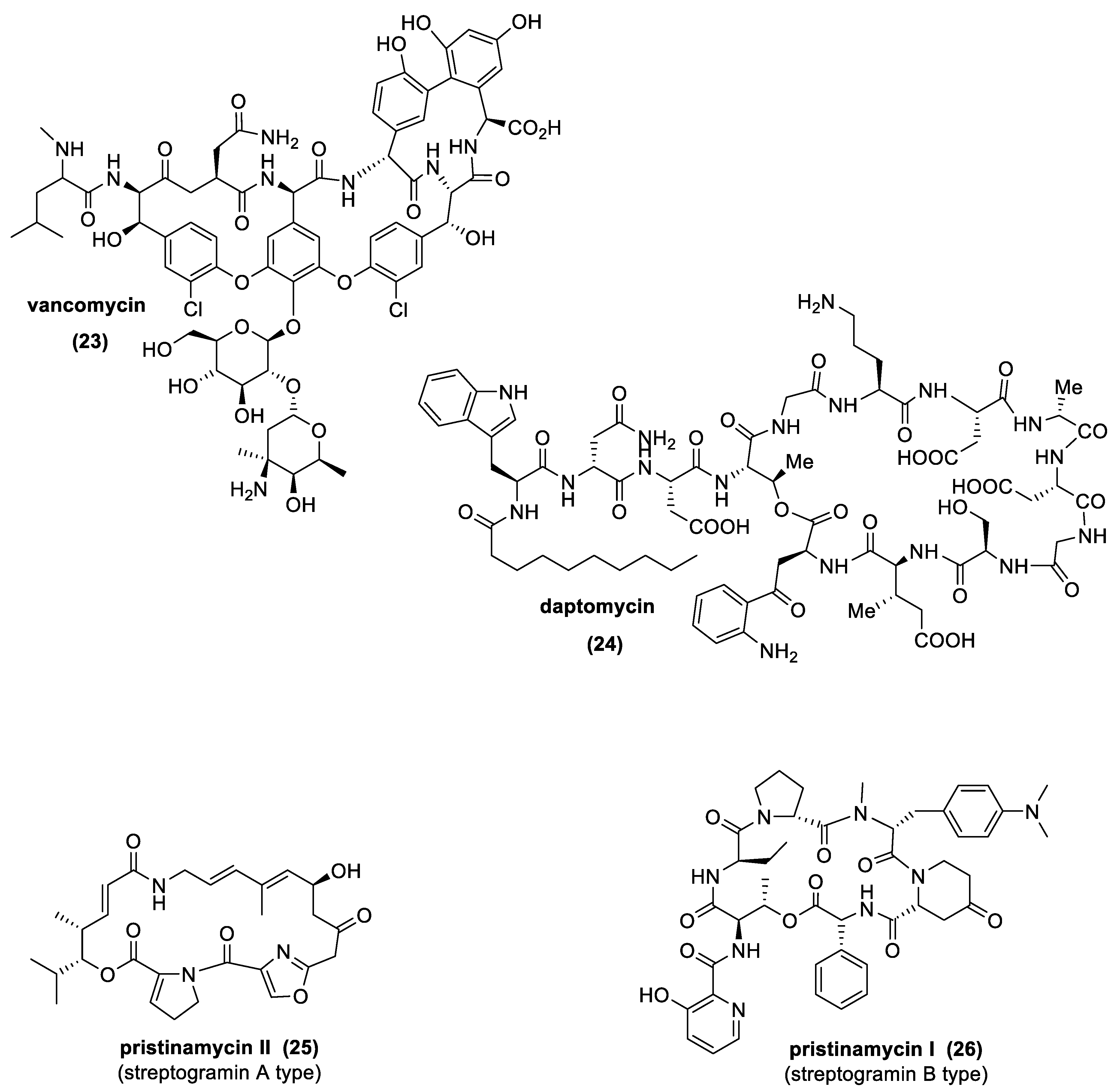
3.7. Antibiotic Peptides
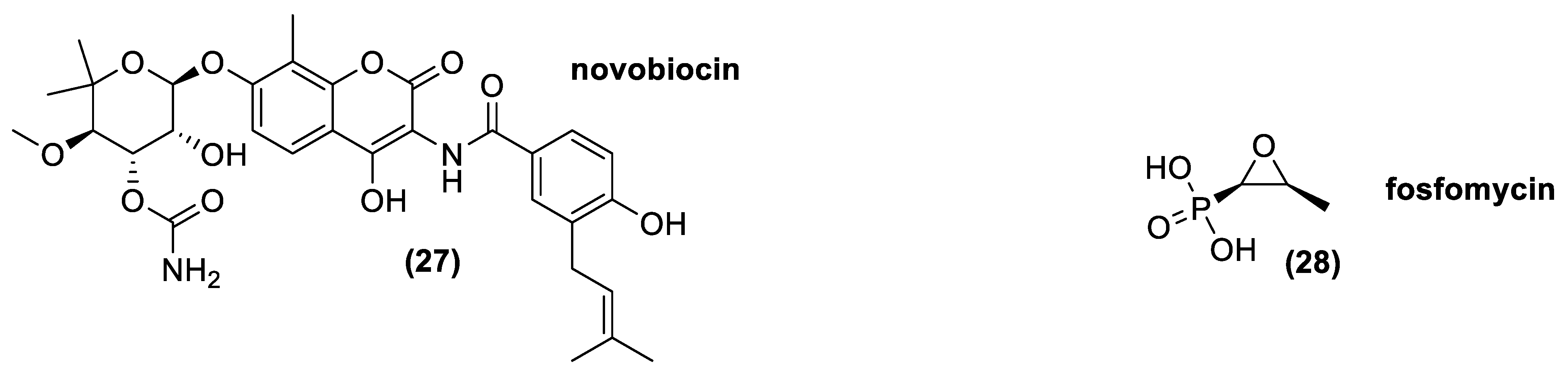
3.8. Aminocumarines
3.9. Epoxides
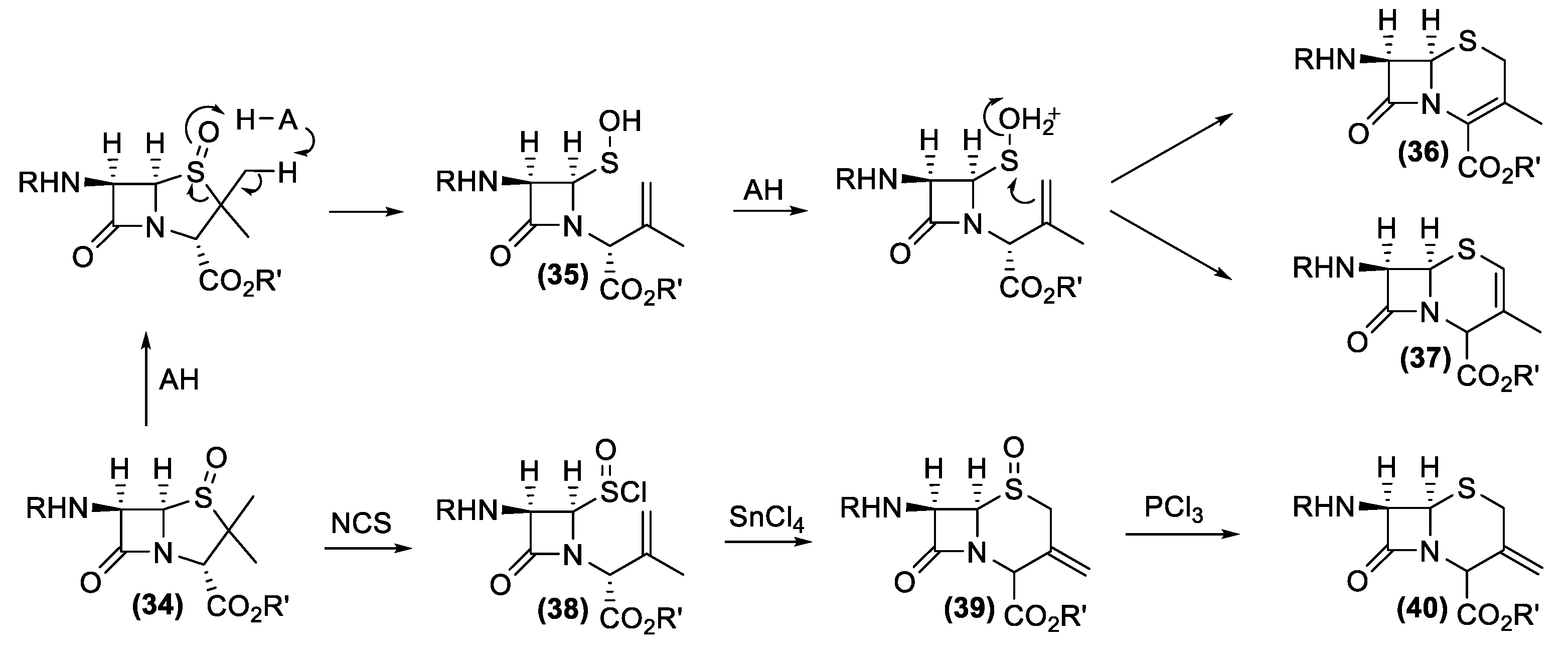
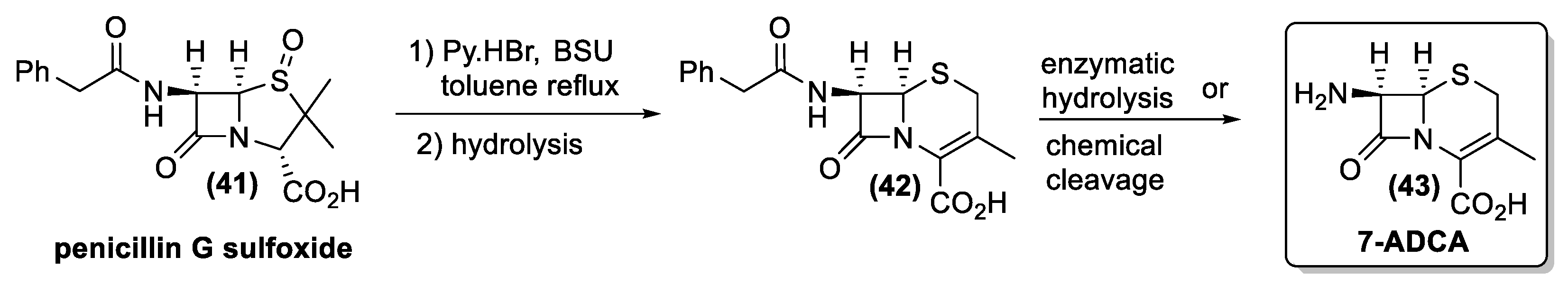
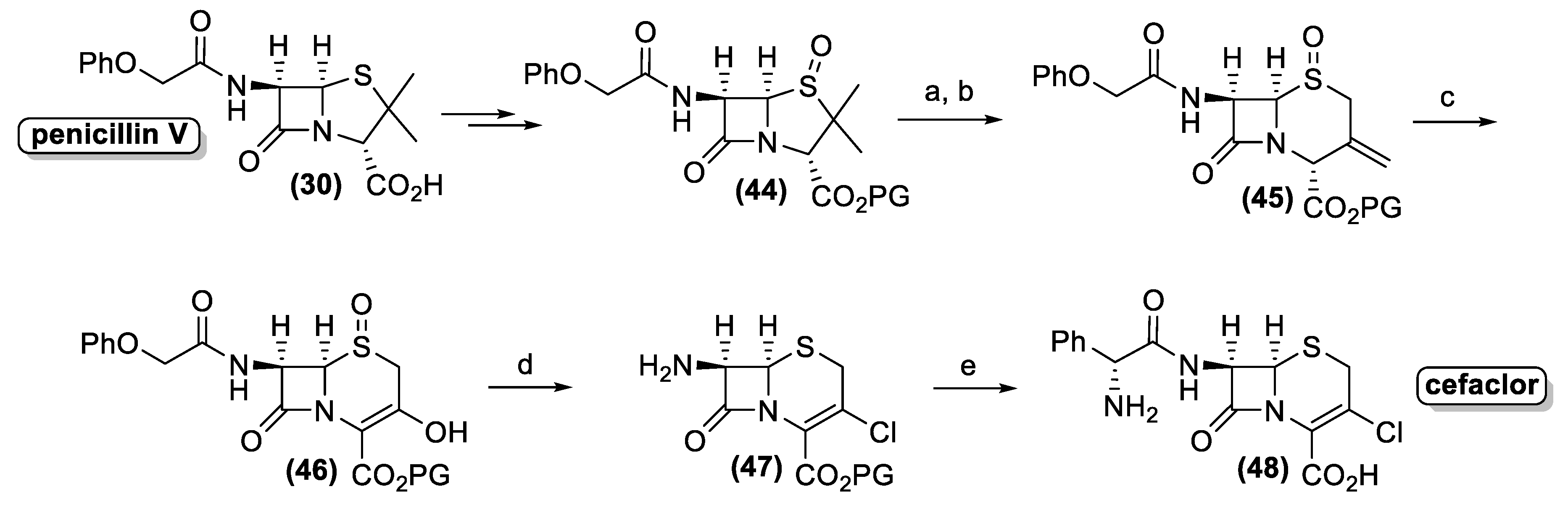
4. Antibiotics from Actinomycetes: From Isolation to Chemical Characterization, Total Synthesis, and Industrial Production
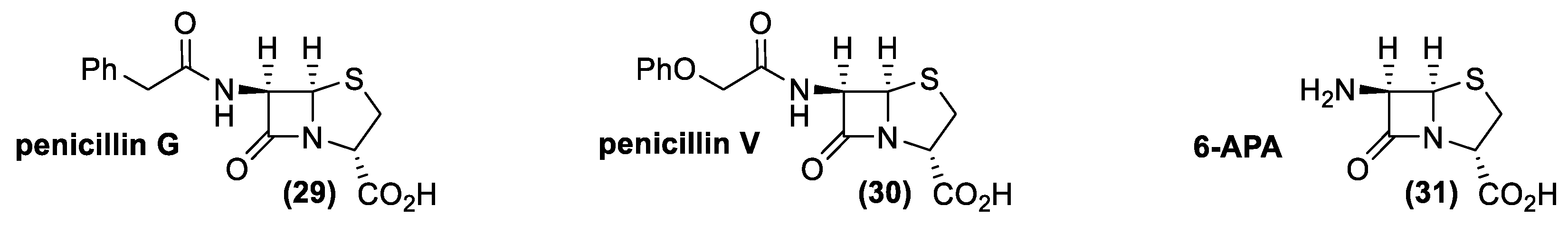
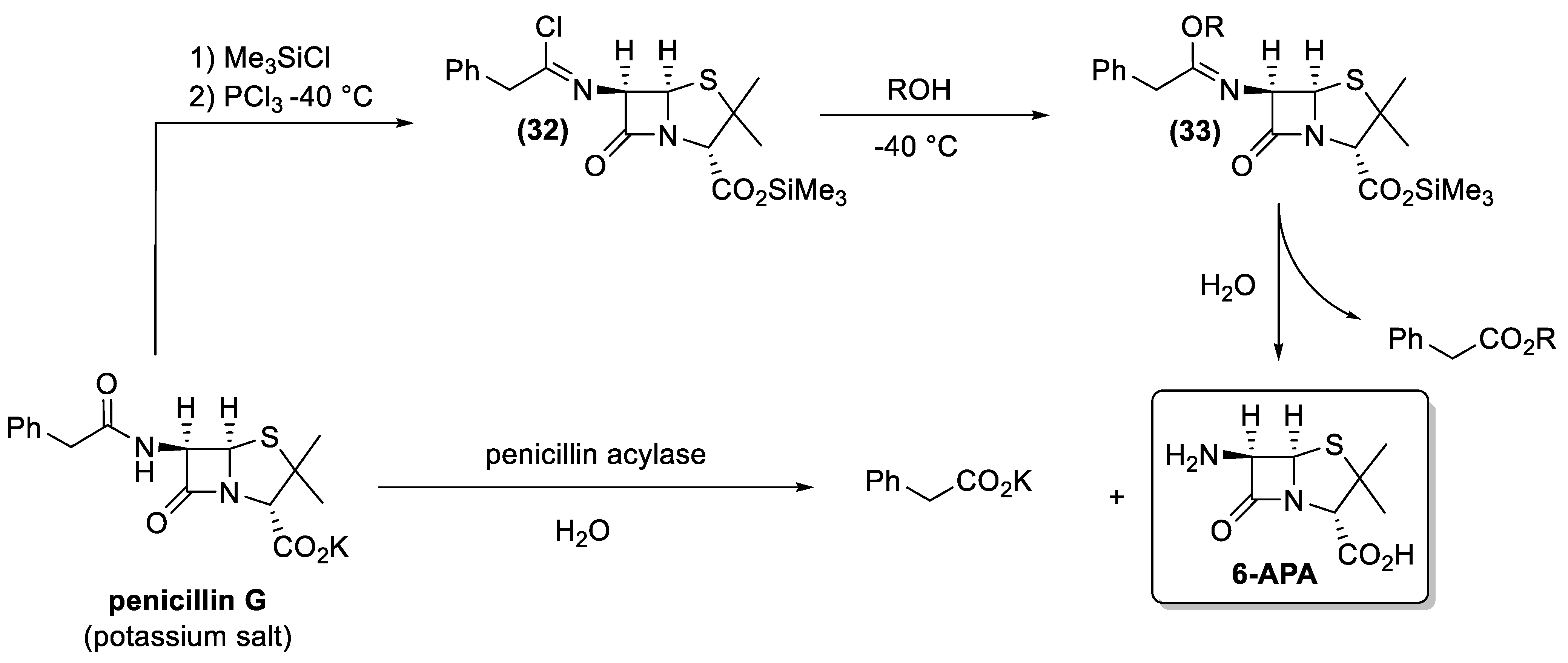
4.1. Penicillin and Cephalosporin
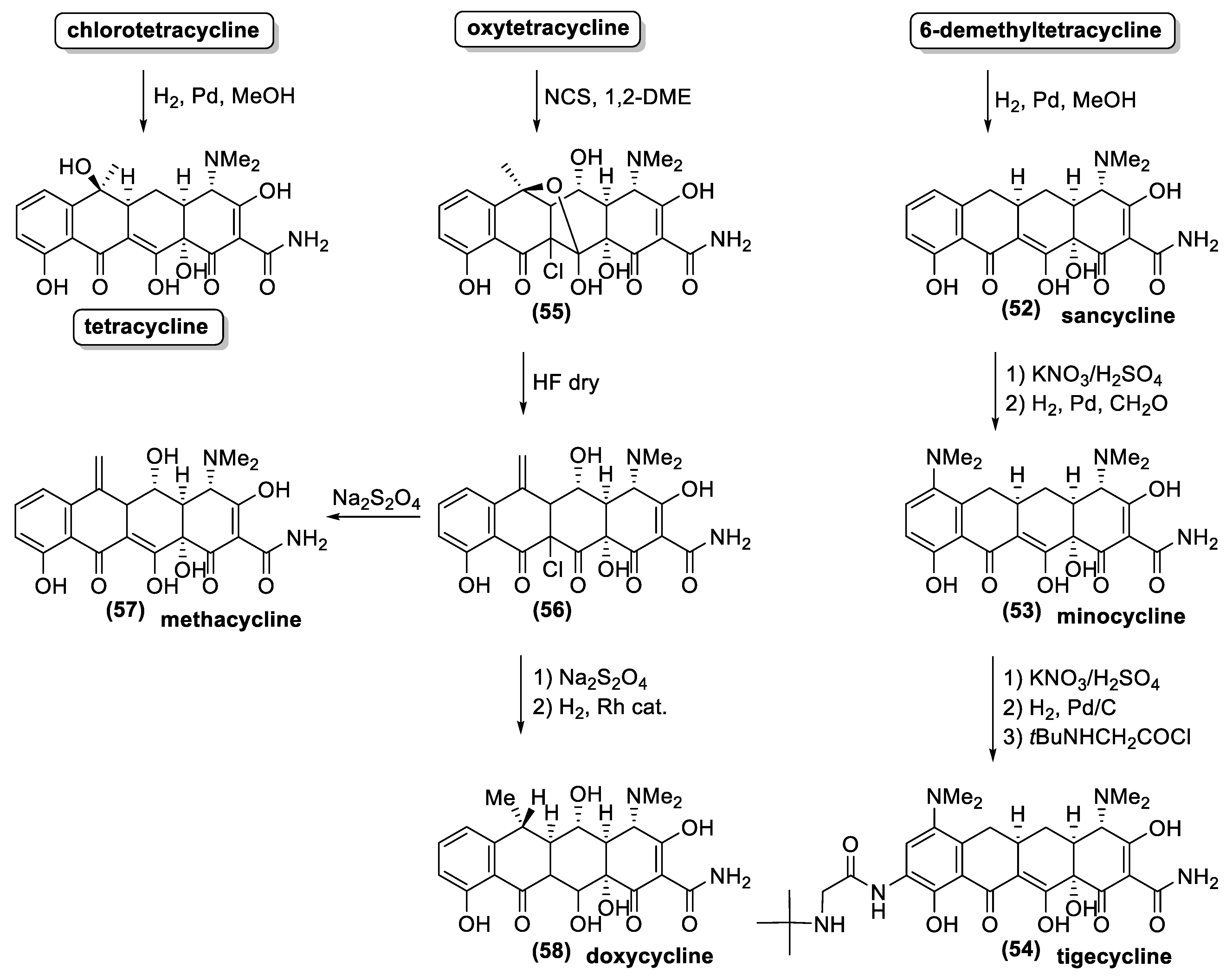
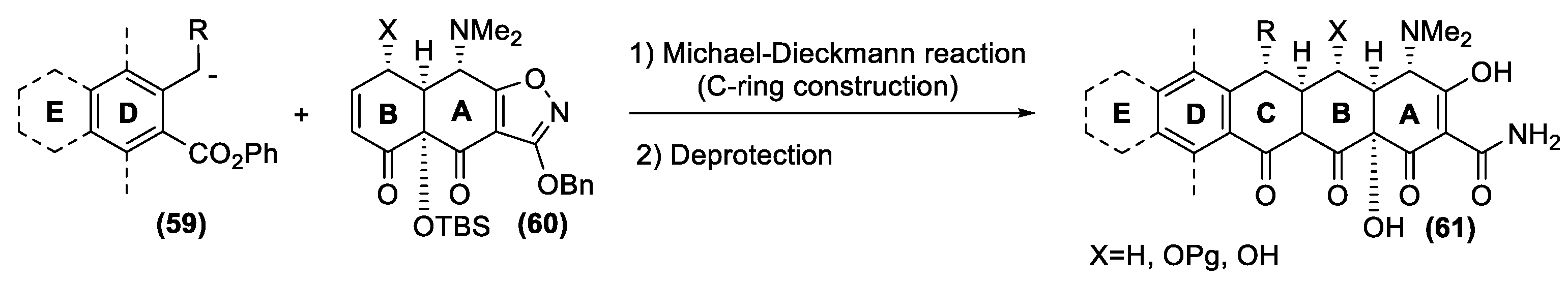
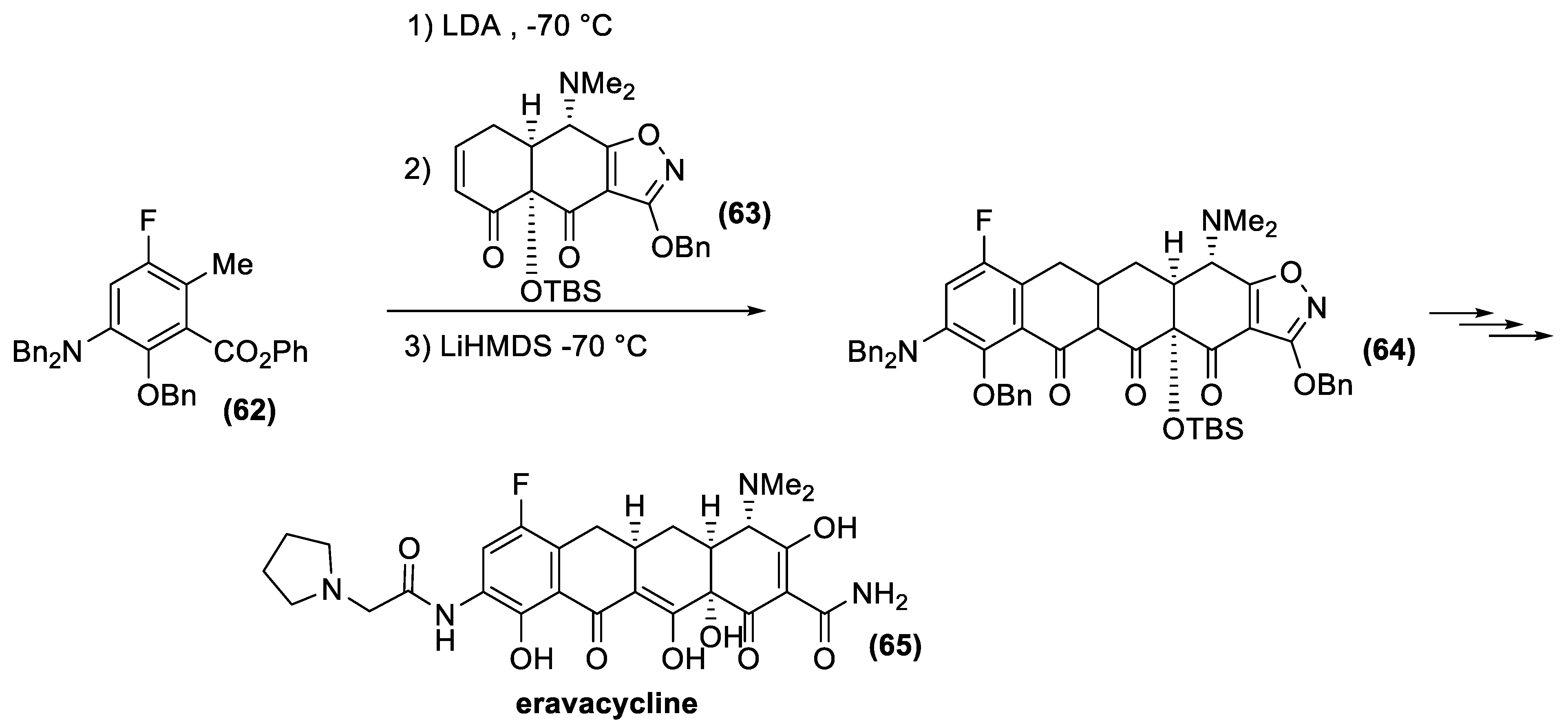
4.2. Tetracyclines
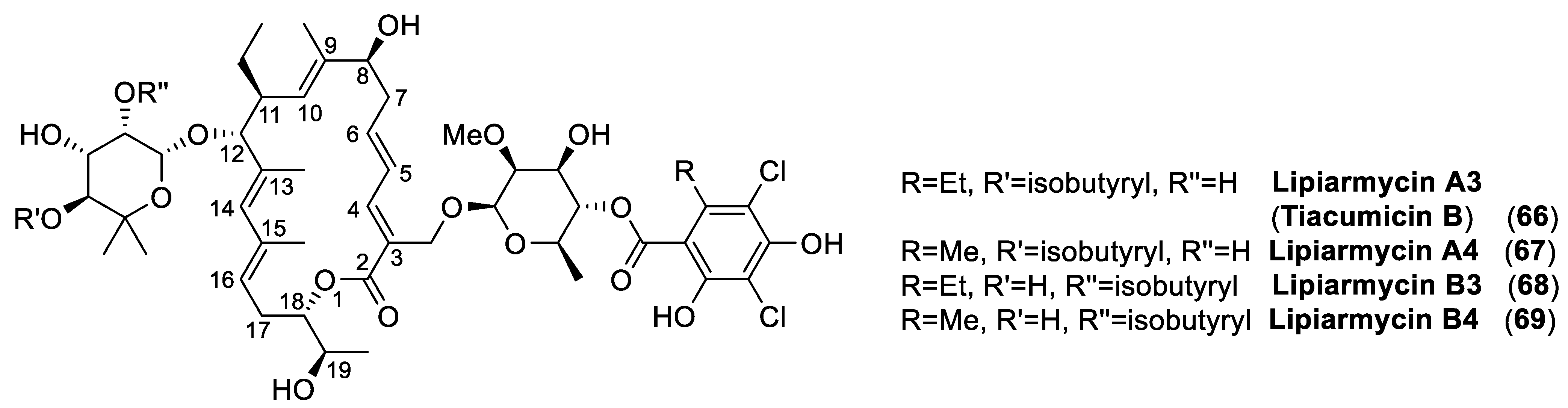
4.3. Lipiarmycins
5. Actinomycetes Are still Considered a Source of Bioactive Compounds
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- American Chemical Society International Historic Chemical Landmarks. Discovery and Development of Penicillin. Available online: http://www.acs.org/content/acs/en/education/whatischemistry/landmarks/flemingpenicillin.html (accessed on 28 April 2020).
- Aminov, R. A brief history of the antibiotic era: Lessons learned and challenges for the future. Front. Microbiol. 2010, 1, 134. [Google Scholar] [CrossRef] [Green Version]
- Grasso, L.L.; Martino, D.C.; Alduina, R. Production of antibacterial compounds from Actinomycetes. In Actinobacteria-Basics and Biotechnological Applications; Dhanasekaran, D., Jiang, Y., Eds.; InTech: Rijeka, Croatia, 2016; p. Ch. 07. [Google Scholar]
- Ventola, C.L. The antibiotic resistance crisis: Part 1: Causes and threats. Pharm. Ther. 2015, 40, 277–283. [Google Scholar]
- Chang, Q.; Wang, W.; Regev-Yochay, G.; Lipsitch, M.; Hanage, W.P. Antibiotics in agriculture and the risk to human health: How worried should we be? Evol. Appl. 2015, 8, 240–247. [Google Scholar] [CrossRef] [Green Version]
- Buttimer, C.; McAuliffe, O.; Ross, R.P.; Hill, C.; O’Mahony, J.; Coffey, A. Bacteriophages and bacterial plant diseases. Front. Microbiol. 2017, 8, 34. [Google Scholar] [CrossRef] [Green Version]
- Ventola, C.L. The antibiotic resistance crisis: Part 2: Management strategies and new agents. Pharm. Ther. 2015, 40, 344. [Google Scholar]
- Llewelyn, M.J.; Fitzpatrick, J.M.; Darwin, E.; Tonkin-Crine, S.; Gorton, C.; Paul, J.; Peto, T.E.A.; Yardley, L.; Hopkins, S.; Walker, A.S. The antibiotic course has had its day. BMJ 2017, 358, j3418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ventura, M.; Canchaya, C.; Tauch, A.; Chandra, G.; Fitzgerald, G.F.; Chater, K.F.; van Sinderen, D. Genomics of actinobacteria: Tracing the evolutionary history of an ancient phylum. Microbiol. Mol. Biol. R 2007, 71, 495–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barka, E.A.; Vatsa, P.; Sanchez, L.; Gaveau-Vaillant, N.; Jacquard, C.; Klenk, H.-P.; Clément, C.; Ouhdouch, Y.; van Wezel, G.P. Taxonomy, physiology, and natural products of actinobacteria. Microbiol. Mol. Biol. R. 2016, 80, 1–43. [Google Scholar] [CrossRef] [Green Version]
- Atlas, R.M. Principles of Microbiology, 2nd ed.; WCB McGrill-Hill: New York, NY, USA, 1997; ISBN 0815108893. [Google Scholar]
- Nodwell, J.R. Antimicrobials: Expressing antibiotic gene clusters. Nat. Microbiol. 2017, 2, 17061. [Google Scholar] [CrossRef] [PubMed]
- Craney, A.; Ahmed, S.; Nodwell, J. Towards a new science of secondary metabolism. J. Antibiot. 2013, 66, 387–400. [Google Scholar] [CrossRef] [PubMed]
- Thomashow, L.S.; Bonsall, R.F.; Weller, D.M. Detection of antibiotics produced by soil and rhizosphere microbes in situ. In Secondary Metabolites in Soil Ecology; Karlovsky, P., Ed.; Springer: Berlin/Heidelberg, Germany, 2008; pp. 23–36. [Google Scholar]
- Sánchez, L.; Brana, A. Cell density influences antibiotic biosynthesis in Streptomyces clavuligerus. Microbiology 1996, 142, 1209–1220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mlot, C. Antibiotics in nature: Beyond biological warfare. Science 2009, 324, 1637–1639. [Google Scholar] [CrossRef] [PubMed]
- Aminov, R.I. The role of antibiotics and antibiotic resistance in nature. Environ. Microbiol. 2009, 11, 2970–2988. [Google Scholar] [CrossRef] [PubMed]
- Linares, J.F.; Gustafsson, I.; Baquero, F.; Martinez, J. Antibiotics as intermicrobial signaling agents instead of weapons. Proc. Natl. Acad. Sci. USA 2006, 103, 19484–19489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casertano, M.; Menna, M.; Imperatore, C. The ascidian-derived metabolites with antimicrobial properties. Antibiotics 2020, 9, 510. [Google Scholar] [CrossRef] [PubMed]
- Stincone, P.; Brandelli, A. Marine bacteria as source of antimicrobial compounds. Crit. Rev. Biotechnol. 2020, 40, 306–319. [Google Scholar] [CrossRef] [PubMed]
- Noller, H.F. Structure of ribosomal RNA. Annu. Rev. Biochem. 1984, 53, 119–162. [Google Scholar] [CrossRef] [PubMed]
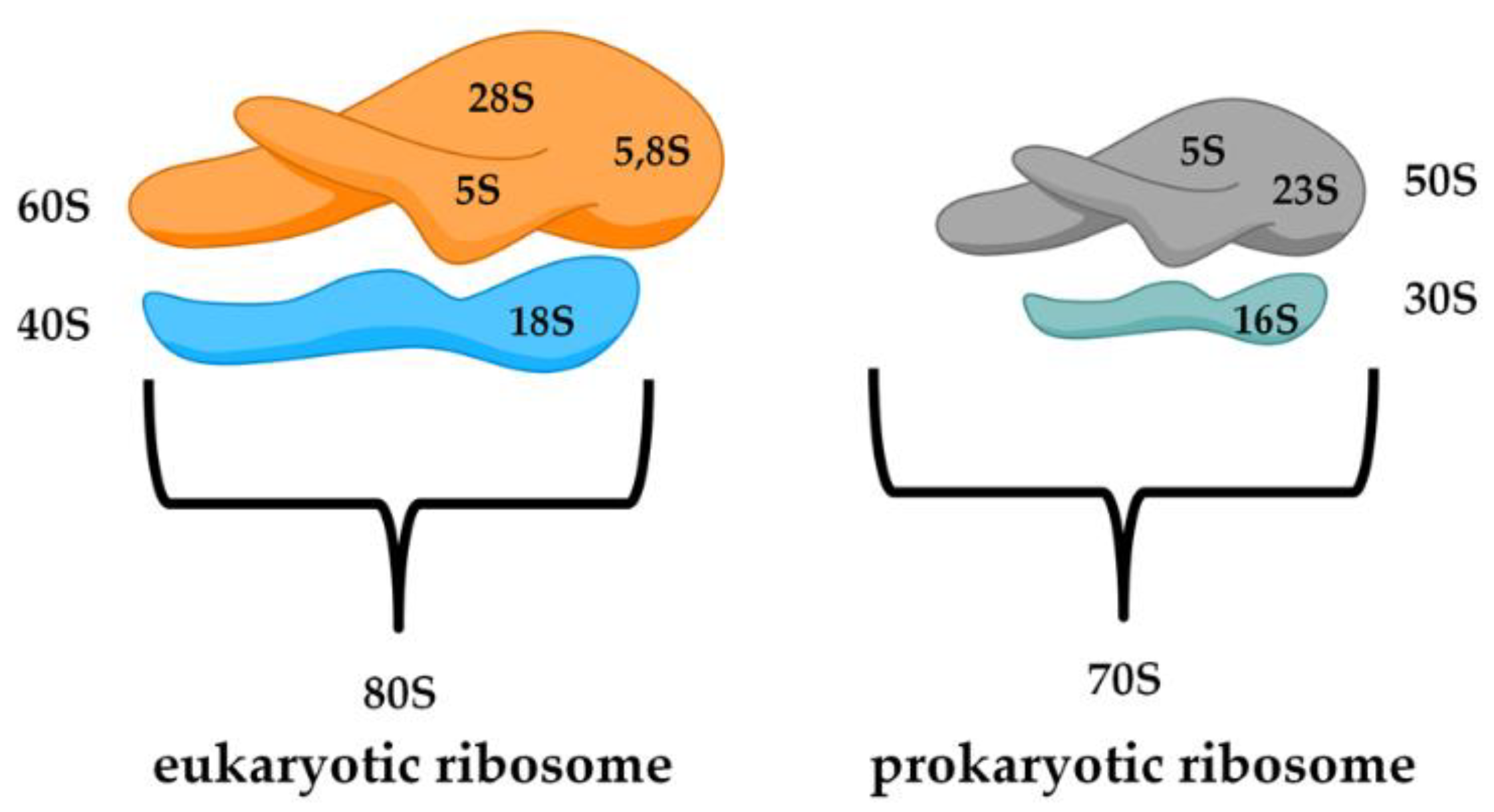
- Lambert, T. Antibiotics that affect the ribosome. Rev. Sci. Tech. OIE 2012, 31, 57. [Google Scholar] [CrossRef] [Green Version]
- Böttger, E.C.; Springer, B.; Prammananan, T.; Kidan, Y.; Sander, P. Structural basis for selectivity and toxicity of ribosomal antibiotics. EMBO Rep. 2001, 2, 318–323. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Villa, D.; Aguilar, M.R.; Rojo, L. Folic acid antagonists: Antimicrobial and immunomodulating mechanisms and applications. Int. J. Mol. Sci. 2019, 20, 4996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silver, L.L. Appropriate targets for antibacterial drugs. Csh. Perspect. Med. 2016, 6, a030239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruer, S.; Pinotsis, N.; Steadman, D.; Waksman, G.; Remaut, H. Virulence-targeted antibacterials: Concept, promise, and susceptibility to resistance mechanisms. Chem. Biol. Drug Des. 2015, 86, 379–399. [Google Scholar] [CrossRef] [PubMed]
- Davin-Regli, A.; Bolla, J.-M.; James, C.E.; Lavigne, J.-P.; Chevalier, J.; Garnotel, E.; Molitor, A. Membrane permeability and regulation of drug “influx and efflux” in enterobacterial pathogens. Curr. Drug Targets 2008, 9, 750–759. [Google Scholar] [CrossRef] [PubMed]
- Sandegren, L.; Andersson, D.I. Bacterial gene amplification: Implications for the evolution of antibiotic resistance. Nat. Rev. Microbiol. 2009, 7, 578–588. [Google Scholar] [CrossRef]
- Morar, M.; Wright, G.D. The genomic enzymology of antibiotic resistance. Annu. Rev. Genet. 2010, 44, 25–51. [Google Scholar] [CrossRef] [PubMed]
- Wondrack, L.; Massa, M.; Yang, B.; Sutcliffe, J. Clinical strain of Staphylococcus aureus inactivates and causes efflux of macrolides. Antimicrob. Agents Chemother. 1996, 40, 992–998. [Google Scholar] [CrossRef] [Green Version]
- Matsuoka, M.; Endou, K.; Kobayashi, H.; Inoue, M.; Nakajima, Y. A plasmid that encodes three genes for resistance to macrolide antibiotics in Staphylococcus aureus. FEMS Microbiol. Lett. 1998, 167, 221–227. [Google Scholar] [CrossRef]
- Yang, W.; Moore, I.F.; Koteva, K.P.; Bareich, D.C.; Hughes, D.W.; Wright, G.D. Tetx is a flavin-dependent monooxygenase conferring resistance to tetracycline antibiotics. J. Biol. Chem. 2004, 279, 52346–52352. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.-M.; Rock, C.O. Membrane lipid homeostasis in bacteria. Nat. Rev. Microbiol. 2008, 6, 222–233. [Google Scholar] [CrossRef]
- Pinho, M.G.; Kjos, M.; Veening, J.-W. How to get (a)round: Mechanisms controlling growth and division of coccoid bacteria. Nat. Rev. Microbiol. 2013, 11, 601–614. [Google Scholar] [CrossRef] [Green Version]
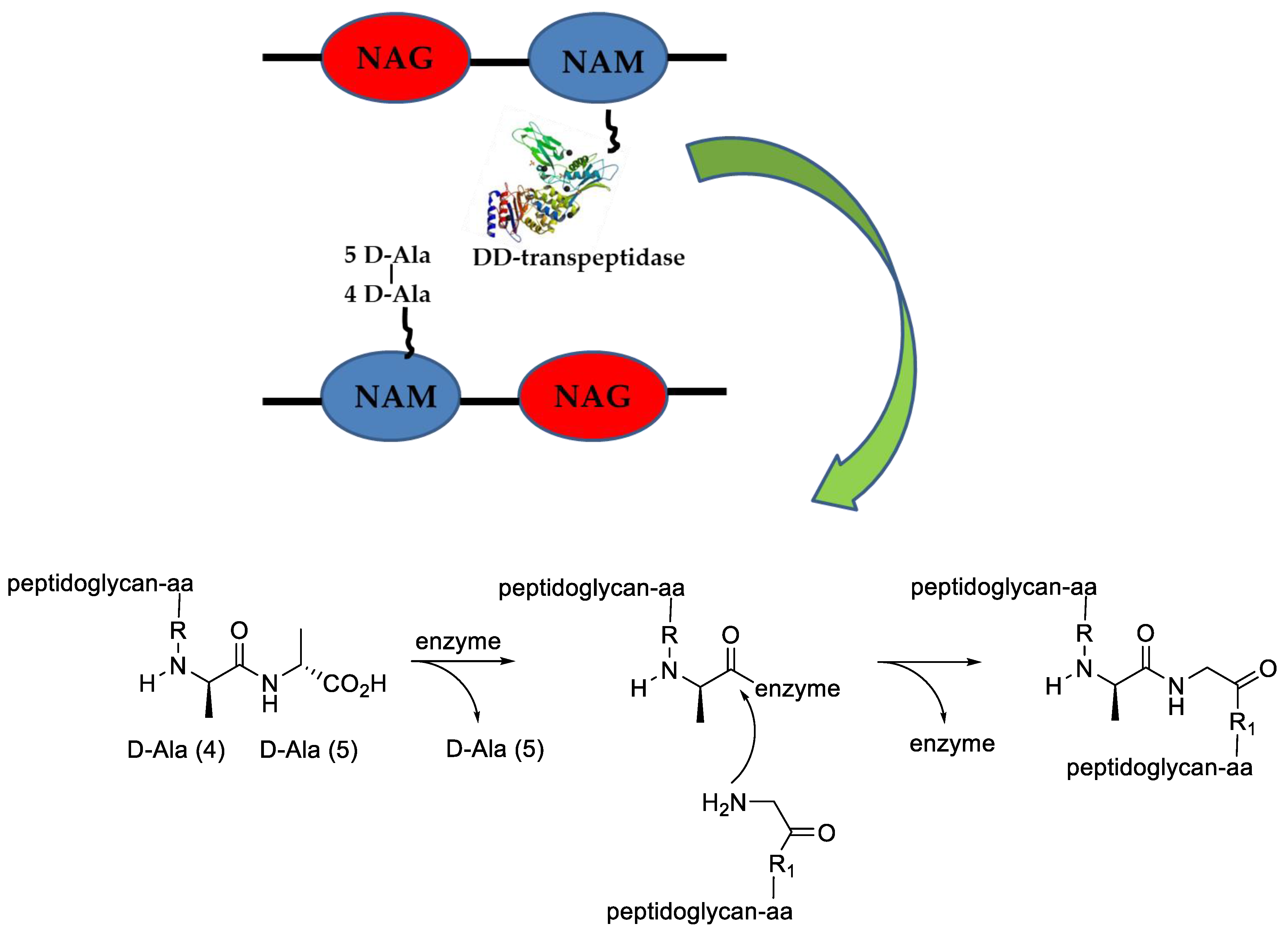
- Vollmer, W.; Blanot, D.; De Pedro, M.A. Peptidoglycan structure and architecture. FEMS Microbiol. Rev. 2008, 32, 149–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sauvage, E.; Herman, R.; Petrella, S.; Duez, C.; Bouillenne, F.; Frère, J.-M.; Charlier, P. Crystal structure of the Actinomadura R39 DD-peptidase reveals new domains in penicillin-binding proteins. J. Biol. Chem. 2005, 280, 31249–31256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalhoff, A.; Janjic, N.; Echols, R. Redefining penems. Biochem. Pharmacol. 2006, 71, 1085–1095. [Google Scholar] [CrossRef] [PubMed]
- Strains Producing Special Compounds or Enzymes-DSMZ. Available online: https://www.dsmz.de/fileadmin/Bereiche/ChiefEditors/SpecificCatalogues/Production.pdf (accessed on 28 April 2020).
- DSMZ-Production, Transformation, Enzyme Studies. Available online: https://www.dsmz.de/fileadmin/Bereiche/Microbiology/Dateien/dsmz-production-strains.pdf (accessed on 28 April 2020).
- Nagarajan, R.; Boeck, L.D.; Gorman, M.; Hamill, R.L.; Higgens, C.E.; Hoehn, M.M.; Stark, W.M.; Whitney, J.G. Beta-lactam antibiotics from Streptomyces. J. Am. Chem. Soc. 1971, 93, 2308–2310. [Google Scholar] [CrossRef] [PubMed]
- Stapley, E.O.; Jackson, M.; Hernandez, S.; Zimmerman, S.B.; Currie, S.A.; Mochales, S.; Mata, J.M.; Woodruff, H.B.; Hendlin, D. Cephamycins, a new family of β-lactam antibiotics I. Production by Actinomycetes, including Streptomyces lactamdurans sp. N. Antimicrob. Agents Chemother. 1972, 2, 122–131. [Google Scholar] [CrossRef] [Green Version]
- Higgens, C.; Hamill, R.; Sands, T.; Hoehn, M.; Davis, N.; Nagarajan, R.; Boeck, L. The occurrence of deacetoxy-cephalosporin C in fungi and Streptomycetes. J. Antibiot. 1974, 27, 298–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakao, Y.; Kitano, K.; Kintaka, K.; Suzuki, S.; Katamoto, K.; Nara, K. Production of Deacetoxycephalosporin C. U.S. Patent US3979260, 7 September 1976. [Google Scholar]
- Shomura, T.; Watanabe, H.; Ogawa, Y.; Ohba, K.; Kondo, Y.; Kojima, M.; Inoje, S.; Niida, T. Process for Preparing a Cephamycin Type Antibiotic Substance. U.S. Patent US3974035A, 10 August 1976. [Google Scholar]
- Liras, P.; Demain, A. Chapter 16 Enzymology of β-Lactam Compounds with Cephem Structure Produced by Actinomycete; Academic Press: Cambridge, MA, USA, 2009; Volume 458, pp. 401–429. [Google Scholar]
- Nozaki, Y.; Kitano, K.; Imada, A. Blocked mutants in the biosynthesis of carbapenem antibiotics from Streptomyces griseus subsp. Cryophilus. Agric. Biol. Chem. 1984, 48, 37–44. [Google Scholar] [CrossRef]
- Williamson, J.M.; Inamine, E.; Wilson, K.E.; Douglas, A.W.; Liesch, J.M.; Albers-Schönberg, G. Biosynthesis of the beta-lactam antibiotic, thienamycin, by Streptomyces cattleya. J. Biol. Chem. 1985, 260, 4637–4647. [Google Scholar] [CrossRef]
- Núñez, L.E.; Méndez, C.; Braña, A.F.; Blanco, G.; Salas, J.A. The biosynthetic gene cluster for the β-lactam carbapenem thienamycin in Streptomyces cattleya. Chem. Biol. 2003, 10, 301–311. [Google Scholar] [CrossRef] [Green Version]
- Aoki, H.; Okuhara, M. Natural beta-lactam antibiotics. Annu. Rev. Microbiol. 1980, 34, 159–181. [Google Scholar] [CrossRef] [PubMed]
- Imada, A.; Nozaki, Y.; Kintaka, K.; Okonogi, K.; Kitano, K.; Harada, S. C-19393 S2 and H2, new carbapenem antibiotics. J. Antibiot. 1980, 33, 1417–1424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomita, K.; Tsukiura, H.; Kawaguchi, H. Fermentation Process for Producing Nocardicins A and B. U.S. Patent US4320199A, 16 March 1982. [Google Scholar]
- Maslow, M.J.; Portal-Celhay, C. 27-rifamycins. In Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases, 8th ed.; Bennett, J.E., Dolin, R., Blaser, M.J., Eds.; Content Repository Only!: Philadelphia, PA, USA, 2015; pp. 339–349.e333. [Google Scholar]
- Neuman, M.; De Simone, C. Vademecum degli Antibiotici ed Agenti Chemioterapici Antinfettivi; Marrapese, 1994. [Google Scholar]
- Lancini, G.; Grandi, M. Biosynthesis of ansamycins. In Biosynthesis; Springer: Berlin/Heidelberg, Germany, 1981; pp. 12–40. [Google Scholar]
- Wilson, M.C.; Gulder, T.A.M.; Mahmud, T.; Moore, B.S. Shared biosynthesis of the saliniketals and rifamycins in Salinispora arenicola is controlled by the sare1259-encoded cytochrome P450. J. Am. Chem. Soc. 2010, 132, 12757–12765. [Google Scholar] [CrossRef] [Green Version]
- Williams, J.; Sefton, A. Comparison of macrolide antibiotics. J. Antimicrob. Chemother. 1993, 31, 11–26. [Google Scholar] [CrossRef]
- Saribas, Z.; Tunckanat, F.; Pinar, A. Prevalence of erm genes encoding macrolide-lincosamide-streptogramin (MLS) resistance among clinical isolates of Staphylococcus aureus in a Turkish University hospital. Clin. Microbiol. Infect. 2006, 12, 797–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arthur, M.; Autissier, D.; Courvalin, P. Analysis of the nucleotide sequence of the ereB gene encoding the erythromydn esterase type II. Nucleic Acid Res. 1986, 14, 4987–4999. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, T.; Dougherty, T.J.; Magee, T.V. 7.18-macrolide antibiotics. In Comprehensive Medicinal Chemistry II; Taylor, J.B., Triggle, D.J., Eds.; Elsevier: Oxford, UK, 2007; pp. 519–566. [Google Scholar]
- Matsuhashi, Y.; Ogawa, H.; Nagaoka, K. The enzymatic interconversion between midecamycin A1 and A3. J. Antibiot. 1979, 32, 777–779. [Google Scholar] [CrossRef] [PubMed]
- Horan, A.C.; Brodski, B.C. Micromonospora rosaria sp. Nov., nom. Rev., the rosaramicin producer. Int. J. Syst. Evol. Microbiol. 1986, 36, 478–480. [Google Scholar] [CrossRef] [Green Version]
- Weber, J.M.; Wierman, C.; Hutchinson, C.R. Genetic analysis of erythromycin production in Streptomyces erythreus. J. Bacteriol. 1985, 164, 425–433. [Google Scholar] [CrossRef] [Green Version]
- Miller, E.S.; Woese, C.R.; Brenner, S. Description of the erythromycin-producing bacterium Arthrobacter sp. Strain NRRL B-3381 as Aeromicrobium erythreum gen. Nov., sp. Nov. Int. J. Syst. Evol. Microbiol. 1991, 41, 363–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Kim, S.G.; Blenis, J. Rapamycin: One drug, many effects. Cell Metab. 2014, 19, 373–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spížek, J.; Řezanka, T. Lincomycin, clindamycin and their applications. Appl. Microbiol. Biotechnol. 2004, 64, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Lee, M.-J.; Choi, Y.-E.; Chun, G.-T.; Jeong, Y.-S. Optimization of cultivation medium and fermentation parameters for lincomycin production by Streptomyces lincolnensis. Biotechnol. Bioprocess Eng. 2014, 19, 1014–1021. [Google Scholar] [CrossRef]
- Melancon, C.E., III. Biochemistry: Elusive source of sulfur unravelled. Nature 2015, 518, 45–46. [Google Scholar] [CrossRef] [PubMed]
- Spížek, J.; Řezanka, T. Lincomycin, cultivation of producing strains and biosynthesis. Appl. Microbiol. Biot. 2004, 63, 510–519. [Google Scholar] [CrossRef] [PubMed]
- Spížek, J.; Řezanka, T. Lincosamides: Chemical structure, biosynthesis, mechanism of action, resistance, and applications. Biochem. Pharmacol. 2017, 133, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Bibikova, M.; Singal, E.; Ivanitskaia, L.; Zhdanovich, I. Production of lincomycin by Micromonospora halophytica culture. Antibiot. Khimioterapiia Antibiot. Chemoterapy 1989, 34, 723–726. [Google Scholar]
- Kuznetsov, V.; Bushueva, O. Effect of the temperature of preservation of lyophilized actinomycete spores (producers of streptomycin and lincomycin) on their viability and variability. Antibiotiki 1974, 19, 690. [Google Scholar]
- Nelson, M.L.; Levy, S.B. The history of the tetracyclines. Ann. N. Y. Acad. Sci. 2011, 1241, 17–32. [Google Scholar] [CrossRef] [PubMed]
- Gualerzi, C.O.; Brandi, L.; Fabbretti, A.; Pon, C.L. Antibiotics: Targets, Mechanisms and Resistance; John Wiley & Sons: Hoboken, NJ, USA, 2013. [Google Scholar]
- Alexander, G.; Joseph, L. Production of Tetracycline and Substituted Tetracyclines. U.S. Patent US2712517A, 5 July 1955. [Google Scholar]
- Innes, C.M.J.; Allan, E.J. Induction, growth and antibiotic production of Streptomyces viridifaciens l-form bacteria. J. Appl. Microbiol. 2001, 90, 301–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lotvin, J.A.; Ryan, M.J.; Strathy, N. Cloning of the Biosynthetic Pathway Genes for Chlortetracycline Production from Streptomyces aureofaciens & Their Expression in Steptomyces lividans. EP0468220A3, 29 January 1992. [Google Scholar]
- Li, H.; Ye, R.; Lin, G.; Zhu, D.; Mao, Q. Protein expression analysis of a high-demeclocycline producing strain of Streptomyces aureofaciens and the roles of CtcH and CtcJ in demeclocycline biosynthesis. Bioresour. Bioprocess. 2016, 3, 46. [Google Scholar] [CrossRef] [Green Version]
- Grill, M.F.; Maganti, R.K. Neurotoxic effects associated with antibiotic use: Management considerations. Br. J. Clin. Pharmacol. 2011, 72, 381–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taber, H.W.; Mueller, J.P.; Miller, P.F.; Arrow, A.S. Bacterial uptake of aminoglycoside antibiotics. Microbiol. Rev. 1987, 51, 439–457. [Google Scholar] [CrossRef]
- Hancock, R.E.; Farmer, S.W.; Li, Z.S.; Poole, K. Interaction of aminoglycosides with the outer membranes and purified lipopolysaccharide and OmpF porin of Escherichia coli. Antimicrob. Agents Chemother. 1991, 35, 1309–1314. [Google Scholar] [CrossRef] [Green Version]
- Joshi, T.; Voo, Z.X.; Graham, B.; Spiccia, L.; Martin, L.L. Real-time examination of aminoglycoside activity towards bacterial mimetic membranes using quartz crystal microbalance with dissipation monitoring (QCM-D). Biochim. Biophys. Acta BBA Biomembr. 2015, 1848, 385–391. [Google Scholar] [CrossRef] [Green Version]
- Kotra, L.P.; Haddad, J.; Mobashery, S. Aminoglycosides: Perspectives on mechanisms of action and resistance and strategies to counter resistance. Antimicrob. Agents Chemother. 2000, 44, 3249–3256. [Google Scholar] [CrossRef] [Green Version]
- Dunkle, J.A.; Vinal, K.; Desai, P.M.; Zelinskaya, N.; Savic, M.; West, D.M.; Conn, G.L.; Dunham, C.M. Molecular recognition and modification of the 30S ribosome by the aminoglycoside-resistance methyltransferase NpmA. Proc. Natl. Acad. Sci. USA 2014, 111, 6275–6280. [Google Scholar] [CrossRef] [Green Version]
- Wachino, J.-I.; Arakawa, Y. Exogenously acquired 16S rRNA methyltransferases found in aminoglycoside-resistant pathogenic gram-negative bacteria: An update. Drug Resist. Update 2012, 15, 133–148. [Google Scholar] [CrossRef] [PubMed]
- Hotta, K.; Takahashi, A.; Okami, Y.; Umezawa, H. Relationship between antibiotic resistance and antibiotic productivity in Actinomycetes which produce aminoglycoside antibiotics. J. Antibiot. 1983, 36, 1789–1791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piwowarski, J.M.; Shaw, P.D. Streptomycin resistance in a streptomycin-producing microorganism. Antimicrob. Agents Chemother. 1979, 16, 176–182. [Google Scholar] [CrossRef] [Green Version]
- Huddleston, A.S.; Cresswell, N.; Neves, M.; Beringer, J.E.; Baumberg, S.; Thomas, D.I.; Wellington, E. Molecular detection of streptomycin-producing Streptomycetes in brazilian soils. Appl. Environ. Microbiol. 1997, 63, 1288–1297. [Google Scholar] [CrossRef] [Green Version]
- Shapiro, S. Regulation of Secondary Metabolism in Actinomycetes; Taylor & Francis: Oxford, UK, 1989. [Google Scholar]
- Kharel, M.K.; Basnet, D.B.; Lee, H.C.; Liou, K.; Woo, J.S.; Kim, B.-G.; Sohng, J.K. Isolation and characterization of the tobramycin biosynthetic gene cluster from Streptomyces tenebrarius. FEMS Microbiol. Lett. 2004, 230, 185–190. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, R.; Islas, L.; Obregon, A.-M.; Escalante, L.; Sanchez, S. Gentamicin formation in Micromonospora purpurea: Stimulatory effect of ammonium. J. Antibiot. 1995, 48, 479–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Q.; Huang, F.; Leadlay, P.F.; Spencer, J.B. The neomycin biosynthetic gene cluster of Streptomyces fradiae NCIMB 8233: Genetic and biochemical evidence for the roles of two glycosyltransferases and a deacetylase. Org. Biomol. Chem. 2008, 6, 3306–3314. [Google Scholar] [CrossRef] [PubMed]
- Hancock, R.E.W.; Chapple, D.S. Peptide antibiotics. Antimicrob. Agents Chemother. 1999, 43, 1317–1323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richard, H.B. Biosynthesis and genetic engineering of lipopeptide antibiotics related to daptomycin. Curr. Top. Med. Chem. 2008, 8, 618–638. [Google Scholar] [CrossRef]
- Mukhtar, T.A.; Wright, G.D. Streptogramins, oxazolidinones, and other inhibitors of bacterial protein synthesis. Chem. Rev. 2005, 105, 529–542. [Google Scholar] [CrossRef]
- Yim, G.; Thaker, M.N.; Koteva, K.; Wright, G. Glycopeptide antibiotic biosynthesis. J. Antibiot. 2014, 67, 31–41. [Google Scholar] [CrossRef] [Green Version]
- Marahiel, M.A. Working outside the protein-synthesis rules: Insights into non-ribosomal peptide synthesis. J. Pept. Sci. 2009, 15, 799–807. [Google Scholar] [CrossRef]
- Strieker, M.; Tanović, A.; Marahiel, M.A. Nonribosomal peptide synthetases: Structures and dynamics. Curr. Opin. Struct. Biol. 2010, 20, 234–240. [Google Scholar] [CrossRef]
- Martínez-Núñez, M.A.; y López, V.E.L. Nonribosomal peptides synthetases and their applications in industry. Sustain. Chem. Process. 2016, 4, 13. [Google Scholar] [CrossRef]
- Binda, E.; Marinelli, F.; Marcone, G.L. Old and new glycopeptide antibiotics: Action and resistance. Antibiotics 2014, 3, 572–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, H.-K.; Park, Y. Glycopeptide antibiotics: Structure and mechanisms of action. J. Bacteriol. Virol. 2015, 45, 67–78. [Google Scholar] [CrossRef] [Green Version]
- Straus, S.K.; Hancock, R.E. Mode of action of the new antibiotic for gram-positive pathogens daptomycin: Comparison with cationic antimicrobial peptides and lipopeptides. BBA Biomembr. 2006, 1758, 1215–1223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahne, D.; Leimkuhler, C.; Lu, W.; Walsh, C. Glycopeptide and lipoglycopeptide antibiotics. Chem. Rev. 2005, 105, 425–448. [Google Scholar] [CrossRef] [PubMed]
- Pootoolal, J.; Neu, J.; Wright, G.D. Glycopeptide antibiotic resistance. Annu. Rev. Pharmacol. 2002, 42, 381–408. [Google Scholar] [CrossRef] [PubMed]
- Mast, Y.; Wohlleben, W. Streptogramins—Two are better than one! Int. J. Med. Microbiol. 2014, 304, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.H.; Lee, K.W.; Cha, K.H. New Streptomyces filamentosus Variant and Method for Producing Daptomycin Using Same. WO2015093839A1, 25 June 2015. [Google Scholar]
- Baltz, R.H. Daptomycin: Mechanisms of action and resistance, and biosynthetic engineering. Curr. Opin. Chem. Biol. 2009, 13, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Penn, J.; Li, X.; Whiting, A.; Latif, M.; Gibson, T.; Silva, C.J.; Brian, P.; Davies, J.; Miao, V.; Wrigley, S.K.; et al. Heterologous production of daptomycin in Streptomyces lividans. J. Ind. Microbiol. Biot. 2006, 33, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Miao, V.; Coeffet-LeGal, M.-F.; Brian, P.; Brost, R.; Penn, J.; Whiting, A.; Martin, S.; Ford, R.; Parr, I.; Bouchard, M. Daptomycin biosynthesis in Streptomyces roseosporus: Cloning and analysis of the gene cluster and revision of peptide stereochemistry. Microbiology 2005, 151, 1507–1523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Crécy-Lagard, V.; Saurin, W.; Thibaut, D.; Gil, P.; Naudin, L.; Crouzet, J.; Blanc, V. Streptogramin B biosynthesis in Streptomyces pristinaespiralis and Streptomyces virginiae: Molecular characterization of the last structural peptide synthetase gene. Antimicrob. Agents Chemother. 1997, 41, 1904–1909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Food and Drug Administration. Determination That ALBAMYCIN (Novobiocin Sodium) Capsule, 250 Milligrams, Was Withdrawn from Sale for Reasons of Safety or Effectiveness; The Federal Register, 2011.
- Steffensky, M.; Mühlenweg, A.; Wang, Z.-X.; Li, S.-M.; Heide, L. Identification of the novobiocin biosynthetic gene cluster of Streptomyces spheroides NCIB 11891. Antimicrob. Agents Chemother. 2000, 44, 1214–1222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, T.; Ishida, Y.; Otoguro, M.; Hatano, K.; Labeda, D.; Price, N.P.; Suzuki, K.-i. Reclassification of Streptomyces caeruleus as a synonym of Actinoalloteichus cyanogriseus and reclassification of Streptomyces spheroides and Streptomyces laceyi as later synonyms of Streptomyces niveus. Int. J. Syst. Evol. Microbiol. 2008, 58, 2812–2814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maxwell, A. The interaction between coumarin drugs and DNA gyrase. Mol. Microbiol. 1993, 9, 681–686. [Google Scholar] [CrossRef] [PubMed]
- Anderle, C.; Stieger, M.; Burrell, M.; Reinelt, S.; Maxwell, A.; Page, M.; Heide, L. Biological activities of novel gyrase inhibitors of the aminocoumarin class. Antimicrob. Agents Chemother. 2008, 52, 1982–1990. [Google Scholar] [CrossRef] [Green Version]
- Schnitzer, R.J.; Hawking, F. (Eds.) Experimental Chemotherapy; Academic Press: Cambridge, MA, USA, 1963; Volume 1. [Google Scholar]
- Vickers, A.A.; Chopra, I.; O’neill, A.J. Intrinsic novobiocin resistance in Staphylococcus saprophyticus. Antimicrob. Agents Chemother. 2007, 51, 4484–4485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silver, L.L. Fosfomycin: Mechanism and resistance. Csh. Perspect. Med. 2017, 7, a025262. [Google Scholar] [CrossRef] [Green Version]
- Takahata, S.; Ida, T.; Hiraishi, T.; Sakakibara, S.; Maebashi, K.; Terada, S.; Muratani, T.; Matsumoto, T.; Nakahama, C.; Tomono, K. Molecular mechanisms of fosfomycin resistance in clinical isolates of Escherichia coli. Int. J. Antimicrob. Agents 2010, 35, 333–337. [Google Scholar] [CrossRef]
- Kuzuyama, T. Fosfomycin biosynthesis and self-resistance mechanism of the producing organism Streptomyces wedmorensis. Actinomycetologica 1998, 12, 71–74. [Google Scholar] [CrossRef]
- Rogers, T.O.; Birnbaum, J. Biosynthesis of fosfomycin by Streptomyces fradiae. Antimicrob. Agents Chemother. 1974, 5, 121–132. [Google Scholar] [CrossRef] [Green Version]
- Hidaka, T.; Goda, M.; Kuzuyama, T.; Takei, N.; Hidaka, M.; Seto, H. Cloning and nucleotide sequence of fosfomycin biosynthetic genes of Streptomyces wedmorensis. MGG 1995, 249, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Hendlin, D.; Stapley, E.; Jackson, M.; Wallick, H.; Miller, A.; Wolf, F.; Miller, T.; Chaiet, L.; Kahan, F.; Foltz, E. Phosphonomycin, a new antibiotic produced by strains of Streptomyces. Science 1969, 166, 122–123. [Google Scholar] [CrossRef] [PubMed]
- Woodyer, R.D.; Shao, Z.; Thomas, P.M.; Kelleher, N.L.; Blodgett, J.A.; Metcalf, W.W.; van der Donk, W.A.; Zhao, H. Heterologous production of fosfomycin and identification of the minimal biosynthetic gene cluster. Chem. Biol. 2006, 13, 1171–1182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolaou, K.C.; Vourloumis, D.; Winssinger, N.; Baran, P.S. The art and science of total synthesis at the dawn of the twenty-first century. Angew. Chem. Int. Ed. 2000, 39, 44–122. [Google Scholar] [CrossRef]
- Wright, P.M.; Seiple, I.B.; Myers, A.G. The evolving role of chemical synthesis in antibacterial drug discovery. Angew. Chem. Int. Ed. 2014, 53, 8840–8869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheehan, J.C.; Henery-Logan, K.R. The total synthesis of penicillin V. J. Am. Chem. Soc. 1957, 79, 1262–1263. [Google Scholar] [CrossRef]
- Newton, G.G.F.; Abraham, E.P. Cephalosporin C, a new antibiotic containing sulphur and D-α-aminoadipic acid. Nature 1955, 175, 548. [Google Scholar] [CrossRef] [PubMed]
- Weissenburger, H.; Van Der Hoeven, M. An efficient nonenzymatic conversion of benzylpenicillin to 6-aminopenicillanic acid. Recueil des Travaux Chimiques des Pays-Bas 1970, 89, 1081–1084. [Google Scholar] [CrossRef]
- Wegman, M.A.; Janssen, M.H.A.; van Rantwijk, F.; Sheldon, R.A. Towards biocatalytic synthesis of β-lactam antibiotics. Adv. Synth. Catal. 2001, 343, 559–576. [Google Scholar] [CrossRef]
- Parmar, A.; Kumar, H.; Marwaha, S.S.; Kennedy, J.F. Advances in enzymatic transformation of penicillins to 6-aminopenicillanic acid (6-APA). Biotechnol. Adv. 2000, 18, 289–301. [Google Scholar] [CrossRef]
- Arroyo, M.; de la Mata, I.; Acebal, C.; Pilar Castillón, M. Biotechnological applications of penicillin acylases: State-of-the-art. Appl. Microbiol. Biotechnol. 2003, 60, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Cooper, R.D.G.; Hatfield, L.D.; Spry, D.O. Chemical interconversion of the Beta-lactam antibiotics. Acc. Chem. Res. 1973, 6, 32–40. [Google Scholar] [CrossRef]
- Morin, R.B.; Jackson, B.G.; Mueller, R.A.; Lavagnino, E.R.; Scanlon, W.B.; Andrews, S.L. Chemistry of cephalosporin antibiotics. XV. Transformations of penicillin sulfoxide. Synthesis of cephalosporin compounds. J. Am. Chem. Soc. 1969, 91, 1401–1407. [Google Scholar] [CrossRef]
- Kukolja, S.; Lammert, S.R.; Gleissner, M.R.B.; Ellis, A.I. Azetidinone antibiotics. 17. A rearrangement of penicillin sulfoxides to 3-methylenecephams via sulfinyl intermediates. J. Am. Chem. Soc. 1976, 98, 5040–5041. [Google Scholar] [CrossRef] [PubMed]
- Verweij, J.; de Vroom, E. Industrial transformations of penicillins and cephalosporins. Recueil des Travaux Chimiques des Pays-Bas 1993, 112, 66–81. [Google Scholar] [CrossRef]
- Dürckheimer, W.; Blumbach, J.; Lattrell, R.; Scheunemann, K.H. Recent developments in the field of β-lactam antibiotics. Angew. Chem. Int. Ed. 1985, 24, 180–202. [Google Scholar] [CrossRef]
- Dürckheimer, W. Tetracyclines: Chemistry, biochemistry, and structure-activity relations. Angew. Chem. Int. Ed. 1975, 14, 721–734. [Google Scholar] [CrossRef] [PubMed]
- Kuniaki, T.; Takuji, Y.; Hiroki, G.; Yoshiya, O.; Masaaki, T. The first total synthesis of natural (−)-tetracycline. Chem. Lett. 2000, 29, 646–647. [Google Scholar] [CrossRef]
- Bauer, A.; Brönstrup, M. Industrial natural product chemistry for drug discovery and development. Nat. Prod. Rep. 2014, 31, 35–60. [Google Scholar] [CrossRef]
- Charest, M.G.; Lerner, C.D.; Brubaker, J.D.; Siegel, D.R.; Myers, A.G. A convergent enantioselective route to structurally diverse 6-deoxytetracycline antibiotics. Science 2005, 308, 395–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burke, M.D. Flexible tetracycline synthesis yields promising antibiotics. Nat. Chem. Biol. 2009, 5, 77–79. [Google Scholar] [CrossRef] [PubMed]
- Ronn, M.; Zhu, Z.; Hogan, P.C.; Zhang, W.-Y.; Niu, J.; Katz, C.E.; Dunwoody, N.; Gilicky, O.; Deng, Y.; Hunt, D.K.; et al. Process R&D of eravacycline: The first fully synthetic fluorocycline in clinical development. Org. Process Res. Dev. 2013, 17, 838–845. [Google Scholar] [CrossRef]
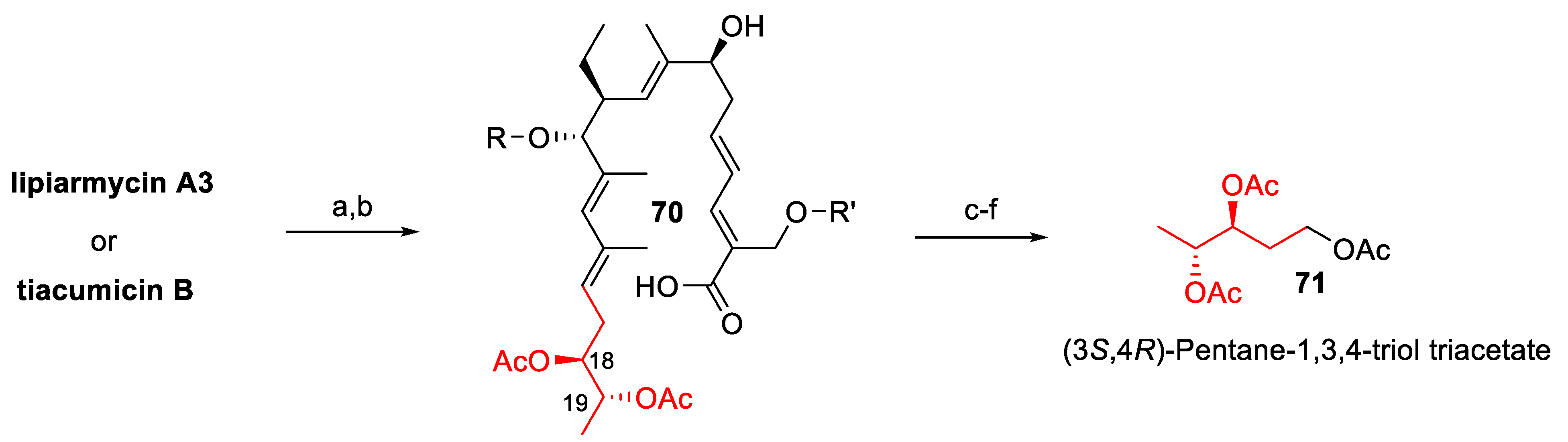
- Erb, W.; Zhu, J. From natural product to marketed drug: The tiacumicin odyssey. Nat. Prod. Rep. 2013, 30, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Roulland, E. Tiacumicin B: An antibiotic of prime importance and a natural product with an inspiring complex structure. Synthesis 2018, 50, 4189–4200. [Google Scholar] [CrossRef]
- Dorst, A.; Gademann, K. Chemistry and biology of the clinically used macrolactone antibiotic fidaxomicin. Helv. Chim. Acta 2020, 103, e2000038. [Google Scholar] [CrossRef]
- Parenti, F.; Pagani, H.; Beretta, G. Lipiarmycin, a new antibiotic from Actinoplanes I. Description of the producer strain and fermentation studies. J. Antibiot. 1975, 28, 247–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coronelli, C.; White, R.J.; Lancini, G.C.; Parenti, F. Lipiarmycin, a new antibiotic from Actinoplanes. II. Isolation, chemical, biological and biochemical characterization. J. Antibiot. 1975, 28, 253–259. [Google Scholar] [CrossRef] [Green Version]
- Somma, S.; Pirali, G.; White, R.; Parenti, F. Lipiarmycin, a new antibiotic from Actinoplanes III. Mechanism of action. J. Antibiot. 1975, 28, 543–549. [Google Scholar] [CrossRef] [Green Version]
- Arnone, A.; Nasini, G.; Cavalleri, B. Structure elucidation of the macrocyclic antibiotic lipiarmycin. J. Chem. Soc. Perkin Trans. 1987, 1, 1353–1359. [Google Scholar] [CrossRef]
- Cavalleri, B.; Arnone, A.; Dimodugno, E.; Nasini, G.; Goldstein, B.P. Structure and biological-activity of lipiarmycin-B. J. Antibiot. 1988, 41, 308–315. [Google Scholar] [CrossRef] [Green Version]
- Omura, S.; Imamura, N.; Oiwa, R.; Kuga, H.; Iwata, R.; Masuma, R.; Iwai, Y. Clostomicins, new antibiotics produced by Micromonospora echinospora subsp. Armeniaca subsp. Nov. I. Production, isolation, and physical-chemical and biological properties. J. Antibiot. 1986, 39, 1407–1412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theriault, R.J.; Karwowski, J.P.; Jackson, M.; Girolami, R.L.; Sunga, G.N.; Vojtko, C.M.; Coen, L.J. Tiacumicins, a novel complex of 18-membered macrolide antibiotics. 1. Taxonomy, fermentation and antibacterial activity. J. Antibiot. 1987, 40, 567–574. [Google Scholar] [CrossRef]
- Swanson, R.N.; Hardy, D.J.; Shipkowitz, N.L.; Hanson, C.W.; Ramer, N.C.; Fernandes, P.B.; Clement, J.J. In vitro and in vivo evaluation of tiacumicins B and C against Clostridium difficile. Antimicrob. Agents Chemother. 1991, 35, 1108–1111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lancaster, J.W.; Matthews, S.J. Fidaxomicin: The newest addition to the armamentarium against Clostridium difficile infections. Clin. Ther. 2012, 34, 1–13. [Google Scholar] [CrossRef]
- Shue, Y.-K.; Hwang, C.-K.; Chiu, Y.-H.; Romero, A.; Babakhani, F.; Sears, P.; Okumu, F. 18-Membered Macrocycles and Analogs Thereof. WO 2006/085838, 17 August 2006. [Google Scholar]
- Kurabachew, M.; Lu, S.H.J.; Krastel, P.; Schmitt, E.K.; Suresh, B.L.; Goh, A.; Knox, J.E.; Ma, N.L.; Jiricek, J.; Beer, D.; et al. Lipiarmycin targets rna polymerase and has good activity against multidrug-resistant strains of Mycobacterium tuberculosis. J. Antimicrob. Chemother. 2008, 62, 713–719. [Google Scholar] [CrossRef] [PubMed]
- Bedeschi, A.; Fonte, P.; Fronza, G.; Fuganti, C.; Serra, S. The co-identity of lipiarmycin A3 and tiacumicin B. Nat. Prod. Commun. 2014, 9, 237–240. [Google Scholar] [CrossRef] [Green Version]
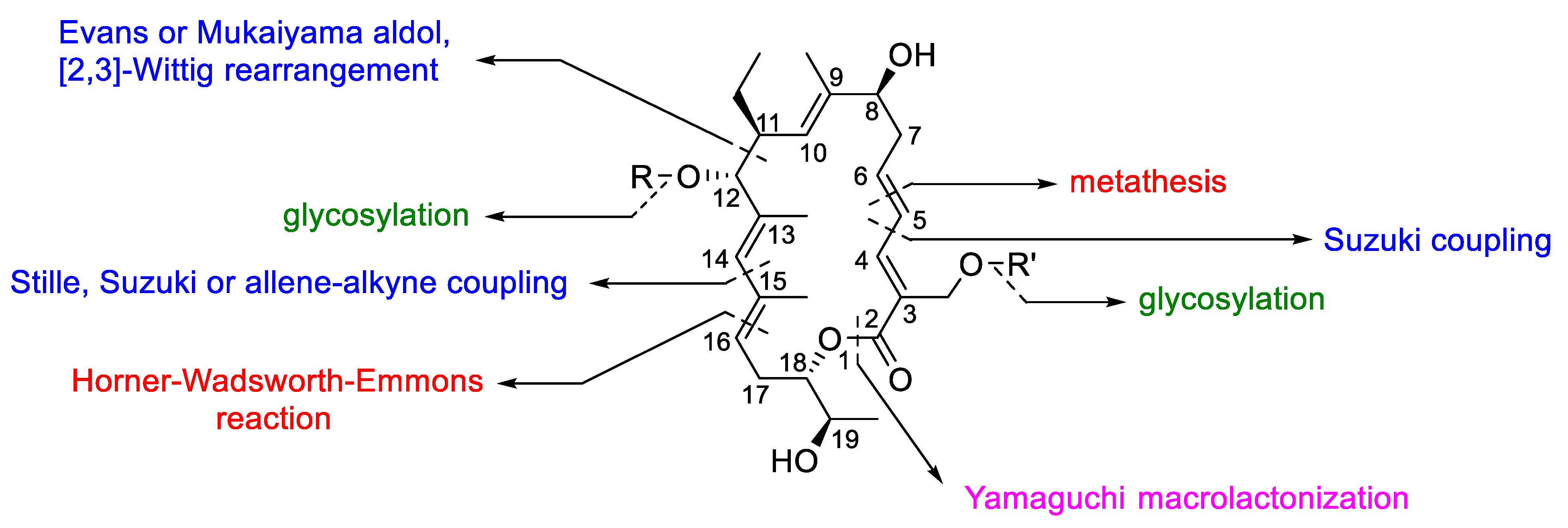
- Erb, W.; Grassot, J.-M.; Linder, D.; Neuville, L.; Zhu, J. Enantioselective synthesis of putative lipiarmycin aglycon related to fidaxomicin/tiacumicin B. Angew. Chem. Int. Ed. 2015, 54, 1929–1932. [Google Scholar] [CrossRef] [PubMed]
- Glaus, F.; Altmann, K.-H. Total synthesis of the tiacumicin B (lipiarmycin A3/fidaxomicin) aglycone. Angew. Chem. Int. Ed. 2015, 54, 1937–1940. [Google Scholar] [CrossRef] [PubMed]
- Miyatake-Ondozabal, H.; Kaufmann, E.; Gademann, K. Total synthesis of the protected aglycon of fidaxomicin (tiacumicin B, lipiarmycin A3). Angew. Chem. Int. Ed. 2015, 54, 1933–1936. [Google Scholar] [CrossRef] [Green Version]
- Kaufmann, E.; Hattori, H.; Miyatake-Ondozabal, H.; Gademann, K. Total synthesis of the glycosylated macrolide antibiotic fidaxomicin. Org. Lett. 2015, 17, 3514–3517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serra, S.; Malpezzi, L.; Bedeschi, A.; Fuganti, C.; Fonte, P. Final demonstration of the co-identity of lipiarmycin A3 and tiacumicin B (fidaxomicin) through single crystal x-ray analysis. Antibiotics 2017, 6, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeanne-Julien, L.; Masson, G.; Astier, E.; Genta-Jouve, G.; Servajean, V.; Beau, J.-M.; Norsikian, S.; Roulland, E. Synthesis of a tiacumicin B protected aglycone. Org. Lett. 2017, 19, 4006–4009. [Google Scholar] [CrossRef]
- Jeanne-Julien, L.; Masson, G.; Astier, E.; Genta-Jouve, G.; Servajean, V.; Beau, J.-M.; Norsikian, S.; Roulland, E. Study of the construction of the tiacumicin B aglycone. J. Org. Chem. 2018, 83, 921–929. [Google Scholar] [CrossRef]
- Hattori, H.; Kaufmann, E.; Miyatake-Ondozabal, H.; Berg, R.; Gademann, K. Total synthesis of tiacumicin A. Total synthesis, relay synthesis, and degradation studies of fidaxomicin (tiacumicin B, lipiarmycin A3). J. Org. Chem. 2018, 83, 7180–7205. [Google Scholar] [CrossRef] [PubMed]
- Norsikian, S.; Tresse, C.; François-Eude, M.; Jeanne-Julien, L.; Masson, G.; Servajean, V.; Genta-Jouve, G.; Beau, J.-M.; Roulland, E. Total synthesis of tiacumicin B: Implementing hydrogen bond directed acceptor delivery for highly selective β-glycosylations. Angew. Chem. Int. Ed. 2020, 59, 6612–6616. [Google Scholar] [CrossRef] [PubMed]
- Dorst, A.; Berg, R.; Gertzen, C.G.W.; Schafle, D.; Zerbe, K.; Gwerder, M.; Schnell, S.D.; Sander, P.; Gohlke, H.; Gademann, K. Semisynthetic analogs of the antibiotic fidaxomicin-design, synthesis, and biological evaluation. ACS Med. Chem. Lett. 2020, 11, 2414–2420. [Google Scholar] [CrossRef] [PubMed]
- Dorst, A.; Shchelik, I.S.; Schäfle, D.; Sander, P.; Gademann, K. Synthesis and biological evaluation of iodinated fidaxomicin antibiotics. Helv. Chim. Acta 2020, 103, e2000130. [Google Scholar] [CrossRef]
- Genilloud, O. Actinomycetes: Still a source of novel antibiotics. Nat. Prod. Rep. 2017, 34, 1203–1232. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.B.; Phillips, J.W.; Wang, J. Highly sensitive target-based whole-cell antibacterial discovery strategy by antisense RNA silencing. Curr. Opin. Drug Disc. 2007, 10, 160. [Google Scholar]
- Thaker, M.N.; Wang, W.; Spanogiannopoulos, P.; Waglechner, N.; King, A.M.; Medina, R.; Wright, G.D. Identifying producers of antibacterial compounds by screening for antibiotic resistance. Nat. Biotechnol. 2013, 31, 922–927. [Google Scholar] [CrossRef]
- Rappé, M.S.; Giovannoni, S.J. The uncultured microbial majority. Annu. Rev. Microbiol. 2003, 57, 369–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardenas, E.; Tiedje, J.M. New tools for discovering and characterizing microbial diversity. Curr. Opin. Biotechnol. 2008, 19, 544–549. [Google Scholar] [CrossRef] [PubMed]
- Van Wezel, G.P.; McDowall, K.J. The regulation of the secondary metabolism of Streptomyces: New links and experimental advances. Nat. Prod. Rep. 2011, 28, 1311–1333. [Google Scholar] [CrossRef] [PubMed]
- Denamur, E.; Matic, I. Evolution of mutation rates in bacteria. Mol. Microbiol. 2006, 60, 820–827. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Behiels, E.; Devreese, B. Toxin–antitoxin systems: Their role in persistence, biofilm formation, and pathogenicity. Pathog. Dis. 2014, 70, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Page, R.; Peti, W. Toxin-antitoxin systems in bacterial growth arrest and persistence. Nat. Chem. Biol. 2016, 12, 208–214. [Google Scholar] [CrossRef]
- Dilip, C.V.; Mulaje, S.; Mohalkar, R. A review on Actinomycetes and their biotechnological application. Int. J. Pharm. Sci. Res. 2013, 4, 1730. [Google Scholar] [CrossRef]

























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
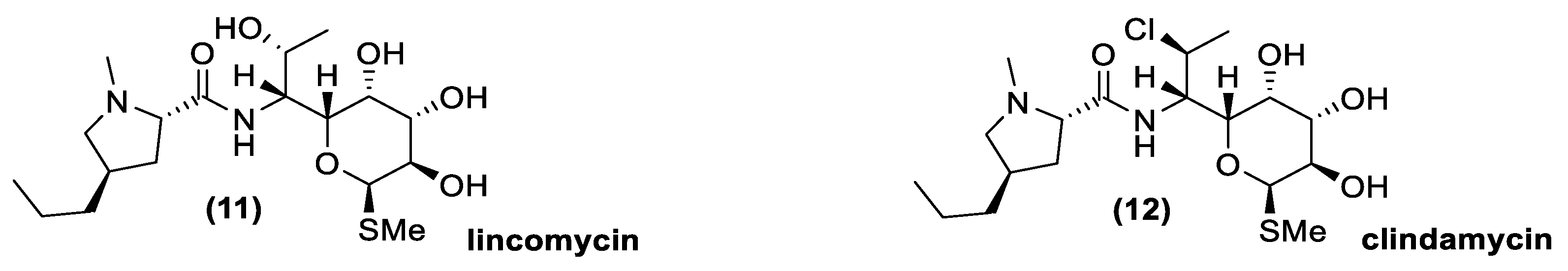{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| β-Lactams | Producer Microorganism 1,2 | |
|---|---|---|
| Penicillin | Penicillin N | S. lipmanii; S. microflavus [38,39]; S. griseus subsp. Cryophilus [46]; S. cattleya [48] |
| Cephalosporines | Cephamycins | S. clavuligerus [38,39]; S. griseus; S. lactamdurans [41]; S. chartreusis [42,43,44]; S. cinnamonensis; S. fimbriatus; S. halstedii; S. rochei; S. viridochromogenes [41]; S. wadayamensis; S. Jumonjineneis; S. lipmanii; S. panayaensis; Amycolatopsis lactamdurans [45]; S. cattleya [48]; S. heteromorphus [49] |
| Cephalosporines | S. microflavus; S. lipmanii; S. clavuligerus [38,39,40,42]; S. hygroscopicus [43] | |
| Carbapenems | Thienamycins | S. cattleya [45,47]; S. griseus subsp. Cryophilus [46,50]; S. penemifaciens [48]; S. flavogriseus; S. olivaceus; S. cremeus subsp. auratilis; S. flavoviridis [49] |
| Monobactam | Nocardicins | Nocardia uniformis [45]; Nocardia uniformis subsp. Tsuyamanensis; Streptomyces alcalophilus sp. nov. [49]; Actinosynnema mirum [48,49]; Microtetraspora caesia [51] |
| PBP inhibitors | Clavulanic acid | S. clavuligerus; S. jumonjinesis; S. katsuharamanus [45] |
| Olivanic acid | S. olivaceus [46]; Streptomyces griseus subsp. cryophilus [49] | |
| Tetracycline Antibiotics | Producer Microorganism 1,2 |
|---|---|
| Tetracycline | S. aureofaciens; S. avellaneus; S. lusitanus; S.viridifaciens [38,39,74,75] |
| Chlortetracycline | S. lusitanus; S. aureofaciens; S. lividans3 [38,39,74,75,76,77] |
| Oxytetracycline | S. alboflavus; Streptomyces albofaciens; Streptomyces erumpens; S. griseus; S. platensis; S. rimosus subsp. rimosus; S. varsoviensis [38,39,74] |
| 6-demethyltetracycline | S. aureofaciens3 [77] |
| Aminoglycoside Antibiotics | Producer Microorganism 1,2 |
|---|---|
| Streptomycin | S. griseus; S. bikiniensis; S. streptomycinii; S. ornatus; S. humidus; S. subrutilus; S. mashuensis; S. glaucescens; S. griseocarneus; Streptomyces rimosus subsp. rimosus; S. galbus [38,39,53,85,86,87] |
| Neomycin | S. fradiae; S. catenulae; S. chrestomyceticus; S. albogriseolus [38,39,53,85,88,91] |
| Tobramycin | Streptoalloteichus hindustanus; S. tenebrarius; S. cremeus [38,39,53,88,89] |
| Kanamycin | S. kanamyceticus [38,39,53] |
| Paromomycym | S. rimosus var. paromomycinus; S. catenulae; S. chrestomyceticus; S. rimosus; S. fradiae [38,39,53,85] |
| Gentamycin | Micromonospora purpurea; Micromonospora pallida; micromonospora echinospora [38,39,53,85,90] |
| Peptide Antibiotics | Producer Microorganism 1,2 | |
|---|---|---|
| Glycopeptides | vancomycin | Amycolatopsis orientalis subsp. Orientalis [38,39] |
| Lipopeptides | daptomycin | Streptomyces sp. KCTC12267BP [105]; S. roseosporus [106,108]; S. lividans 3 [107] |
| Streptogramins | Streptogramins A and B | S. halstedii [38]; S. pristinaespiralis; S. virginiae [109] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Simeis, D.; Serra, S. Actinomycetes: A Never-Ending Source of Bioactive Compounds—An Overview on Antibiotics Production. Antibiotics 2021, 10, 483. https://doi.org/10.3390/antibiotics10050483
De Simeis D, Serra S. Actinomycetes: A Never-Ending Source of Bioactive Compounds—An Overview on Antibiotics Production. Antibiotics. 2021; 10(5):483. https://doi.org/10.3390/antibiotics10050483
Chicago/Turabian StyleDe Simeis, Davide, and Stefano Serra. 2021. "Actinomycetes: A Never-Ending Source of Bioactive Compounds—An Overview on Antibiotics Production" Antibiotics 10, no. 5: 483. https://doi.org/10.3390/antibiotics10050483
APA StyleDe Simeis, D., & Serra, S. (2021). Actinomycetes: A Never-Ending Source of Bioactive Compounds—An Overview on Antibiotics Production. Antibiotics, 10(5), 483. https://doi.org/10.3390/antibiotics10050483