
Pan-Resistome Insights into the Multidrug Resistance of Acinetobacter baumannii

 , ,
, ,  , , ,
, , ,  ,
, 

 and
and 
Abstract
:1. Introduction
2. Results
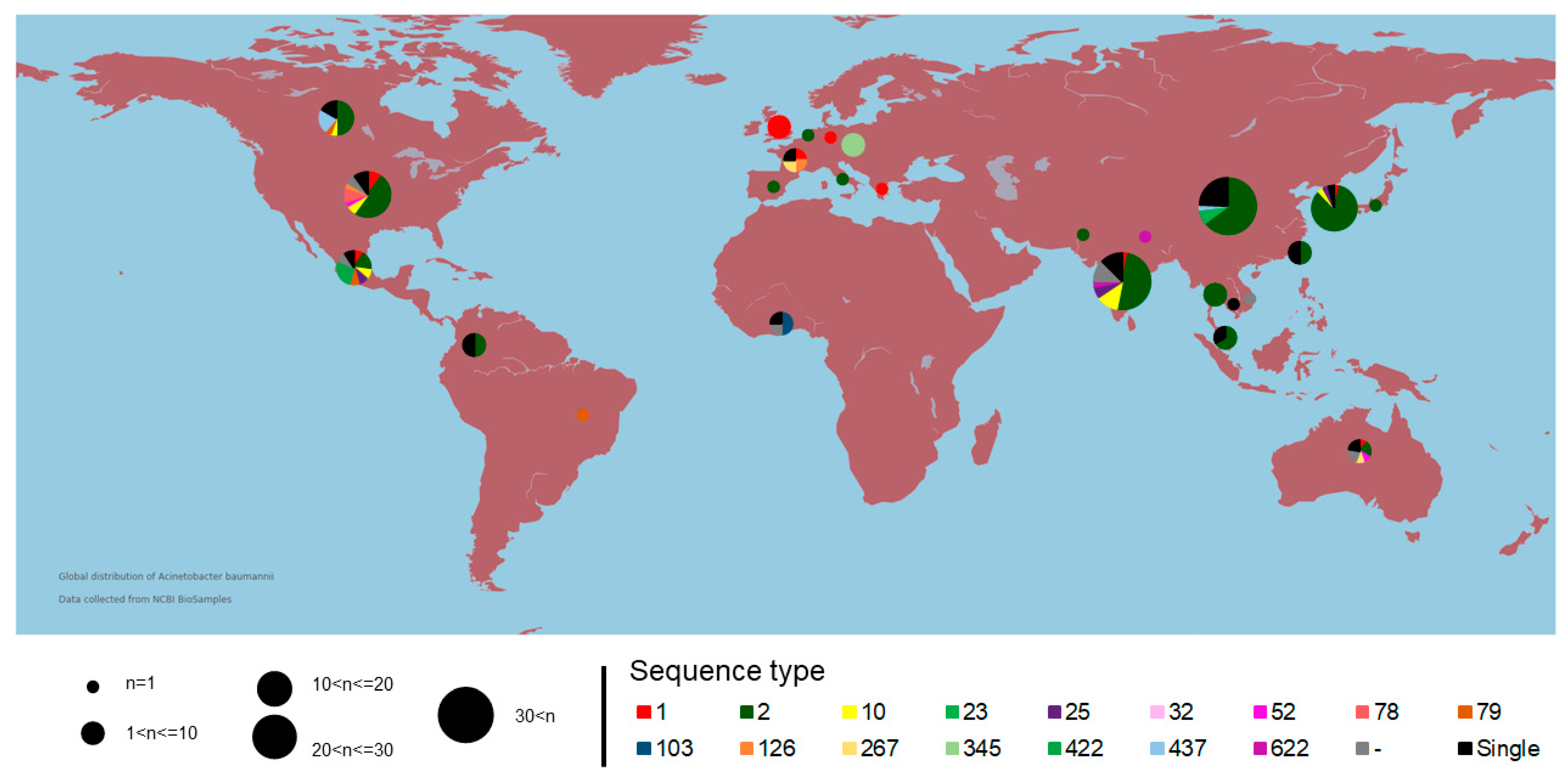
2.1. Genomic Analysis and Geographic Distribution
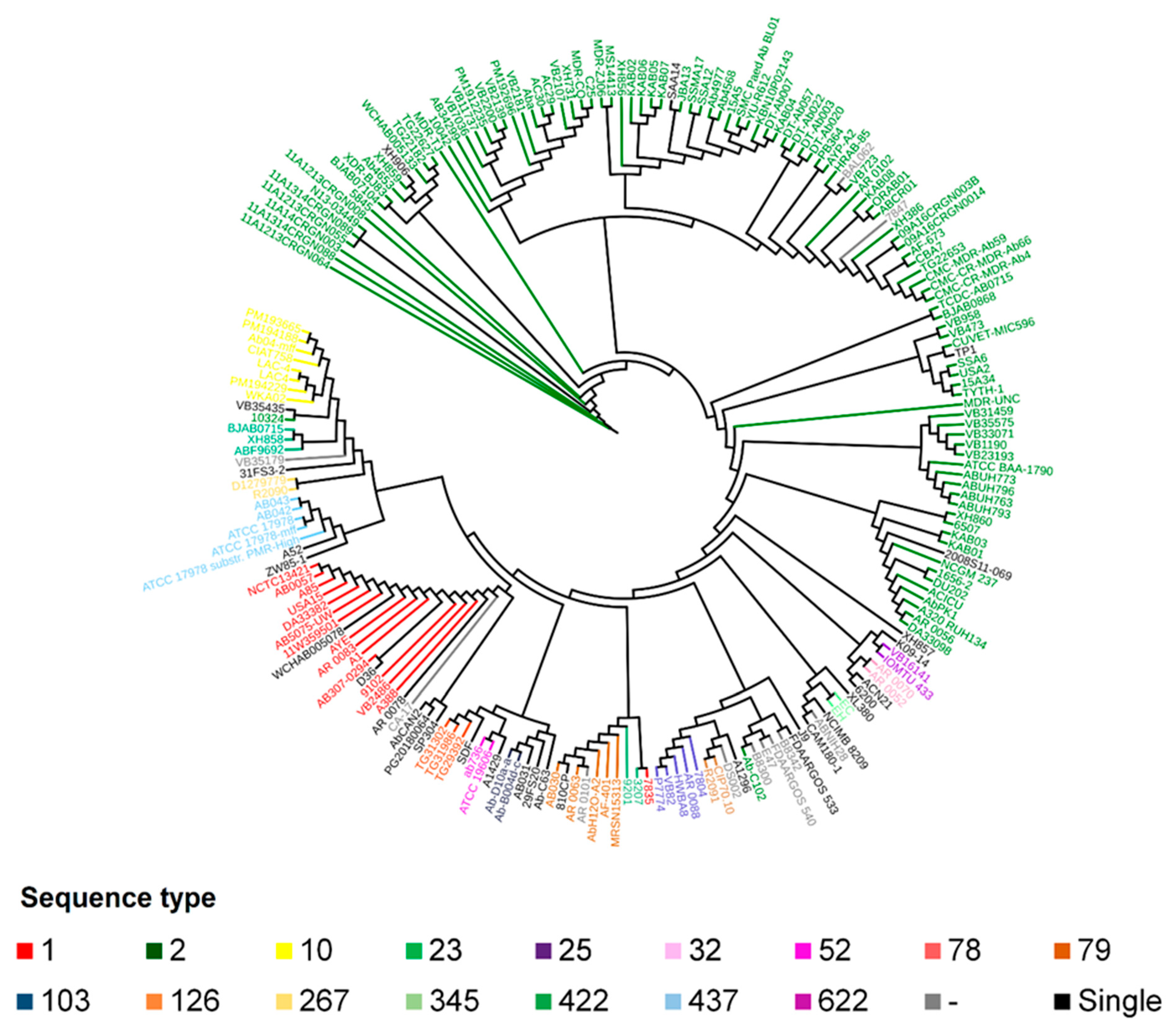
2.2. Phylogeny and Phylogenomics
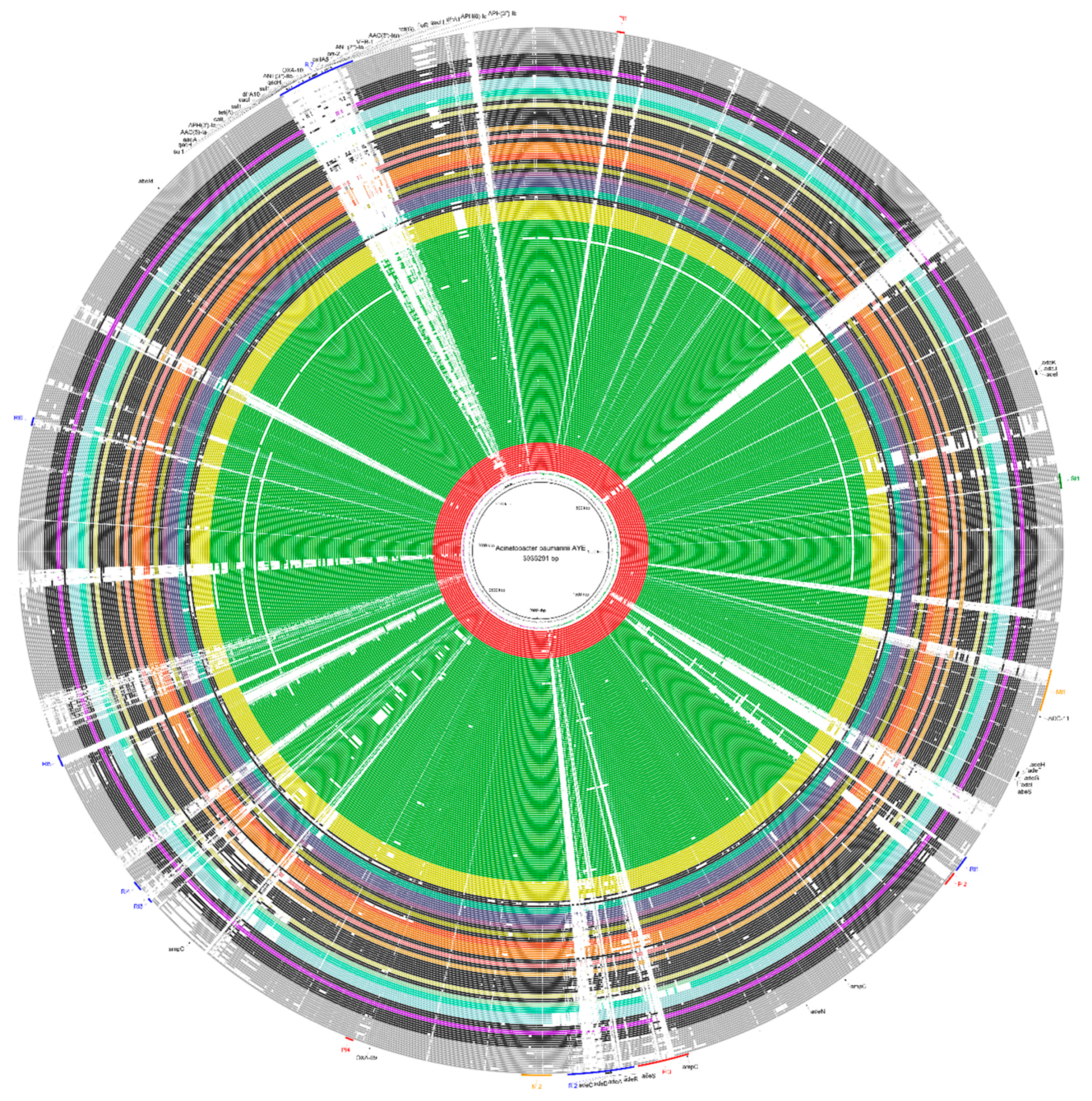
2.3. Genomic Plasticity
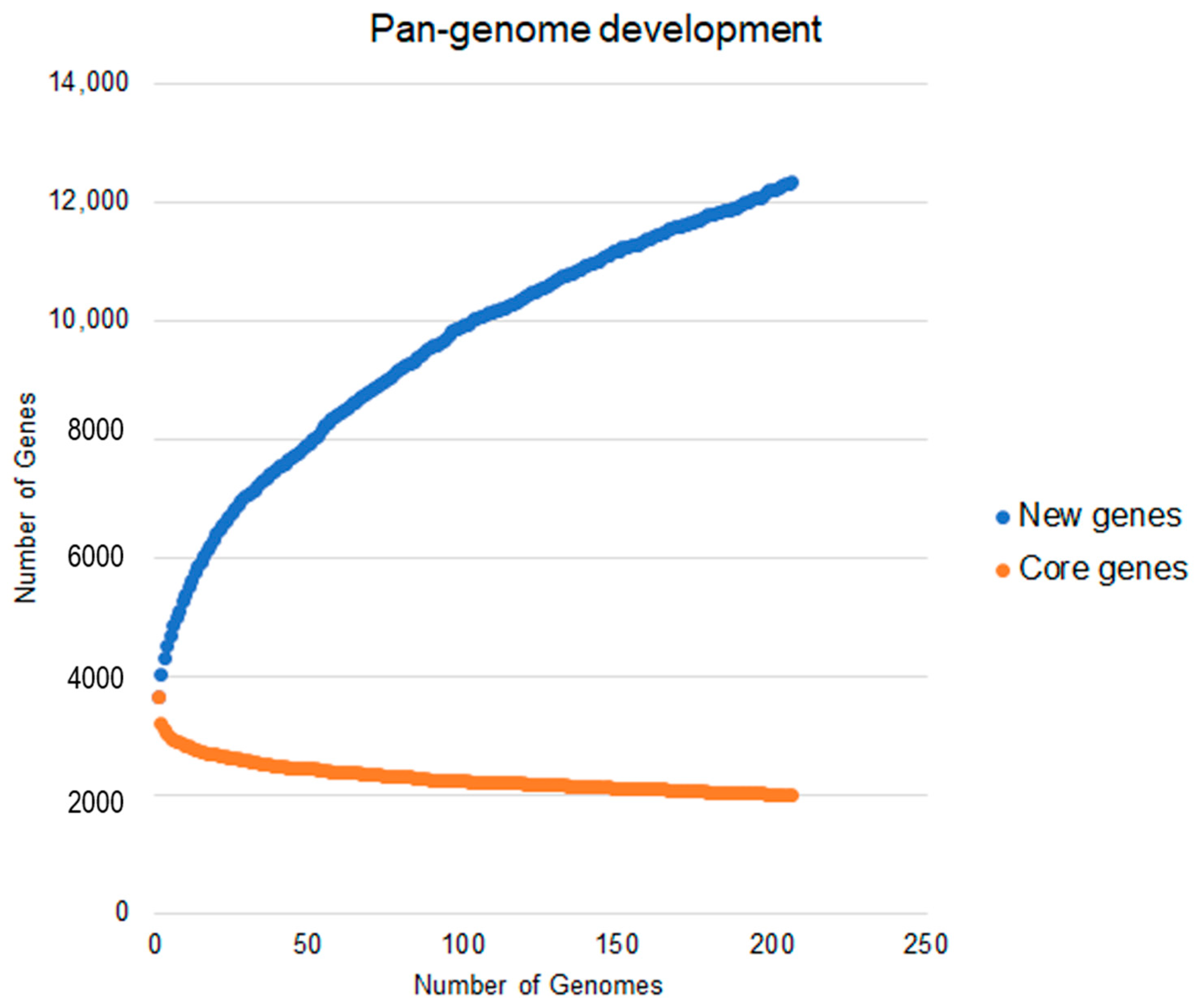
2.4. Analysis of the Pan-Genome for Understanding This Species
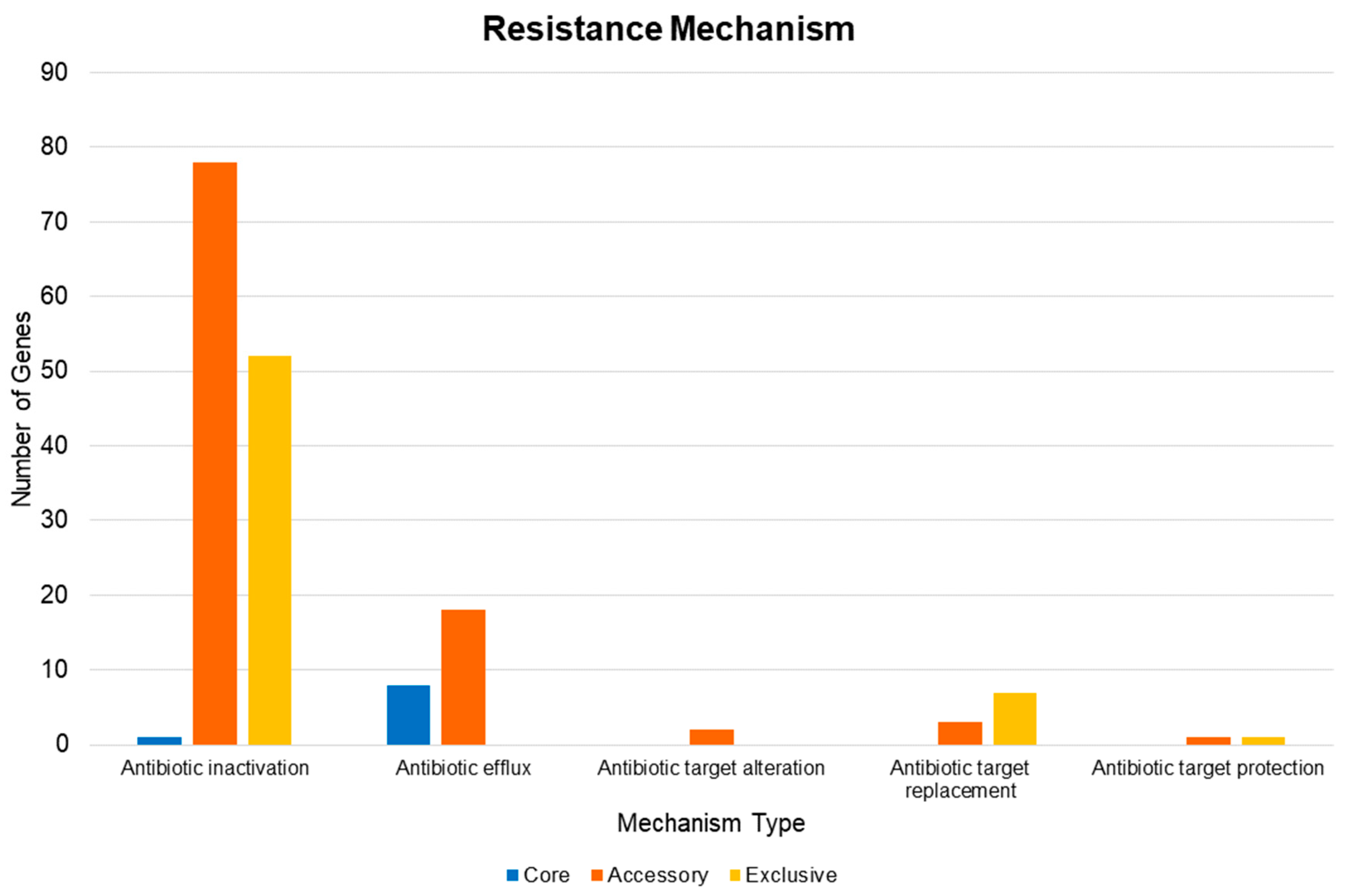
2.5. Pan-Resistome Characterization of Acinetobacter Baumannii
3. Discussion
3.1. Similarity Analysis, Geographic Distribution, and Phylogenomic Reconstruction
3.2. Genomic Plasticity in Acinetobacter Baumannii
3.3. Functional Characterization through Pan-Genome Analysis
3.4. Resistome of Acinetobacter Baumannii
4. Materials and Methods
4.1. Genomes Database, Annotation, and Data Retrieval
4.2. Multilocus Sequence Typing and Phylogeny
4.3. Resistance Genes Profile
4.4. Genomic Islands Analysis
4.5. Pan-Genome and Pan-Resistome Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Howard, A.; O’Donoghue, M.; Feeney, A.; Sleator, R.D. Acinetobacter Baumannii-An Emerging Opportunistic Pathogen. Virulence 2012, 3, 243–250. [Google Scholar] [CrossRef]
- Nowak, P.; Paluchowska, P. Acinetobacter Baumannii: Biology and Drug Resistance—Role of Carbapenemases. Folia Histochem. Cytobiol. 2016, 54, 61–74. [Google Scholar] [CrossRef] [Green Version]
- Antunes, L.C.S.; Visca, P.; Towner, K.J. Acinetobacter Baumannii: Evolution of a Global Pathogen. Pathog. Dis. 2014, 71, 292–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooks, L.E.; Ul-Hasan, S.; Chan, B.K.; Sistrom, M.J. Quantifying the Evolutionary Conservation of Genes Encoding Multidrug Efflux Pumps in the ESKAPE Pathogens to Identify Antimicrobial Drug Targets. mSystems 2018, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, Z.; Chen, Y.; Ong, E.; He, Y. Antibiotic Resistance Determinant-Focused Acinetobacter Baumannii Vaccine Designed Using Reverse Vaccinology. Int. J. Mol. Sci. 2017, 18, 458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Towner, K. The Genus Acinetobacter. In The Prokaryotes: A Handbook on the Biology of Bacteria Volume 6: Proteobacteria: Gamma Subclass; Dworkin, M., Falkow, S., Rosenberg, E., Schleifer, K.-H., Stackebrandt, E., Eds.; Springer: New York, NY, USA, 2006; pp. 746–758. ISBN 978-0-387-30746-6. [Google Scholar]
- Li, X.-Z.; Nikaido, H. Efflux-Mediated Drug Resistance in Bacteria. Drugs 2004, 64, 159–204. [Google Scholar] [CrossRef] [PubMed]
- Peleg, A.Y.; de Breij, A.; Adams, M.D.; Cerqueira, G.M.; Mocali, S.; Galardini, M.; Nibbering, P.H.; Earl, A.M.; Ward, D.V.; Paterson, D.L.; et al. The Success of Acinetobacter Species; Genetic, Metabolic and Virulence Attributes. PLoS ONE 2012, 7, e46984. [Google Scholar] [CrossRef]
- Peleg, A.Y.; Seifert, H.; Paterson, D.L. Acinetobacter Baumannii: Emergence of a Successful Pathogen. Clin. Microbiol. Rev. 2008, 21, 538–582. [Google Scholar] [CrossRef] [Green Version]
- Gandham, P. A Review on Multidrug-Resistant Acinetobacter Baumannii. Int. J. Curr. Microbiol. Appl. Sci. 2014, 3, 5. [Google Scholar]
- Alcock, B.P.; Raphenya, A.R.; Lau, T.T.Y.; Tsang, K.K.; Bouchard, M.; Edalatmand, A.; Huynh, W.; Nguyen, A.-L.V.; Cheng, A.A.; Liu, S.; et al. CARD 2020: Antibiotic Resistome Surveillance with the Comprehensive Antibiotic Resistance Database. Nucleic Acids Res. 2020, 48, D517–D525. [Google Scholar] [CrossRef]
- Sultan, I.; Rahman, S.; Jan, A.T.; Siddiqui, M.T.; Mondal, A.H.; Haq, Q.M.R. Antibiotics, Resistome and Resistance Mechanisms: A Bacterial Perspective. Front. Microbiol. 2018, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, H.; Lo, N.W.-S.; Loo, J.F.-C.; Lin, X.; Yim, A.K.-Y.; Tsui, S.K.-W.; Lau, T.C.-K.; Ip, M.; Chan, T.-F. Comparative Transcriptomics of Multidrug-Resistant Acinetobacter Baumannii in Response to Antibiotic Treatments. Sci. Rep. 2018, 8, 3515. [Google Scholar] [CrossRef] [PubMed]
- Gaiarsa, S.; Bitar, I.; Comandatore, F.; Corbella, M.; Piazza, A.; Scaltriti, E.; Villa, L.; Postiglione, U.; Marone, P.; Nucleo, E.; et al. Can Insertion Sequences Proliferation Influence Genomic Plasticity? Comparative Analysis of Acinetobacter Baumannii Sequence Type 78, a Persistent Clone in Italian Hospitals. Front. Microbiol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Jeannot, K.; Diancourt, L.; Vaux, S.; Thouverez, M.; Ribeiro, A.; Coignard, B.; Courvalin, P.; Brisse, S. Molecular Epidemiology of Carbapenem Non-Susceptible Acinetobacter Baumannii in France. PLoS ONE 2014, 9. [Google Scholar] [CrossRef]
- Bastardo, A.; Ravelo, C.; Romalde, J.L. Phylogeography of Yersinia Ruckeri Reveals Effects of Past Evolutionary Events on the Current Strain Distribution and Explains Variations in the Global Transmission of Enteric Redmouth (ERM) Disease. Front. Microbiol. 2015, 6. [Google Scholar] [CrossRef] [Green Version]
- Bastardo, A.; Balboa, S.; Romalde, J.L. From the Gene Sequence to the Phylogeography through the Population Structure: The Cases of Yersinia Ruckeri and Vibrio Tapetis; IntechOpen: Rijeka, Croatia, 2017; ISBN 978-953-51-2950-9. [Google Scholar]
- Vijayakumar, S.; Mathur, P.; Kapil, A.; Das, B.K.; Ray, P.; Gautam, V.; Sistla, S.; Parija, S.C.; Walia, K.; Ohri, V.C.; et al. Molecular Characterization & Epidemiology of Carbapenem-Resistant Acinetobacter Baumannii Collected across India. Indian J. Med. Res. 2019, 149, 240–246. [Google Scholar]
- Azevedo, F.K.S.F.; Dutra, V.; Nakazato, L.; Pepato, M.A.; Sousa, A.T.H.I.; Azevedo, C.C.S.F.; Souto, F.J.D. New Sequence Types of Acinetobacter Baumannii in Two Emergency Hospitals in the Central-West Region of Brazil. Rev. Soc. Bras. Med. Trop. 2019, 52, e20190077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez, C.H.; Balderrama Yarhui, N.; Nastro, M.; Nuñez Quezada, T.; Castro Cañarte, G.; Magne Ventura, R.; Ugarte Cuba, T.; Valenzuela, N.; Roach, F.; Mota, M.I.; et al. Molecular Epidemiology of Carbapenem-Resistant Acinetobacter Baumannii in South America. J. Med. Microbiol. 2016, 65, 1088–1091. [Google Scholar] [CrossRef]
- Jain, C.; Rodriguez-R, L.M.; Phillippy, A.M.; Konstantinidis, K.T.; Aluru, S. High Throughput ANI Analysis of 90K Prokaryotic Genomes Reveals Clear Species Boundaries. Nat. Commun. 2018, 9, 5114. [Google Scholar] [CrossRef] [Green Version]
- Goris, J.; Konstantinidis, K.T.; Klappenbach, J.A.; Coenye, T.; Vandamme, P.; Tiedje, J.M.Y. 2007 DNA–DNA Hybridization Values and Their Relationship to Whole-Genome Sequence Similarities. Int. J. Syst. Evol. Microbiol. 2007, 57, 81–91. [Google Scholar] [CrossRef] [Green Version]
- Belén, A.; Pavón, I.; Maiden, M.C.J. Multilocus Sequence Typing. Methods Mol. Biol. 2009, 551, 129–140. [Google Scholar]
- Busse, H.-J. Review of the Taxonomy of the Genus Arthrobacter, Emendation of the Genus Arthrobacter Sensu Lato, Proposal to Reclassify Selected Species of the Genus Arthrobacter in the Novel Genera Glutamicibacter Gen. Nov., Paeniglutamicibacter Gen. Nov., Pseudoglutamicibacter Gen. Nov., Paenarthrobacter Gen. Nov. and Pseudarthrobacter Gen. Nov., and Emended Description of Arthrobacter Roseus. Int. J. Syst. Evol. Microbiol. 2016, 66, 9–37. [Google Scholar]
- Kishi, L.T.; Fernandes, C.C.; Omori, W.P.; Campanharo, J.C.; de Macedo Lemos, E.G. Reclassification of the Taxonomic Status of SEMIA3007 Isolated in Mexico B-11A Mex as Rhizobium Leguminosarum Bv. Viceae by Bioinformatic Tools. BMC Microbiol. 2016, 16. [Google Scholar] [CrossRef] [Green Version]
- Martens, T.; Heidorn, T.; Pukall, R.; Simon, M.; Tindall, B.J.; Brinkhoff, T. Reclassification of Roseobacter Gallaeciensis Ruiz-Ponte et al. 1998 as Phaeobacter Gallaeciensis Gen. Nov., Comb. Nov., Description of Phaeobacter Inhibens Sp. Nov., Reclassification of Ruegeria Algicola (Lafay et al. 1995) Uchino et al. 1999 as Marinovum Algicola Gen. Nov., Comb. Nov., and Emended Descriptions of the Genera Roseobacter, Ruegeria and Leisingera. Int. J. Syst. Evol. Microbiol. 2006, 56, 1293–1304. [Google Scholar]
- Tindall, B.J.; Rosselló-Móra, R.; Busse, H.-J.; Ludwig, W.; Kämpfer, P. Notes on the Characterization of Prokaryote Strains for Taxonomic Purposes. Int. J. Syst. Evol. Microbiol. 2010, 60, 249–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlos Guimaraes, L.; Benevides de Jesus, L.; Vinicius Canario Viana, M.; Silva, A.; Thiago Juca Ramos, R.; de Castro Soares, R.; Azevedo, V. Inside the Pan-Genome-Methods and Software Overview. Curr. Genomics 2015, 16, 245–252. [Google Scholar] [CrossRef] [Green Version]
- Imperi, F.; Antunes, L.C.S.; Blom, J.; Villa, L.; Iacono, M.; Visca, P.; Carattoli, A. The Genomics of Acinetobacter Baumannii: Insights into Genome Plasticity, Antimicrobial Resistance and Pathogenicity. IUBMB Life 2011, 63, 1068–1074. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Zhou, X.; Chen, Z.; Deng, X.; Gehring, A.; Ou, H.; Zhang, L.; Shi, X. PRAP: Pan Resistome Analysis Pipeline. BMC Bioinform. 2020, 21, 20. [Google Scholar] [CrossRef]
- LIU, Y.; LIU, X. Detection of AmpC β-Lactamases in Acinetobacter Baumannii in the Xuzhou Region and Analysis of Drug Resistance. Exp. Ther. Med. 2015, 10, 933–936. [Google Scholar] [CrossRef] [Green Version]
- Corvec, S.; Caroff, N.; Espaze, E.; Giraudeau, C.; Drugeon, H.; Reynaud, A. AmpC Cephalosporinase Hyperproduction in Acinetobacter Baumannii Clinical Strains. J. Antimicrob. Chemother. 2003, 52, 629–635. [Google Scholar] [CrossRef] [Green Version]
- Kazmierczak, K.M.; Rabine, S.; Hackel, M.; McLaughlin, R.E.; Biedenbach, D.J.; Bouchillon, S.K.; Sahm, D.F.; Bradford, P.A. Multiyear, Multinational Survey of the Incidence and Global Distribution of Metallo-β-Lactamase-Producing Enterobacteriaceae and Pseudomonas Aeruginosa. Antimicrob. Agents Chemother. 2016, 60, 1067–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shapiro, B.J.; Polz, M.F. Microbial Speciation. Cold Spring Harb Perspect Biol. 2015, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gadagkar, S.R.; Rosenberg, M.S.; Kumar, S. Inferring Species Phylogenies from Multiple Genes: Concatenated Sequence Tree versus Consensus Gene Tree. J. Exp. Zool. Part B Mol. Dev. Evol. 2005, 304B, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Sahl, J.W.; Gillece, J.D.; Schupp, J.M.; Waddell, V.G.; Driebe, E.M.; Engelthaler, D.M.; Keim, P. Evolution of a Pathogen: A Comparative Genomics Analysis Identifies a Genetic Pathway to Pathogenesis in Acinetobacter. PLoS ONE 2013, 8, e54287. [Google Scholar] [CrossRef] [Green Version]
- Mussi, M.A.; Limansky, A.S.; Relling, V.; Ravasi, P.; Arakaki, A.; Actis, L.A.; Viale, A.M. Horizontal Gene Transfer and Assortative Recombination within the Acinetobacter Baumannii Clinical Population Provide Genetic Diversity at the Single CarO Gene, Encoding a Major Outer Membrane Protein Channel. J. Bacteriol. 2011, 193, 4736–4748. [Google Scholar] [CrossRef] [Green Version]
- Valenzuela, J.K.; Thomas, L.; Partridge, S.R.; van der Reijden, T.; Dijkshoorn, L.; Iredell, J. Horizontal Gene Transfer in a Polyclonal Outbreak of Carbapenem-Resistant Acinetobacter Baumannii. J. Clin. Microbiol. 2007, 45, 453–460. [Google Scholar] [CrossRef] [Green Version]
- Soares, S.C.; Geyik, H.; Ramos, R.T.J.; de Sá, P.H.C.G.; Barbosa, E.G.V.; Baumbach, J.; Figueiredo, H.C.P.; Miyoshi, A.; Tauch, A.; Silva, A.; et al. GIPSy: Genomic Island Prediction Software. J. Biotechnol. 2016, 232, 2–11. [Google Scholar] [CrossRef]
- Wang, H.; Wang, J.; Yu, P.; Ge, P.; Jiang, Y.; Xu, R.; Chen, R.; Liu, X. Identification of Antibiotic Resistance Genes in the Multidrug-Resistant Acinetobacter Baumannii Strain, MDR-SHH02, Using Whole-Genome Sequencing. Int. J. Mol. Med. 2017, 39, 364–372. [Google Scholar] [CrossRef] [Green Version]
- Jani, M.; Mathee, K.; Azad, R.K. Identification of Novel Genomic Islands in Liverpool Epidemic Strain of Pseudomonas Aeruginosa Using Segmentation and Clustering. Front. Microbiol. 2016, 7. [Google Scholar] [CrossRef]
- Lery, L.M.; Frangeul, L.; Tomas, A.; Passet, V.; Almeida, A.S.; Bialek-Davenet, S.; Barbe, V.; Bengoechea, J.A.; Sansonetti, P.; Brisse, S.; et al. Comparative Analysis of Klebsiella Pneumoniae Genomes Identifies a Phospholipase D Family Protein as a Novel Virulence Factor. BMC Biol. 2014, 12, 41. [Google Scholar] [CrossRef]
- Medini, D.; Donati, C.; Tettelin, H.; Masignani, V.; Rappuoli, R. The Microbial Pan-Genome. Curr. Opin. Genet. Dev. 2005, 15, 589–594. [Google Scholar] [CrossRef]
- Tettelin, H.; Masignani, V.; Cieslewicz, M.J.; Donati, C.; Medini, D.; Ward, N.L.; Angiuoli, S.V.; Crabtree, J.; Jones, A.L.; Durkin, A.S.; et al. Genome Analysis of Multiple Pathogenic Isolates of Streptococcus Agalactiae: Implications for the Microbial Pan-Genome. Proc Natl. Acad. Sci. USA 2005, 102, 13950–13955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Auria, G.; Jiménez-Hernández, N.; Peris-Bondia, F.; Moya, A.; Latorre, A. Legionella Pneumophila Pangenome Reveals Strain-Specific Virulence Factors. BMC Genom. 2010, 11, 181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soares, S.C.; Silva, A.; Trost, E.; Blom, J.; Ramos, R.; Carneiro, A.; Ali, A.; Santos, A.R.; Pinto, A.C.; Diniz, C.; et al. The Pan-Genome of the Animal Pathogen Corynebacterium Pseudotuberculosis Reveals Differences in Genome Plasticity between the Biovar Ovis and Equi Strains. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [Green Version]
- Hurtado, R.; Carhuaricra, D.; Soares, S.; Viana, M.V.C.; Azevedo, V.; Maturrano, L.; Aburjaile, F. Pan-Genomic Approach Shows Insight of Genetic Divergence and Pathogenic-Adaptation of Pasteurella Multocida. Gene 2018, 670, 193–206. [Google Scholar] [CrossRef] [PubMed]
- Freschi, L.; Vincent, A.T.; Jeukens, J.; Emond-Rheault, J.-G.; Kukavica-Ibrulj, I.; Dupont, M.-J.; Charette, S.J.; Boyle, B.; Levesque, R.C. The Pseudomonas Aeruginosa Pan-Genome Provides New Insights on Its Population Structure, Horizontal Gene Transfer, and Pathogenicity. Genome Biol. Evol. 2019, 11, 109–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaiswal, A.K.; Tiwari, S.; Jamal, S.B.; de Castro Oliveira, L.; Alves, L.G.; Azevedo, V.; Ghosh, P.; Oliveira, C.J.F.; Soares, S.C. The Pan-Genome of Treponema Pallidum Reveals Differences in Genome Plasticity between Subspecies Related to Venereal and Non-Venereal Syphilis. BMC Genom. 2020, 21, 33. [Google Scholar] [CrossRef]
- Blaustein, R.A.; McFarland, A.G.; Ben Maamar, S.; Lopez, A.; Castro-Wallace, S.; Hartmann, E.M. Pangenomic Approach to Understanding Microbial Adaptations within a Model Built Environment, the International Space Station, Relative to Human Hosts and Soil. mSystems 2019, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rocha, E.P. Evolutionary Patterns in Prokaryotic Genomes. Curr. Opin. Microbiol. 2008, 11, 454–460. [Google Scholar] [CrossRef] [PubMed]
- Klotz, L.-O.; Steinbrenner, H. Cellular Adaptation to Xenobiotics: Interplay between Xenosensors, Reactive Oxygen Species and FOXO Transcription Factors. Redox Biol. 2017, 13, 646–654. [Google Scholar] [CrossRef]
- Patterson, A.D.; Gonzalez, F.J.; Idle, J.R. Xenobiotic Metabolism–A View through the metabolometer. Chem. Res. Toxicol. 2010, 23, 851–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergogne-Bérézin, E. Acinetobacter Spp., Saprophytic Organisms of Increasing Pathogenic Importance. Zentralbl. Bakteriol. 1994, 281, 389–405. [Google Scholar] [CrossRef]
- Doughari, H.J.; Ndakidemi, P.A.; Human, I.S.; Benade, S. The Ecology, Biology and Pathogenesis of Acinetobacter Spp.: An Overview. Microbes Environ. 2011, 26, 101–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassan, A.; Naz, A.; Obaid, A.; Paracha, R.Z.; Naz, K.; Awan, F.M.; Muhmmad, S.A.; Janjua, H.A.; Ahmad, J.; Ali, A. Pangenome and Immuno-Proteomics Analysis of Acinetobacter Baumannii Strains Revealed the Core Peptide Vaccine Targets. BMC Genomics 2016, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangas, E.L.; Rubio, A.; Álvarez-Marín, R.; Labrador-Herrera, G.; Pachón, J.; Pachón-Ibáñez, M.E.; Divina, F.; Pérez-Pulido, A.J. Pangenome of Acinetobacter Baumannii Uncovers Two Groups of Genomes, One of Them with Genes Involved in CRISPR/Cas Defence Systems Associated with the Absence of Plasmids and Exclusive Genes for Biofilm Formation. Microb. Genom. 2019, 5. [Google Scholar] [CrossRef]
- Evans, B.A.; Amyes, S.G.B. OXA β-Lactamases. Clin. Microbiol. Rev. 2014, 27, 241–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdi, S.N.; Ghotaslou, R.; Ganbarov, K.; Mobed, A.; Tanomand, A.; Yousefi, M.; Asgharzadeh, M.; Kafil, H.S. Acinetobacter Baumannii Efflux Pumps and Antibiotic Resistance. Infect. Drug Resist. 2020, 13, 423–434. [Google Scholar] [CrossRef] [Green Version]
- Moubareck, C.A.; Halat, D.H. Insights into Acinetobacter Baumannii: A Review of Microbiological, Virulence, and Resistance Traits in a Threatening Nosocomial Pathogen. Antibiotics 2020, 9, 119. [Google Scholar] [CrossRef] [Green Version]
- Elham, B.; Fawzia, A. Colistin Resistance in Acinetobacter Baumannii Isolated from Critically Ill Patients: Clinical Characteristics, Antimicrobial Susceptibility and Outcome. Afr. Health Sci. 2019, 19, 2400–2406. [Google Scholar] [CrossRef]
- Genteluci, G.L.; Gomes, D.B.C.; Souza, M.J.; de Carvalho, K.R.; Villas-Bôas, M.H.S.; Genteluci, G.L.; Gomes, D.B.C.; Souza, M.J.; de Carvalho, K.R.; Villas-Bôas, M.H.S. Emergence of Polymyxin B-Resistant Acinetobacter Baumannii in Hospitals in Rio de Janeiro. J. Bras. Patol. Med. Lab. 2016, 52, 91–95. [Google Scholar] [CrossRef]
- Pruitt, K.D.; Tatusova, T.; Maglott, D.R. NCBI Reference Sequences (RefSeq): A Curated Non-Redundant Sequence Database of Genomes, Transcripts and Proteins. Nucleic Acids Res. 2007, 35, D61–D65. [Google Scholar] [CrossRef] [Green Version]
- Seemann, T. Prokka: Rapid Prokaryotic Genome Annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Lagesen, K.; Hallin, P.; Rødland, E.A.; Stærfeldt, H.-H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and Rapid Annotation of Ribosomal RNA Genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef] [PubMed]
- Jolley, K.A.; Maiden, M.C. BIGSdb: Scalable Analysis of Bacterial Genome Variation at the Population Level. BMC Bioinform. 2010, 11, 595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diancourt, L.; Passet, V.; Nemec, A.; Dijkshoorn, L.; Brisse, S. The Population Structure of Acinetobacter Baumannii: Expanding Multiresistant Clones from an Ancestral Susceptible Genetic Pool. PLoS ONE 2010, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A. Rambaut/Figtree; GitHub Repository. 2020. Available online: https://github.com/rambaut/figtree.git (accessed on 18 May 2021).
- Mount, D.W. Using the Basic Local Alignment Search Tool (BLAST). Cold Spring Harb. Protoc. 2007, 2007. [Google Scholar] [CrossRef]
- Rodrigues, D.L.N. PanViTa Tool; GitHub Repository. 2020. Available online: https://github.com/dlnrodrigues/panvita (accessed on 18 May 2021).
- Adams, M.D.; Goglin, K.; Molyneaux, N.; Hujer, K.M.; Lavender, H.; Jamison, J.J.; MacDonald, I.J.; Martin, K.M.; Russo, T.; Campagnari, A.A.; et al. Comparative Genome Sequence Analysis of Multidrug-Resistant Acinetobacter Baumannii. J. Bacteriol. 2008, 190, 8053–8064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallenet, D.; Nordmann, P.; Barbe, V.; Poirel, L.; Mangenot, S.; Bataille, E.; Dossat, C.; Gas, S.; Kreimeyer, A.; Lenoble, P.; et al. Comparative Analysis of Acinetobacters: Three Genomes for Three Lifestyles. PLoS ONE 2008, 3, e1805. [Google Scholar] [CrossRef]
- Alikhan, N.-F.; Petty, N.K.; Ben Zakour, N.L.; Beatson, S.A. BLAST Ring Image Generator (BRIG): Simple Prokaryote Genome Comparisons. BMC Genomics 2011, 12, 402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic Orthology Inference for Comparative Genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New Perspectives on Genomes, Pathways, Diseases and Drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Definition | Mechanism | Antibiotic |
|---|---|---|---|
| adeK | The outer membrane factor protein in the adeIJK multidrug efflux complex | Antibiotic efflux | Phenicol, rifamycin, penem, diaminopyrimidine, tetracycline, carbapenem, macrolide, lincosamide, floroquinolone, cephalosporin |
| adeJ | An RND efflux protein that acts as the inner membrane transporter of the AdeIJK efflux complex | Antibiotic efflux | Diaminopyrimidine, phenicol, tetracycline, rifamycin, carbapenem, penem, fluoroquinolone, macrolide, cephalosporin, lincosamide |
| adeI | The membrane fusion protein of the AdeIJK multidrug efflux complex | Antibiotic efflux | Phenicol, rifamycin, penem, diaminopyrimidine, tetracycline, carbapenem, macrolide, lincosamide, floroquinolone, cephalosporin |
| adeF | The membrane fusion protein of the multidrug efflux complex AdeFGH | Antibiotic efflux | Tetracycline, fluoroquinolone |
| adeG | The inner membrane transporter of the AdeFGH multidrug efflux complex. | Antibiotic efflux | Tetracycline, fluoroquinolone |
| adeL | A regulator of AdeFGH in Acinetobacter baumannii. AdeL mutations are associated with AdeFGH overexpression and multidrug resistance. | Antibiotic efflux | Tetracycline, fluoroquinolone |
| ampC | AmpC type beta-lactamases are commonly isolated from extended-spectrum cephalosporin-resistant Gram-negative bacteria. | Antibiotic inactivation | Cephalosporins |
| adeN | AdeN is a repressor of AdeIJK, an RND-type efflux pump in Acinetobacter baumannii. Its inactivation increases the expression of AdeJ. | Antibiotic efflux | Carbapenem, diaminopyrimidine, rifamycin, penem, tetracycline antibiotic, phenicol, lincosamide, fluoroquinolone, cephalosporin, macrolide |
| abeM | AbeM is a multidrug efflux pump found in Acinetobacter baumannii. | Antibiotic efflux | Acridine dye, fluoroquinolone antibiotic, triclosan |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodrigues, D.L.N.; Morais-Rodrigues, F.; Hurtado, R.; dos Santos, R.G.; Costa, D.C.; Barh, D.; Ghosh, P.; Alzahrani, K.J.; Soares, S.C.; Ramos, R.; et al. Pan-Resistome Insights into the Multidrug Resistance of Acinetobacter baumannii. Antibiotics 2021, 10, 596. https://doi.org/10.3390/antibiotics10050596
Rodrigues DLN, Morais-Rodrigues F, Hurtado R, dos Santos RG, Costa DC, Barh D, Ghosh P, Alzahrani KJ, Soares SC, Ramos R, et al. Pan-Resistome Insights into the Multidrug Resistance of Acinetobacter baumannii. Antibiotics. 2021; 10(5):596. https://doi.org/10.3390/antibiotics10050596
Chicago/Turabian StyleRodrigues, Diego Lucas Neres, Francielly Morais-Rodrigues, Raquel Hurtado, Roselane Gonçalves dos Santos, Daniela Camargos Costa, Debmalya Barh, Preetam Ghosh, Khalid J. Alzahrani, Siomar Castro Soares, Rommel Ramos, and et al. 2021. "Pan-Resistome Insights into the Multidrug Resistance of Acinetobacter baumannii" Antibiotics 10, no. 5: 596. https://doi.org/10.3390/antibiotics10050596
APA StyleRodrigues, D. L. N., Morais-Rodrigues, F., Hurtado, R., dos Santos, R. G., Costa, D. C., Barh, D., Ghosh, P., Alzahrani, K. J., Soares, S. C., Ramos, R., Góes-Neto, A., Azevedo, V., & Aburjaile, F. F. (2021). Pan-Resistome Insights into the Multidrug Resistance of Acinetobacter baumannii. Antibiotics, 10(5), 596. https://doi.org/10.3390/antibiotics10050596