
Revisiting Antibiotic Resistance: Mechanistic Foundations to Evolutionary Outlook



Abstract
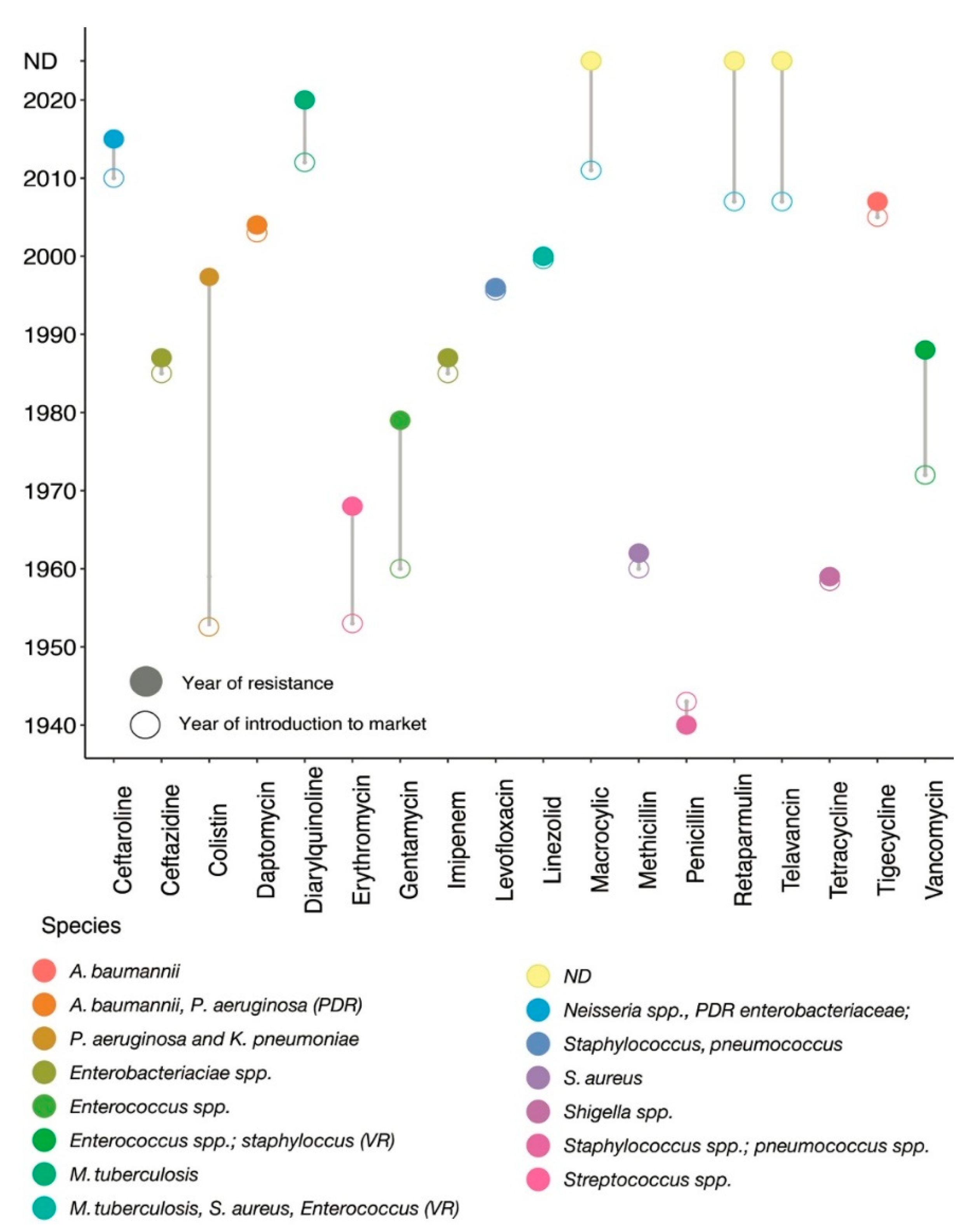
:1. Introduction
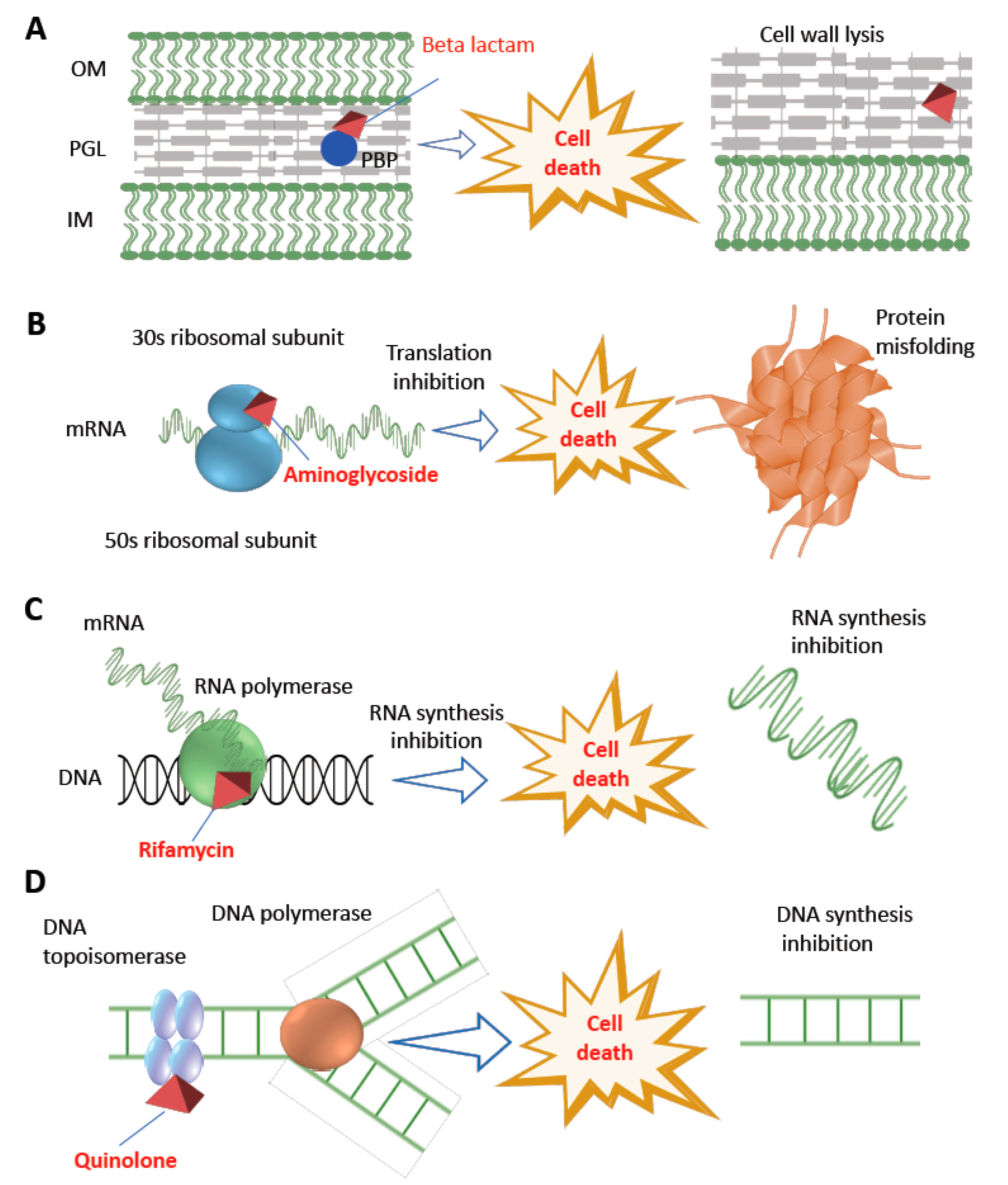
2. Antibiotics and Their Impact on Bacterial Cellular Perturbation
3. Bacteria Employ Diverse Devices to Resist Antibiotics and Spread Antibiotic Resistance
3.1. Natural Defense Systems to Antibiotics
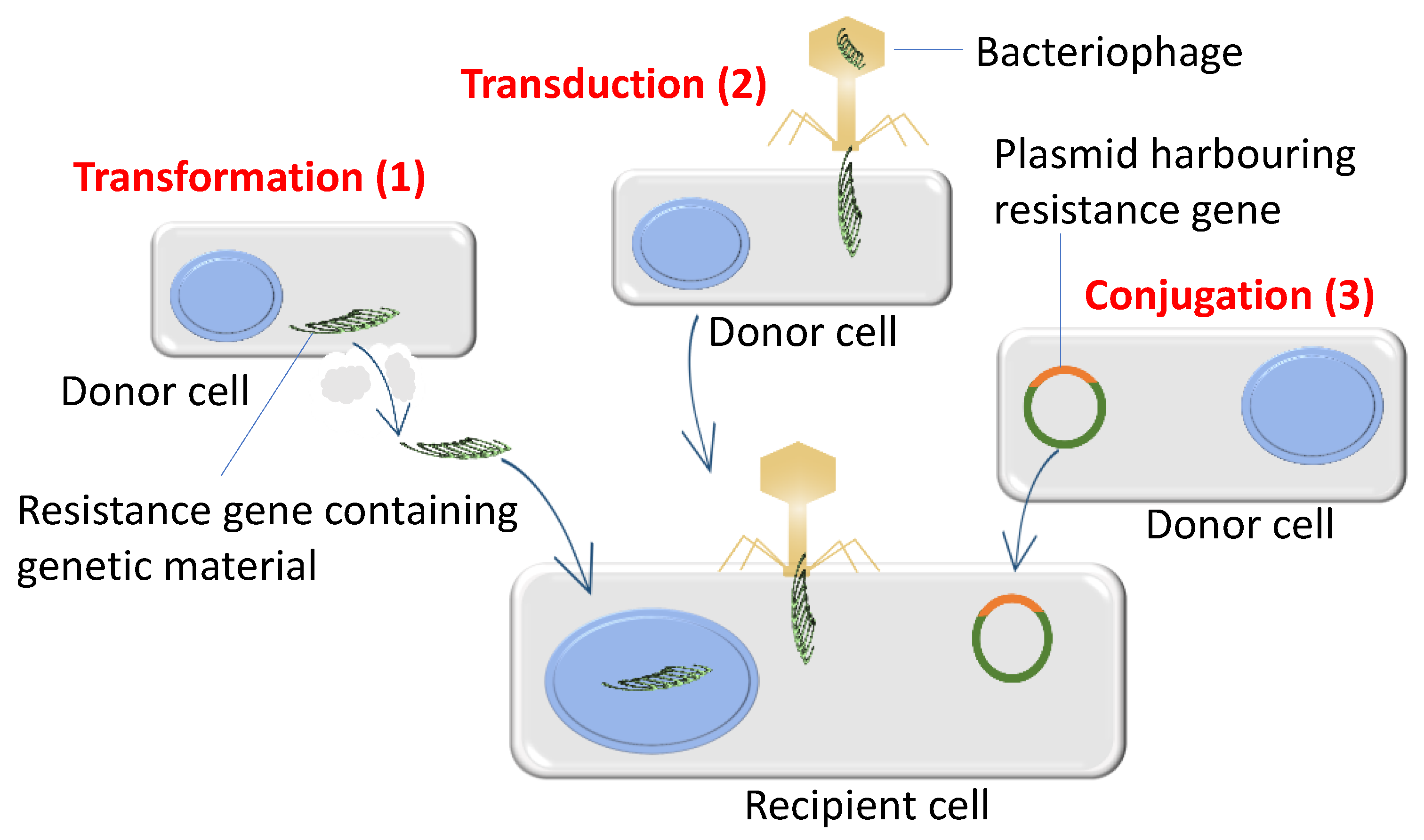
3.2. Bacteria Acquire High Levels of Antibiotic Resistance through De Novo Mutations and Horizontal Gene Transfer
4. Bacterial Resistance to Multiple Antibiotics via Diverse Biochemical Mechanisms
4.1. Disabling Antibiotic Activities through Enzymatic Modification
4.2. Bypassing Antibiotic Interactions by Altering Target Sites
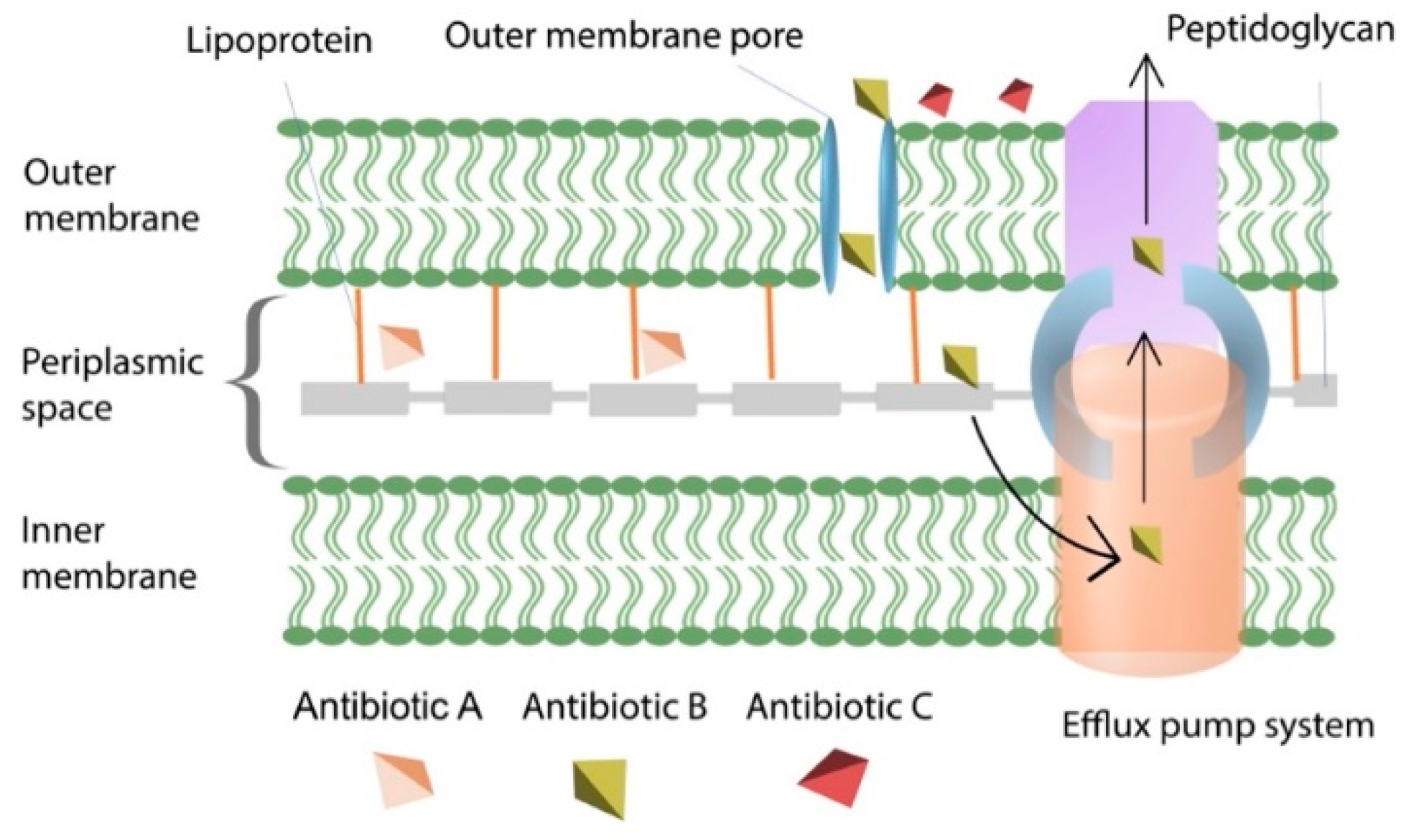
4.3. Efflux Pumps Reduce Intracellular Antibiotic Concentration
4.4. Alteration of Membrane Permeability Prevents the Penetration of Antibiotics into Bacterial Cells
5. Evolution of Antibiotic Resistance Is a Result of Natural Selection
6. Horizontal Gene Transfer and Bacterial Recombination Promotes Evolutionary Adaptation in Antibiotic Environments
7. Factors Affecting Evolutionary Dynamics of Resistance in Bacteria
8. Future Landscapes
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Katz, L.; Baltz, R.H. Natural product discovery: Past, present, and future. J. Ind. Microbiol. Biotechnol. 2016, 43, 155–176. [Google Scholar] [CrossRef]
- Boucher, H.W.; Talbot, G.H.; Bradley, J.S.; Edwards, J.E.; Gilbert, D.; Rice, L.B.; Scheld, M.; Spellberg, B.; Bartlett, J. Bad bugs, no drugs: No ESKAPE! An update from the Infectious Diseases Society of America. Clin. Infect. Dis. 2009, 48, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Lesho, E.P.; Laguio-Vila, M. The Slow-Motion Catastrophe of Antimicrobial Resistance and Practical Interventions for All Prescribers. Mayo Clin. Proc. 2019, 94, 1040–1047. [Google Scholar] [CrossRef]
- Giske, C.G.; Monnet, D.L.; Cars, O.; Carmeli, Y. Clinical and economic impact of common multidrug-resistant gram-negative bacilli. Antimicrob. Agents Chemother. 2008, 52, 813–821. [Google Scholar] [CrossRef] [Green Version]
- Årdal, C.; Balasegaram, M.; Laxminarayan, R.; McAdams, D.; Outterson, K.; Rex, J.H.; Sumpradit, N. Antibiotic development—Economic, regulatory and societal challenges. Nat. Rev. Microbiol. 2020, 18, 267–274. [Google Scholar] [CrossRef]
- O’Neill, J. Tackling Drug-Resistant Infections Globally: Final Report and Recommendations: The Review on Antimicrobial Resistance; Wellcome Trust: London, UK, 2016; Available online: https://amr-review.org (accessed on 12 October 2021).
- Kohanski, M.A.; Dwyer, D.J.; Collins, J.J. How antibiotics kill bacteria: From targets to networks. Nat. Rev. Microbiol. 2010, 8, 423–435. [Google Scholar] [CrossRef] [Green Version]
- Lawrie, R. First clinical use of penicillin. Br. Med. J. 1985, 290, 397. [Google Scholar] [CrossRef] [PubMed]
- Hutchings, M.I.; Truman, A.W.; Wilkinson, B. Antibiotics: Past, present and future. Curr. Opin. Microbiol. 2019, 51, 72–80. [Google Scholar] [CrossRef]
- Levy, S.B. Antibiotic Resistance: Consequences of Inaction. Clin. Infect. Dis. 2001, 33, S124–S129. [Google Scholar] [CrossRef] [PubMed]
- Ventola, C.L. The antibiotic resistance crisis: Part 1: Causes and threats. Pharm. Ther. 2015, 40, 277–283. [Google Scholar]
- Kwon, J.H.; Powderly, W.G. The post-antibiotic era is here. Science 2021, 373, 471. [Google Scholar] [CrossRef] [PubMed]
- Abraham, E.P.; Chain, E. An Enzyme from Bacteria able to Destroy Penicillin. Nature 1940, 146, 837. [Google Scholar] [CrossRef]
- Lobanovska, M.; Pilla, G. Penicillin’s discovery and antibiotic resistance: Lessons for the future? Yale J. Biol. Med. 2017, 90, 135–145. [Google Scholar]
- Rammelkamp, C.H.; Maxon, T. Resistance of Staphylococcus aureus to the Action of Penicillin. Proc. Soc. Exp. Biol. Med. 1942, 51, 386–389. [Google Scholar] [CrossRef]
- Biswas, S.; Brunel, J.M.; Dubus, J.C.; Reynaud-Gaubert, M.; Rolain, J.M. Colistin: An update on the antibiotic of the 21st century. Expert Rev. Anti. Infect. Ther. 2012, 10, 917–934. [Google Scholar] [CrossRef]
- Falagas, M.E.; Rafailidis, P.I.; Matthaiou, D.K. Resistance to polymyxins: Mechanisms, frequency and treatment options. Drug Resist. Updates 2010, 13, 132–138. [Google Scholar] [CrossRef]
- Baquero, F.; Levin, B.R. Proximate and ultimate causes of the bactericidal action of antibiotics. Nat. Rev. Microbiol. 2021, 19, 123–132. [Google Scholar] [CrossRef]
- Davis, B.D. Mechanism of bactericidal action of aminoglycosides. Microbiol. Rev. 1987, 51, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Walsh, C. Antibiotics; American Society of Microbiology: Washington, DC, USA, 2003. [Google Scholar]
- Drlica, K.; Malik, M. Fluoroquinolones: Action and Resistance. Curr. Top. Med. Chem. 2003, 3, 249–282. [Google Scholar] [CrossRef]
- Tomasz, A. The mechanism of the irreversible antimicrobial effects of penicillins: How the beta-lactam antibiotics kill and lyse bacteria. Annu. Rev. Microbiol. 1979, 33, 113–137. [Google Scholar] [CrossRef]
- Kohanski, M.A.; Dwyer, D.J.; Hayete, B.; Lawrence, C.A.; Collins, J.J. A Common Mechanism of Cellular Death Induced by Bactericidal Antibiotics. Cell 2007, 130, 797–810. [Google Scholar] [CrossRef] [Green Version]
- Kohanski, M.A.; Dwyer, D.J.; Wierzbowski, J.; Cottarel, G.; Collins, J.J. Mistranslation of Membrane Proteins and Two-Component System Activation Trigger Antibiotic-Mediated Cell Death. Cell 2008, 135, 679–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smilack, J.D. The Tetracyclines. Mayo Clin. Proc. 1999, 74, 727–729. [Google Scholar] [CrossRef]
- Chopra, I.; Roberts, M. Tetracycline Antibiotics: Mode of Action, Applications, Molecular Biology, and Epidemiology of Bacterial Resistance. Microbiol. Mol. Biol. Rev. 2001, 65, 232–260. [Google Scholar] [CrossRef] [Green Version]
- Smieja, M. Current indications for the use of clindamycin: A critical review. Can. J. Infect. Dis. 1998, 9, 22–28. [Google Scholar] [CrossRef] [Green Version]
- Fyfe, C.; Grossman, T.H.; Kerstein, K.; Sutcliffe, J. Resistance to macrolide antibiotics in public health pathogens. Cold Spring Harb. Perspect. Med. 2016, 6, a025395. [Google Scholar] [CrossRef] [Green Version]
- Brock Madigan, M.T.; Martinko, J.M.; Stahl, D.A.; Clark, D.P. Brock Biology of Microorganisms, 13th ed.; Prentice Hall: Upper Saddle River, NJ, USA, 2009; ISBN 1402472633. [Google Scholar]
- Sköld, O. Sulfonamide resistance: Mechanisms and trends. Drug Resist. Update 2000, 3, 155–160. [Google Scholar] [CrossRef]
- Vannuffel, P.; Cocito, C. Mechanism of action of streptogramins and macrolides. Drugs 1996, 51, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Werner, G.; Klare, I.; Witte, W. Molecular analysis of streptogramin resistance in enterococci. Int. J. Med. Microbiol. 2002, 292, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Pandit, N.; Singla, R.K.; Shrivastava, B. Current Updates on Oxazolidinone and Its Significance. Int. J. Med. Chem. 2012, 2012, 159285. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Wang, X.; Yin, Y.; Li, S.; Zhang, Y.; Wang, Q.; Wang, H. Molecular characteristics of oxazolidinone resistance in enterococci from a multicenter study in China. BMC Microbiol. 2019, 19, 162. [Google Scholar] [CrossRef] [PubMed]
- Schlünzen, F.; Zarivach, R.; Harms, J.; Bashan, A.; Tocilj, A.; Albrecht, R.; Yonath, A.; Franceschi, F. Structural basis for the interaction of antibiotics with the peptidyl transferase centre in eubacteria. Nature 2001, 413, 814–821. [Google Scholar] [CrossRef]
- Leclercq, R. Mechanisms of resistance to macrolides and lincosamides: Nature of the resistance elements and their clinical implications. Clin. Infect. Dis. 2002, 34, 482–492. [Google Scholar] [CrossRef] [Green Version]
- Manna, M.S.; Tamer, Y.T.; Gaszek, I.; Poulides, N.; Ahmed, A.; Wang, X.; Toprak, F.C.R.; Woodard, D.R.; Koh, A.Y.; Williams, N.S.; et al. A trimethoprim derivative impedes antibiotic resistance evolution. Nat. Commun. 2021, 12, 2949. [Google Scholar] [CrossRef]
- Tamer, Y.T.; Gaszek, I.K.; Abdizadeh, H.; Batur, T.A.; Reynolds, K.A.; Atilgan, A.R.; Atilgan, C.; Toprak, E. High-Order Epistasis in Catalytic Power of Dihydrofolate Reductase Gives Rise to a Rugged Fitness Landscape in the Presence of Trimethoprim Selection. Mol. Biol. Evol. 2019, 36, 1533–1550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yocum, R.R.; Rasmussen, J.R.; Strominger, J.L. The mechanism of action of penicillin. Penicillin acylates the active site of Bacillus stearothermophilus D-alanine carboxypeptidase. J. Biol. Chem. 1980, 255, 3977–3986. [Google Scholar] [CrossRef]
- Lowy, F.D. Antimicrobial resistance: The example of Staphylococcus aureus. J. Clin. Investig. 2003, 111, 1265–1273. [Google Scholar] [CrossRef]
- Jacoby, G.A. AmpC β-Lactamases. Clin. Microbiol. Rev. 2009, 22, 161–182. [Google Scholar] [CrossRef] [Green Version]
- Canton, R.; Gonzalez-Alba, J.M.; Galán, J.C. CTX-M Enzymes: Origin and Diffusion. Front. Microbiol. 2012, 3, 110. [Google Scholar] [CrossRef] [Green Version]
- Kakoullis, L.; Papachristodoulou, E.; Chra, P.; Panos, G. Mechanisms of Antibiotic Resistance in Important Gram-Positive and Gram-Negative Pathogens and Novel Antibiotic Solutions. Antibiotics 2021, 10, 415. [Google Scholar] [CrossRef]
- Yotsuji, A.; Mitsuyama, J.; Hori, R.; Yasuda, T.; Saikawa, I.; Inoue, M.; Mitsuhashi, S. Mechanism of action of cephalosporins and resistance caused by decreased affinity for penicillin-binding proteins in Bacteroides fragilis. Antimicrob. Agents Chemother. 1988, 32, 1848–1853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papp-Wallace, K.M.; Endimiani, A.; Taracila, M.A.; Bonomo, R.A. Carbapenems: Past, Present, and Future. Antimicrob. Agents Chemother. 2011, 55, 4943–4960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nordmann, P.; Dortet, L.; Poirel, L. Carbapenem resistance in Enterobacteriaceae: Here is the storm! Trends Mol. Med. 2012, 18, 263–272. [Google Scholar] [CrossRef]
- Marie, Q.A.; Karen, B. Carbapenemases: The Versatile β-Lactamases. Clin. Microbiol. Rev. 2007, 20, 440–458. [Google Scholar] [CrossRef] [Green Version]
- Meletis, G. Carbapenem resistance: Overview of the problem and future perspectives. Ther. Adv. Infect. Dis. 2015, 3, 15–21. [Google Scholar] [CrossRef] [Green Version]
- Bonomo, R.A.; Szabo, D. Mechanisms of Multidrug Resistance in Acinetobacter Species and Pseudomonas aeruginosa. Clin. Infect. Dis. 2006, 43, S49–S56. [Google Scholar] [CrossRef] [Green Version]
- Walsh, T.R. Emerging carbapenemases: A global perspective. Int. J. Antimicrob. Agents 2010, 36, S8–S14. [Google Scholar] [CrossRef]
- Krause, K.M.; Serio, A.W.; Kane, T.R.; Connolly, L.E. Aminoglycosides: An overview. Cold Spring Harb. Perspect. Med. 2016, 6, a027029. [Google Scholar] [CrossRef] [Green Version]
- Garneau-Tsodikova, S.; Labby, K.J. Mechanisms of resistance to aminoglycoside antibiotics: Overview and perspectives. Medchemcomm 2016, 7, 11–27. [Google Scholar] [CrossRef] [Green Version]
- Aldred, K.J.; Kerns, R.J.; Osheroff, N. Mechanism of Quinolone Action and Resistance. Biochemistry 2014, 53, 1565–1574. [Google Scholar] [CrossRef]
- Jacoby, G.A. Mechanisms of Resistance to Quinolones. Clin. Infect. Dis. 2005, 41, S120–S126. [Google Scholar] [CrossRef] [Green Version]
- Wehrli, W. Rifampin: Mechanisms of Action and Resistance. Rev. Infect. Dis. 1983, 5, S407–S411. [Google Scholar] [CrossRef] [PubMed]
- Tupin, A.; Gualtieri, M.; Roquet-Banères, F.; Morichaud, Z.; Brodolin, K.; Leonetti, J.-P. Resistance to rifampicin: At the crossroads between ecological, genomic and medical concerns. Int. J. Antimicrob. Agents 2010, 35, 519–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velkov, T.; Thompson, P.E.; Nation, R.L.; Li, J. Structure-activity relationships of polymyxin antibiotics. J. Med. Chem. 2010, 53, 1898–1916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bingbing, S.; Haiyan, L.; Yu, J.; Lei, S.; Sheng, Y.; Daijie, C.; Mariana, C. New Mutations Involved in Colistin Resistance in Acinetobacter baumannii. mSphere 2021, 5, e00895-19. [Google Scholar] [CrossRef] [Green Version]
- Andrade, F.F.; Silva, D.; Rodrigues, A.; Pina-Vaz, C. Colistin update on its mechanism of action and resistance, present and future challenges. Microorganisms 2020, 8, 1716. [Google Scholar] [CrossRef]
- Laurent, P.; Aurélie, J.; Patrice, N. Polymyxins: Antibacterial Activity, Susceptibility Testing, and Resistance Mechanisms Encoded by Plasmids or Chromosomes. Clin. Microbiol. Rev. 2017, 30, 557–596. [Google Scholar] [CrossRef] [Green Version]
- Randall, C.P.; Mariner, K.R.; Chopra, I.; O’Neill, A.J. The Target of Daptomycin Is Absent from Escherichia coli and Other Gram-Negative Pathogens. Antimicrob. Agents Chemother. 2013, 57, 637–639. [Google Scholar] [CrossRef] [Green Version]
- Mishra, N.N.; Yang, S.-J.; Chen, L.; Muller, C.; Saleh-Mghir, A.; Kuhn, S.; Peschel, A.; Yeaman, M.R.; Nast, C.C.; Kreiswirth, B.N.; et al. Emergence of Daptomycin Resistance in Daptomycin-Naïve Rabbits with Methicillin-Resistant Staphylococcus aureus Prosthetic Joint Infection Is Associated with Resistance to Host Defense Cationic Peptides and mprF Polymorphisms. PLoS ONE 2013, 8, e71151. [Google Scholar]
- Blaskovich, M.A.T.; Hansford, K.A.; Butler, M.S.; Jia, Z.; Mark, A.E.; Cooper, M.A. Developments in Glycopeptide Antibiotics. ACS Infect. Dis. 2018, 4, 715–735. [Google Scholar] [CrossRef] [Green Version]
- Stogios, P.J.; Savchenko, A. Molecular mechanisms of vancomycin resistance. Protein Sci. 2020, 29, 654–669. [Google Scholar] [CrossRef]
- Floss, H.G.; Yu, T.W. Rifamycin—Mode of action, resistance, and biosynthesis. Chem. Rev. 2005, 105, 621–632. [Google Scholar] [CrossRef]
- Campbell, E.A.; Korzheva, N.; Mustaev, A.; Murakami, K.; Nair, S.; Goldfarb, A.; Darst, S.A. Structural mechanism for rifampicin inhibition of bacterial RNA polymerase. Cell 2001, 104, 901–912. [Google Scholar] [CrossRef]
- McClure, W.R.; Cech, C.L. On the mechanism of rifampicin inhibition of RNA synthesis. J. Biol. Chem. 1978, 253, 8949–8956. [Google Scholar] [CrossRef]
- Vakulenko, S.B.; Mobashery, S. Versatility of aminoglycosides and prospects for their future. Clin. Microbiol. Rev. 2003, 16, 430–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunkle, J.A.; Xiong, L.; Mankin, A.S.; Cate, J.H.D. Structures of the Escherichia coli ribosome with antibiotics bound near the peptidyl transferase center explain spectra of drug action. Proc. Natl. Acad. Sci. USA 2010, 107, 17152–17157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nissen, P.; Hansen, J.; Ban, N.; Moore, P.B.; Steitz, T.A. The Structural Basis of Ribosome Activity in Peptide Bond Synthesis. Science 2000, 289, 920–930. [Google Scholar] [CrossRef] [Green Version]
- Menninger, J.R.; Otto, D.P. Erythromycin, carbomycin, and spiramycin inhibit protein synthesis by stimulating the dissocation of peptidyl-tRNA from ribosomes. Antimicrob. Agents Chemother. 1982, 21, 811–818. [Google Scholar] [CrossRef] [Green Version]
- Patel, U.; Yan, Y.P.; Hobbs, F.W.; Kaczmarczyk, J.; Slee, A.M.; Pompliano, D.L.; Kurilla, M.G.; Bobkova, E.V. Oxazolidinones Mechanism of Action: Inhibition of the First Peptide Bond Formation. J. Biol. Chem. 2001, 276, 37199–37205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karimi, R.; Ehrenberg, M. Dissociation Rate of Cognate Peptidyl-tRNA from the A-Site of Hyper-Accurate and Error-Prone Ribosomes. Eur. J. Biochem. 1994, 226, 355–360. [Google Scholar] [CrossRef]
- Falagas, M.E.; Kasiakou, S.K. Colistin: The revival of polymyxins for the management of multidrug-resistant gram-negative bacterial infections. Clin. Infect. Dis. 2005, 40, 1333–1341. [Google Scholar] [CrossRef] [Green Version]
- Gurjar, M. Colistin for lung infection: An update. J. Intensive Care 2015, 3, 3–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ernst, C.M.; Peschel, A. MprF-mediated daptomycin resistance. Int. J. Med. Microbiol. 2019, 309, 359–363. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.; Davies, D. Origins and evolution of antibiotic resistance. Microbiol. Mol. Biol. Rev. 2010, 74, 417–433. [Google Scholar] [CrossRef] [Green Version]
- Andersson, D.I.; Hughes, D. Antibiotic resistance and its cost: Is it possible to reverse resistance? Nat. Rev. Microbiol. 2010, 8, 260–271. [Google Scholar] [CrossRef]
- Langendonk, R.F.; Neill, D.R.; Fothergill, J.L. The Building Blocks of Antimicrobial Resistance in Pseudomonas aeruginosa: Implications for Current Resistance-Breaking Therapies. Front. Cell. Infect. Microbiol. 2021, 11, 307. [Google Scholar] [CrossRef] [PubMed]
- Tenover, F.C. Mechanisms of Antimicrobial Resistance in Bacteria. Am. J. Med. 2006, 119 (Suppl. S3–S10). [Google Scholar] [CrossRef]
- Tsuchido, T.; Takano, M. Senzitization by heat treatment of Escherichia coli K-12 cells to hydrophobic antibacterial compounds. Antimicrob. Agents Chemother. 1988, 32, 1680–1683. [Google Scholar] [CrossRef] [Green Version]
- Blair, J.M.A.; Webber, M.A.; Baylay, A.J.; Ogbolu, D.O.; Piddock, L.J.V. Molecular mechanisms of antibiotic resistance. Nat. Rev. Microbiol. 2015, 13, 42–51. [Google Scholar] [CrossRef]
- Schroeder, J.W.; Yeesin, P.; Simmons, L.A.; Wang, J.D. Sources of spontaneous mutagenesis in bacteria. Crit. Rev. Biochem. Mol. Biol. 2018, 53, 29–48. [Google Scholar] [CrossRef]
- Barbier, F.; Luyt, C.-E. Understanding resistance. Intensive Care Med. 2016, 42, 2080–2083. [Google Scholar] [CrossRef]
- Ruppé, É.; Woerther, P.L.; Barbier, F. Mechanisms of antimicrobial resistance in Gram-negative bacilli. Ann. Intensive Care 2015, 5, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Craig, M.R.; Alvaro, S.M. The evolution of antibiotic resistance. Science 2019, 365, 1082–1083. [Google Scholar] [CrossRef]
- Dubey, G.P.; Ben-Yehuda, S. Intercellular Nanotubes Mediate Bacterial Communication. Cell 2011, 144, 590–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domingues, S.; Nielsen, K.M. Membrane vesicles and horizontal gene transfer in prokaryotes. Curr. Opin. Microbiol. 2017, 38, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Scharn, C.R.; Tenover, F.C.; Goering, R.V. Transduction of staphylococcal cassette chromosome mec elements between strains of Staphylococcus aureus. Antimicrob. Agents Chemother. 2013, 57, 5233–5238. [Google Scholar] [CrossRef] [Green Version]
- Dowson, C.G.; Coffey, T.J.; Kell, C.; Whiley, R.A. Evolution of penicillin resistance in Streptococcus pneumoniae; the role of Streptococcus mitis in the formation of a low affinity PBP2B in S. pneumoniae. Mol. Microbiol. 1993, 9, 635–643. [Google Scholar] [CrossRef]
- Unemo, M.; Golparian, D.; Nicholas, R.; Ohnishi, M.; Gallay, A.; Sednaouie, P. High-level cefixime- and ceftriaxone-resistant Neisseria gonorrhoeae in France: Novel penA mosaic allele in a successful international clone causes treatment failure. Antimicrob. Agents Chemother. 2012, 56, 1273–1280. [Google Scholar] [CrossRef] [Green Version]
- Frost, L.S.; Leplae, R.; Summers, A.O.; Toussaint, A. Mobile genetic elements: The agents of open source evolution. Nat. Rev. Microbiol. 2005, 3, 722–732. [Google Scholar] [CrossRef]
- Alekshun, M.N.; Levy, S.B. Molecular Mechanisms of Antibacterial Multidrug Resistance. Cell 2007, 128, 1037–1050. [Google Scholar] [CrossRef] [Green Version]
- Paterson, D.L.; Bonomo, R.A. Extended-spectrum β-lactamases: A clinical update. Clin. Microbiol. Rev. 2005, 18, 657–686. [Google Scholar] [CrossRef] [Green Version]
- Kumarasamy, K.K.; Toleman, M.A.; Walsh, T.R.; Bagaria, J.; Butt, F.; Balakrishnan, R.; Chaudhary, U.; Doumith, M.; Giske, C.G.; Irfan, S.; et al. Emergence of a new antibiotic resistance mechanism in India, Pakistan, and the UK: A molecular, biological, and epidemiological study. Lancet Infect. Dis. 2010, 10, 597–602. [Google Scholar] [CrossRef]
- Yong, D.; Toleman, M.A.; Giske, C.G.; Cho, H.S.; Sundman, K.; Lee, K.; Walsh, T.R. Characterization of a new metallo-β-lactamase gene, bla NDM-1, and a novel erythromycin esterase gene carried on a unique genetic structure in Klebsiella pneumoniae sequence type 14 from India. Antimicrob. Agents Chemother. 2009, 53, 5046–5054. [Google Scholar] [CrossRef] [Green Version]
- Wozniak, R.A.F.; Waldor, M.K. Integrative and conjugative elements: Mosaic mobile genetic elements enabling dynamic lateral gene flow. Nat. Rev. Microbiol. 2010, 8, 552–563. [Google Scholar] [CrossRef]
- San Millan, A. Evolution of Plasmid-Mediated Antibiotic Resistance in the Clinical Context. Trends Microbiol. 2018, 26, 978–985. [Google Scholar] [CrossRef] [Green Version]
- Dijkshoorn, L.; Nemec, A.; Seifert, H. An increasing threat in hospitals: Multidrug-resistant Acinetobacter baumannii. Nat. Rev. Microbiol. 2007, 5, 939–951. [Google Scholar] [CrossRef] [PubMed]
- Levin, A.S.; Barone, A.A.; Penço, J.; Santos, M.V.; Marinho, I.S.; Arruda, E.A.G.; Manrique, E.I.; Costa, S.F. Intravenous Colistin as Therapy for Nosocomial Infections Caused by Multidrug-Resistant Pseudomonas aeruginosa and Acinetobacter baumannii. Clin. Infect. Dis. 1999, 28, 1008–1011. [Google Scholar] [CrossRef] [Green Version]
- Nikaido, H. Multidrug resistance in bacteria. Annu. Rev. Biochem. 2009, 78, 119–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikaido, H. Multiple antibiotic resistance and efflux. Curr. Opin. Microbiol. 1998, 1, 516–523. [Google Scholar] [CrossRef]
- Laurent, P.; Thierry, L.; Salih, T.; Esthel, R.; Jean-Louis, G.; Patrice, N. Characterization of Class 1 Integrons from Pseudomonas aeruginosa That Contain the blaVIM-2Carbapenem-Hydrolyzing β-Lactamase Gene and of Two Novel Aminoglycoside Resistance Gene Cassettes. Antimicrob. Agents Chemother. 2001, 45, 546–552. [Google Scholar] [CrossRef] [Green Version]
- Jacoby, G.A.; Medeiros, A.A. More extended-spectrum β-lactamases. Antimicrob. Agents Chemother. 1991, 35, 1697–1704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonnet, R. Growing Group of Extended-Spectrum β-Lactamases: The CTX-M Enzymes. Antimicrob. Agents Chemother. 2004, 48, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drlica, K.; Malik, M.; Kerns, R.J.; Zhao, X. Quinolone-mediated bacterial death. Antimicrob. Agents Chemother. 2008, 52, 385–392. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.K.; Mohan, A. Multidrug-resistant tuberculosis: A menace that threatens to destabilize tuberculosis control. Chest 2006, 130, 261–272. [Google Scholar] [CrossRef]
- Goldstein, B.P. Resistance to rifampicin: A review. J. Antibiot. 2014, 67. [Google Scholar] [CrossRef] [Green Version]
- Alangaden, G.J.; Kreiswirth, B.N.; Aouad, A.; Khetarpal, M.; Igno, F.R.; Moghazeh, S.L.; Manavathu, E.K.; Lerner, S.A. Mechanism of resistance to amikacin and kanamycin in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 1998, 42, 1295–1297. [Google Scholar] [CrossRef] [Green Version]
- Zgurskaya, H.I.; López, C.A.; Gnanakaran, S. Permeability Barrier of Gram-Negative Cell Envelopes and Approaches to Bypass It. ACS Infect. Dis. 2016, 1, 512–522. [Google Scholar] [CrossRef] [Green Version]
- Baharoglu, Z.; Mazel, D. Vibrio cholerae triggers SOS and mutagenesis in response to a wide range of antibiotics: A route towards multiresistance. Antimicrob. Agents Chemother. 2011, 55, 2438–2441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foster, P.L. Stress-induced mutagenesis in bacteria. Crit. Rev. Biochem. Mol. Biol. 2007, 42, 373–397. [Google Scholar] [CrossRef]
- Miller, C.; Thomsen, L.E.; Gaggero, C.; Mosseri, R.; Ingmer, H.; Cohen, S.N. SOS response induction by β-lactams and bacterial defense against antibiotic lethality. Science 2004, 305, 1629–1631. [Google Scholar] [CrossRef] [PubMed]
- Blázquez, J.; Couce, A.; Rodríguez-Beltrán, J.; Rodríguez-Rojas, A. Antimicrobials as promoters of genetic variation. Curr. Opin. Microbiol. 2012, 15, 561–569. [Google Scholar] [CrossRef] [PubMed]
- Galhardo, R.S.; Hastings, P.J.; Rosenberg, S.M. Mutation as a stress response and the regulation of evolvability. Crit. Rev. Biochem. Mol. Biol. 2007, 42, 399–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliver, A.; Cantón, R.; Campo, P.; Baquero, F.; Blázquez, J. High frequency of hypermutable Pseudomonas aeruginosa in cystic fibrosis lung infection. Science 2000, 288, 1251–1254. [Google Scholar] [CrossRef]
- Diaz-Diaz, S.; Recacha, E.; Machuca, J.; García-Duque, A.; Docobo-Pérez, F.; Blázquez, J.; Pascual, A.; Rodríguez-Martínez, J.M. Synergistic quinolone sensitization by targeting recA SOS response gene and oxidative stress. Antimicrob. Agents Chemother. 2021, 65, e02004-20. [Google Scholar] [CrossRef]
- Ciofu, O.; Mandsberg, L.F.; Bjarnsholt, T.; Wassermann, T.; Høiby, N. Genetic adaptation of pseudomonas aeruginosa during chronic lung infection of patients with cystic fibrosis: Strong and weak mutators with heterogeneous genetic backgrounds emerge in mucA and/or lasR mutants. Microbiology 2010, 156, 1108–1119. [Google Scholar] [CrossRef] [Green Version]
- Garibyan, L.; Huang, T.; Kim, M.; Wolff, E.; Nguyen, A.; Nguyen, T.; Diep, A.; Hu, K.; Iverson, A.; Yang, H.; et al. Use of the rpoB gene to determine the specificity of base substitution mutations on the Escherichia coli chromosome. DNA Repair 2003, 2, 593–608. [Google Scholar] [CrossRef]
- LeClerc, J.E.; Li, B.; Payne, W.L.; Cebula, T.A. High mutation frequencies among Escherichia coli and Salmonella pathogens. Science 1996, 274, 1208–1211. [Google Scholar] [CrossRef] [PubMed]
- Gogarten, J.P.; Townsend, J.P. Horizontal gene transfer, genome innovation and evolution. Nat. Rev. Microbiol. 2005, 3, 679–687. [Google Scholar] [CrossRef]
- Schönknecht, G.; Chen, W.H.; Ternes, C.M.; Barbier, G.G.; Shrestha, R.P.; Stanke, M.; Bräutigam, A.; Baker, B.J.; Banfield, J.F.; Garavito, R.M.; et al. Gene Transfer from Bacteria and Archaea Facilitated Evolution of an Extremophilic Eukaryote. Science 2013, 339, 1207–1210. [Google Scholar] [CrossRef]
- Lin, M.; Kussell, E. Inferring bacterial recombination rates from large-scale sequencing datasets. Nat. Methods 2019, 16, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Niehus, R.; Mitri, S.; Fletcher, A.G.; Foster, K.R. Migration and horizontal gene transfer divide microbial genomes into multiple niches. Nat. Commun. 2015, 6, 8924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soucy, S.M.; Huang, J.; Gogarten, J.P. Horizontal gene transfer: Building the web of life. Nat. Rev. Genet. 2015, 16, 472–482. [Google Scholar] [CrossRef]
- Iwasaki, W.; Takagi, T. Rapid Pathway Evolution Facilitated by Horizontal Gene Transfers across Prokaryotic Lineages. PLoS Genet. 2009, 5, e1000402. [Google Scholar] [CrossRef] [PubMed]
- Gal-Mor, O.; Finlay, B.B. Pathogenicity islands: A molecular toolbox for bacterial virulence. Cell. Microbiol. 2006, 8, 1707–1719. [Google Scholar] [CrossRef] [PubMed]
- Power, J.J.; Pinheiro, F.; Pompei, S.; Kovacova, V.; Yüksel, M.; Rathmann, I.; Förster, M.; Lässig, M.; Maier, B. Adaptive evolution of hybrid bacteria by horizontal gene transfer. Proc. Natl. Acad. Sci. USA 2021, 118, e2007873118. [Google Scholar] [CrossRef]
- Frazão, N.; Sousa, A.; Lässig, M.; Gordo, I. Horizontal gene transfer overrides mutation in Escherichia coli colonizing the mammalian gut. Proc. Natl. Acad. Sci. USA 2019, 116, 17906–17915. [Google Scholar] [CrossRef] [Green Version]
- Dagan, T.; Martin, W. The tree of one percent. Genome Biol. 2006, 7, 118. [Google Scholar] [CrossRef] [Green Version]
- Woods, L.C.; Gorrell, R.J.; Taylor, F.; Connallon, T.; Kwok, T.; McDonald, M.J. Horizontal gene transfer potentiates adaptation by reducing selective constraints on the spread of genetic variation. Proc. Natl. Acad. Sci. USA 2020, 117, 26868–26875. [Google Scholar] [CrossRef]
- Perron, G.G.; Lee, A.E.G.; Wang, Y.; Huang, W.E.; Barraclough, T.G. Bacterial recombination promotes the evolution of multi-drug-resistance in functionally diverse populations. Proc. R. Soc. B Biol. Sci. 2012, 279, 1477–1484. [Google Scholar] [CrossRef]
- Cooper, T.F. Recombination speeds adaptation by reducing competition between beneficial mutations in populations of Escherichia coli. PLoS Biol. 2007, 5, e225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorenz, M.G.; Wackernagel, W. Bacterial gene transfer by natural genetic transformation in the environment. Microbiol. Rev. 1994, 58, 563–602. [Google Scholar] [CrossRef]
- Bubendorfer, S.; Krebes, J.; Yang, I.; Hage, E.; Schulz, T.F.; Bahlawane, C.; Didelot, X.; Suerbaum, S. Genome-wide analysis of chromosomal import patterns after natural transformation of Helicobacter pylori. Nat. Commun. 2016, 7, 11995. [Google Scholar] [CrossRef] [Green Version]
- Muller, H.J. The relation of recombination to mutational advance. Mutat. Res. Mol. Mech. Mutagen. 1964, 1, 2–9. [Google Scholar] [CrossRef]
- Good, B.H.; McDonald, M.J.; Barrick, J.E.; Lenski, R.E.; Desai, M.M. The dynamics of molecular evolution over 60,000 generations. Nature 2017, 551, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Narra, H.P.; Ochman, H. Of What Use Is Sex to Bacteria? Curr. Biol. 2006, 16, R705–R710. [Google Scholar] [CrossRef] [Green Version]
- Lerminiaux, N.A.; Cameron, A.D.S. Horizontal transfer of antibiotic resistance genes in clinical environments. Can. J. Microbiol. 2018, 65, 34–44. [Google Scholar] [CrossRef]
- Fournier, P.E.; Vallenet, D.; Barbe, V.; Audic, S.; Ogata, H.; Poirel, L.; Richet, H.; Robert, C.; Mangenot, S.; Abergel, C.; et al. Comparative genomics of multidrug resistance in Acinetobacter baumannii. PLoS Genet. 2006, 2, e7. [Google Scholar] [CrossRef] [Green Version]
- Hughes, D.; Andersson, D.I. Evolutionary consequences of drug resistance: Shared principles across diverse targets and organisms. Nat. Rev. Genet. 2015, 16, 459–471. [Google Scholar] [CrossRef] [PubMed]
- Brandis, G.; Wrande, M.; Liljas, L.; Hughes, D. Fitness-compensatory mutations in rifampicin-resistant RNA polymerase. Mol. Microbiol. 2012, 85, 142–151. [Google Scholar] [CrossRef]
- Huseby, D.L.; Pietsch, F.; Brandis, G.; Garoff, L.; Tegehall, A.; Hughes, D. Mutation Supply and Relative Fitness Shape the Genotypes of Ciprofloxacin-Resistant Escherichia coli. Mol. Biol. Evol. 2017, 34, 1029–1039. [Google Scholar] [CrossRef] [Green Version]
- Dunai, A.; Spohn, R.; Farkas, Z.; Lázár, V.; Györkei, Á.; Apjok, G.; Boross, G.; Szappanos, B.; Grézal, G.; Faragó, A.; et al. Rapid decline of bacterial drug-resistance in an antibiotic-free environment through phenotypic reversion. Elife 2019, 8, e47088. [Google Scholar] [CrossRef]
- Shcherbakov, D.; Akbergenov, R.; Matt, T.; Sander, P.; Andersson, D.I.; Böttger, E.C. Directed mutagenesis of mycobacterium smegmatis 16S rRNA to reconstruct the in vivo evolution of aminoglycoside resistance in mycobacterium tuberculosis. Mol. Microbiol. 2010, 77, 830–840. [Google Scholar] [CrossRef] [PubMed]
- Lipsitch, M.; Bergstrom, C.T.; Levin, B.R. The epidemiology of antibiotic resistance in hospitals: Paradoxes and prescriptions. Proc. Natl. Acad. Sci. USA 2000, 97, 1938–1943. [Google Scholar] [CrossRef] [Green Version]
- Drlica, K.; Zhao, X. Mutant selection window hypothesis updated. Clin. Infect. Dis. 2007, 44, 681–688. [Google Scholar] [CrossRef]
- Gullberg, E.; Cao, S.; Berg, O.G.; Ilbäck, C.; Sandegren, L.; Hughes, D.; Andersson, D.I. Selection of resistant bacteria at very low antibiotic concentrations. PLoS Pathog. 2011, 7, e1002158. [Google Scholar] [CrossRef] [Green Version]
- Stanton, I.C.; Murray, A.K.; Zhang, L.; Snape, J.; Gaze, W.H. Evolution of antibiotic resistance at low antibiotic concentrations including selection below the minimal selective concentration. Commun. Biol. 2020, 3, 467. [Google Scholar] [CrossRef] [PubMed]
- Wistrand-Yuen, E.; Knopp, M.; Hjort, K.; Koskiniemi, S.; Berg, O.G.; Andersson, D.I. Evolution of high-level resistance during low-level antibiotic exposure. Nat. Commun. 2018, 9, 1599. [Google Scholar] [CrossRef] [Green Version]
- Andersson, D.I.; Hughes, D. Microbiological effects of sublethal levels of antibiotics. Nat. Rev. Microbiol. 2014, 12, 465–478. [Google Scholar] [CrossRef] [PubMed]
- Oz, T.; Guvenek, A.; Yildiz, S.; Karaboga, E.; Tamer, Y.T.; Mumcuyan, N.; Ozan, V.B.; Senturk, G.H.; Cokol, M.; Yeh, P.; et al. Strength of selection pressure is an important parameter contributing to the complexity of antibiotic resistance evolution. Mol. Biol. Evol. 2014, 31, 2387–2401. [Google Scholar] [CrossRef] [Green Version]
- Markussen, T.; Marvig, R.L.; Gómez-Lozano, M.; Aanæs, K.; Burleigh, A.E.; Høiby, N.; Johansen, H.K.; Molin, S.; Jelsbak, L. Environmental heterogeneity drives within-host diversification and evolution of Pseudomonas aeruginosa. MBio 2014, 5, e01592-14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerrish, P.J.; Lenski, R.E. The fate of competing beneficial mutations in an asexual population. Genetica 1998, 127–144. [Google Scholar] [CrossRef]
- Gifford, D.R.; MacLean, R.C. Evolutionary reversals of antibiotic resistance in experimental populations of pseudomonas aeruginosa. Evolution 2013, 67, 2973–2981. [Google Scholar] [CrossRef]
- Qi, Q.; Toll-Riera, M.; Heilbron, K.; Preston, G.M.; Maclean, R.C. The genomic basis of adaptation to the fitness cost of rifampicin resistance in pseudomonas aeruginosa. Proc. R. Soc. B Biol. Sci. 2016, 283, 20152452. [Google Scholar] [CrossRef] [Green Version]
- Wong, A. Epistasis and the evolution of antimicrobial resistance. Front. Microbiol. 2017, 8, 246. [Google Scholar] [CrossRef] [Green Version]
- Angst, D.C.; Hall, A.R. The cost of antibiotic resistance depends on evolutionary history in Escherichia coli. BMC Evol. Biol. 2013, 13, 163–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinreich, D.M.; Delaney, N.F.; DePristo, M.A.; Hartl, D.L. Darwinian evolution can follow only very few mutational paths to fitter proteins. Science 2006, 312, 111–114. [Google Scholar] [CrossRef] [Green Version]
- Schenk, M.F.; Szendro, I.G.; Krug, J.; de Visser, J.A.G.M. Quantifying the adaptive potential of an antibiotic resistance enzyme. PLoS Genet. 2012, 8, e1002783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez-Beltrán, J.; DelaFuente, J.; León-Sampedro, R.; MacLean, R.C.; San Millán, Á. Beyond horizontal gene transfer: The role of plasmids in bacterial evolution. Nat. Rev. Microbiol. 2021, 19, 347–359. [Google Scholar] [CrossRef]
- Trindade, S.; Sousa, A.; Xavier, K.B.; Dionisio, F.; Ferreira, M.G.; Gordo, I. Positive epistasis drives the acquisition of multidrug resistance. PLoS Genet. 2009, 5, e1000578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durão, P.; Trindade, S.; Sousa, A.; Gordo, I. Multiple resistance at no cost: Rifampicin and streptomycin a dangerous liaison in the spread of antibiotic resistance. Mol. Biol. Evol. 2015, 32, 2675–2680. [Google Scholar] [CrossRef] [Green Version]
- Levin-Reisman, I.; Brauner, A.; Ronin, I.; Balaban, N.Q. Epistasis between antibiotic tolerance, persistence, and resistance mutations. Proc. Natl. Acad. Sci. USA 2019, 116, 14734–14739. [Google Scholar] [CrossRef] [Green Version]
- Lukačišinová, M.; Fernando, B.; Bollenbach, T. Highly parallel lab evolution reveals that epistasis can curb the evolution of antibiotic resistance. Nat. Commun. 2020, 11, 3105. [Google Scholar] [CrossRef]
- Kryazhimskiy, S.; Rice, D.P.; Jerison, E.R.; Desai, M.M. Global epistasis makes adaptation predictable despite sequence-level stochasticity. Science 2014, 344, 1519–1522. [Google Scholar] [CrossRef] [Green Version]
- MacLean, R.C.; Perron, G.G.; Gardner, A. Diminishing Returns From Beneficial Mutations and Pervasive Epistasis Shape the Fitness Landscape for Rifampicin Resistance in Pseudomonas aeruginosa. Genetics 2010, 186, 1345–1354. [Google Scholar] [CrossRef] [Green Version]
- Andersson, D.I.; Levin, B.R. The biological cost of antibiotic resistance. Curr. Opin. Microbiol. 1999, 2, 489–493. [Google Scholar] [CrossRef]
- Maisnier-Patin, S.; Berg, O.G.; Liljas, L.; Andersson, D.I. Compensatory adaptation to the deleterious effect of antibiotic resistance in Salmonella typhimurium. Mol. Microbiol. 2002, 46, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Nagaev, I.; Björkman, J.; Andersson, D.I.; Hughes, D. Biological cost and compensatory evolution in fusidic acid-resistant Staphylococcus aureus. Mol. Microbiol. 2001, 40, 433–439. [Google Scholar] [CrossRef]
- Comas, I.; Borrell, S.; Roetzer, A.; Rose, G.; Malla, B.; Kato-Maeda, M.; Galagan, J.; Niemann, S.; Gagneux, S. Whole-genome sequencing of rifampicin-resistant Mycobacterium tuberculosis strains identifies compensatory mutations in RNA polymerase genes. Nat. Genet. 2012, 44, 106–110. [Google Scholar] [CrossRef] [Green Version]
- Moura de Sousa, J.; Balbontín, R.; Durão, P.; Gordo, I. Multidrug-resistant bacteria compensate for the epistasis between resistances. PLoS Biol. 2017, 15, e2001741. [Google Scholar] [CrossRef] [PubMed]
- Baym, M.; Stone, L.K.; Kishony, R. Multidrug evolutionary strategies to reverse antibiotic resistance. Science 2016, 351, aad3292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chait, R.; Craney, A.; Kishony, R. Antibiotic interactions that select against resistance. Nature 2007, 446, 668–671. [Google Scholar] [CrossRef]
- Michel, J.B.; Yeh, P.J.; Chait, R.; Moellering, R.C.; Kishony, R. Drug interactions modulate the potential for evolution of resistance. Proc. Natl. Acad. Sci. USA 2008, 105, 14918–14923. [Google Scholar] [CrossRef] [Green Version]
- Imamovic, L.; Sommer, M.O.A. Use of collateral sensitivity networks to design drug cycling protocols that avoid resistance development. Sci. Transl. Med. 2013, 5, 204ra132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lázár, V.; Nagy, I.; Spohn, R.; Csörgo, B.; Györkei, Á.; Nyerges, Á.; Horváth, B.; Vörös, A.; Busa-Fekete, R.; Hrtyan, M.; et al. Genome-wide analysis captures the determinants of the antibiotic cross-resistance interaction network. Nat. Commun. 2014, 5, 4352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbosa, C.; Beardmore, R.; Schulenburg, H.; Jansen, G. Antibiotic combination efficacy (ACE) networks for a Pseudomonas aeruginosa model. PLoS Biol. 2018, 16, e2004356. [Google Scholar] [CrossRef] [Green Version]
- Rosenkilde, C.E.H.; Munck, C.; Porse, A.; Linkevicius, M.; Andersson, D.I.; Sommer, M.O.A. Collateral sensitivity constrains resistance evolution of the CTX-M-15 β-lactamase. Nat. Commun. 2019, 10, 618. [Google Scholar] [CrossRef] [Green Version]
- Roemhild, R.; Andersson, D.I. Mechanisms and therapeutic potential of collateral sensitivity to antibiotics. PLoS Pathog. 2021, 17, e1009172. [Google Scholar] [CrossRef] [PubMed]
- Yen, P.; Papin, J.A. History of antibiotic adaptation influences microbial evolutionary dynamics during subsequent treatment. PLOS Biol. 2017, 15, e2001586. [Google Scholar] [CrossRef] [Green Version]
- Nichol, D.; Rutter, J.; Bryant, C.; Hujer, A.M.; Lek, S.; Adams, M.D.; Jeavons, P.; Anderson, A.R.A.; Bonomo, R.A.; Scott, J.G. Antibiotic collateral sensitivity is contingent on the repeatability of evolution. Nat. Commun. 2019, 10, 334. [Google Scholar] [CrossRef] [Green Version]
- Barbosa, C.; Römhild, R.; Rosenstiel, P.; Schulenburg, H. Evolutionary stability of collateral sensitivity to antibiotics in the model pathogen Pseudomonas aeruginosa. Elife 2019, 8, e51481. [Google Scholar] [CrossRef]
- Hughes, D.; Andersson, D.I. Evolutionary Trajectories to Antibiotic Resistance. Annu. Rev. Microbiol. 2017, 71, 579–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garoff, L.; Pietsch, F.; Huseby, D.L.; Lilja, T.; Brandis, G.; Hughes, D. Population Bottlenecks Strongly Influence the Evolutionary Trajectory to Fluoroquinolone Resistance in Escherichia coli. Mol. Biol. Evol. 2020, 37, 1637–1646. [Google Scholar] [CrossRef] [PubMed]
- Vogwill, T.; Phillips, R.L.; Gifford, D.R.; Maclean, R.C. Divergent evolution peaks under intermediate population bottlenecks during bacterial experimental evolution. Proc. R. Soc. B Biol. Sci. 2016, 283, 20160749. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
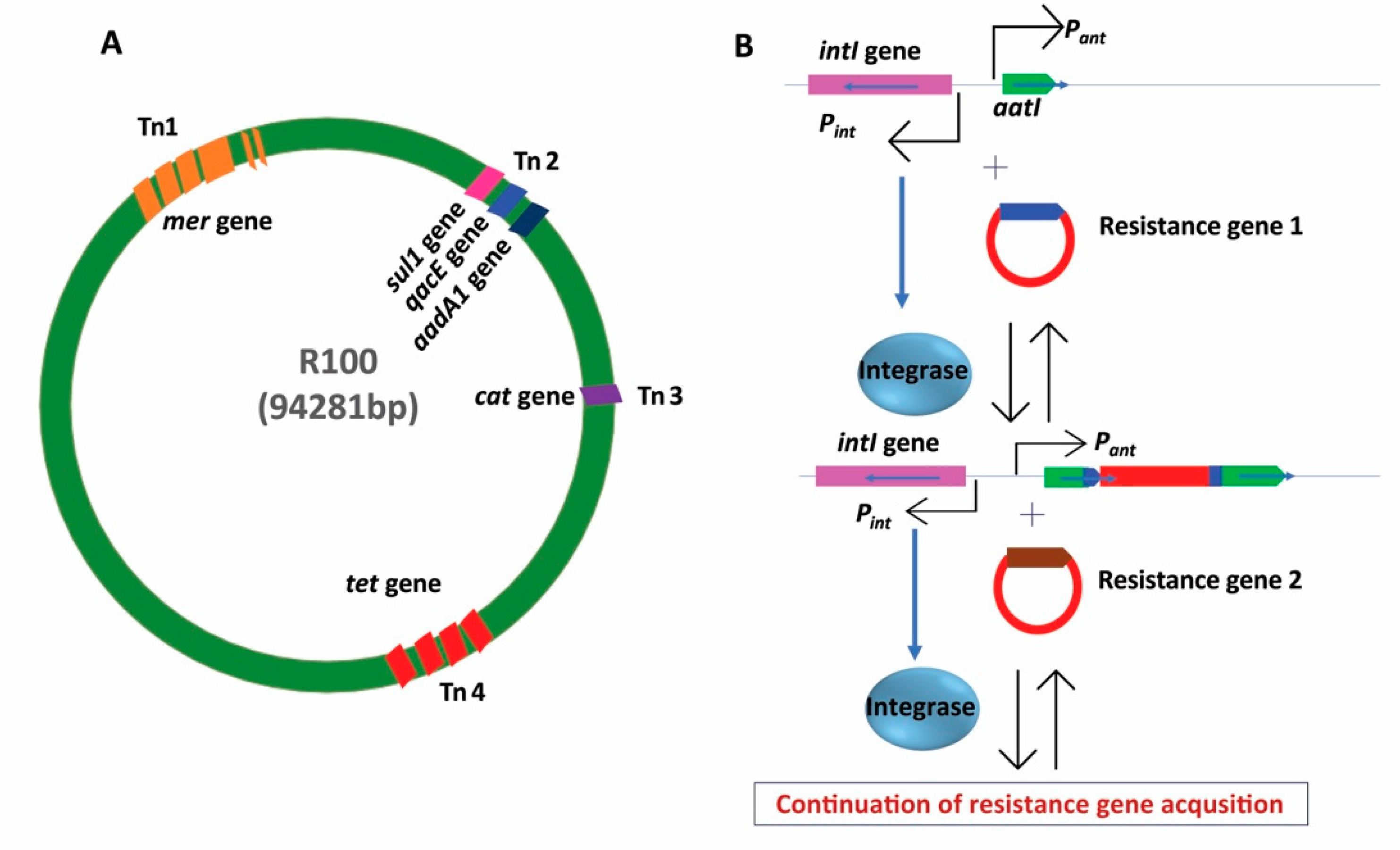{kind=link}
| Bacteriostatic Candidates | Mode of Action | Mechanism of Resistance |
|---|---|---|
| Tetracycline | Reversibly inhibits 30S ribosomal subunit of bacteria [25]. | Efflux system and protecting ribosomes [26]. |
| Macrolides | Reversibly inhibits 50S ribosomal subunit of bacteria [27]. | Methylation of the 23S rRNA, efflux system [28]. |
| Sulphonamides | Inhibits folate synthesizing enzyme dihydropteroate synthase (DHPS) [29]. | By horizontal transfer of dihydropteroate synthase gene [30]. |
| Streptogramins | Reversibly inhibits 50S ribosomal subunit of bacteria [31]. | Acetyltransferases vatD gene expression mediates streptogramin A, wheras vatE and ermB or vgbA gene cluster confers streptogramin B antibiotics [32]. |
| Oxazolidinones | Reversibly inhibits 50S ribosomal subunit of bacteria [33]. | High diversity and coselection of optrA [34]. |
| Lincosamides | Reversibly inhibits 50S ribosomal subunit of bacteria [35]. | Target site modification, efflux system and drug inactivation [36]. |
| Trimethoprim | Occupying the active site of bacterial dihydrofolate reductase (DHFR), thus blocking the activity of the enzyme [37]. | Increase expression of DHFR or decrease the affinity of DHFR to the drug [38]. |
| Bactericide Candidates | Mode of Action | Mechanism of Resistance |
|---|---|---|
| Penicillins | Competitively inhibits the transpeptidase enzyme resulting cross-linking blockage in cell wall [39]. | Beta-lactamase encoded by blaZ, altered PBP2a encoded by mecA [14,40], extended-spectrum-beta-lactamases (ESBLs), AmpC beta-lactamase (i.e., blaAmpC) [41,42,43]. |
| Cephalosporins | Competitively inhibits the transpeptidase enzyme resulting in cross-linking or blockage in cell wall [44]. | AmpC beta-lactamase (i.e., blaAmpC), ESBLs (i.e., blaCTX-M) [41,42]. |
| Carbapenems | Binding with penicillin-binding proteins (PBPs) and inactivation of these proteins leads to cell wall synthesis interruption [45]. | Carbapenemases (i.e., class A serine-carbapenemase including KPCs; class B metallo-carbapenemase including New-Delhi-metallo-beta-lactamases or NDM, Verona-integron-encoded beta-lactamases or VIM, Imepenemase IMP-carbapenemase (also a metallo-beta-lactamase); class D serine carbapenemase such oxacillinase (OXA) [46,47], mutation-derived target enzyme modification [48]; preventing the drug entry by modifying outer membrane permeability [49]; pumping carbapenems out by efflux pump systems [50]. |
| Aminoglycoside | Binding with 30 s ribosomal subunit resulting genetic code misreading followed by interruption of bacterial translation [51]. | Mostly through aminoglycosides modifying enzymes encoded by aac (aminoacetyl-tranferase) and aph (aminophospho-transferase), efflux system, or mutation in rpsL and 16S rRNA [43,52]. |
| Fluoroquinolones | Interrupting bacterial DNA replication by inhibiting topoisomerases [53]. | Target enzyme mutation (DNA gyrase encoded by gyrA and gyrB, and topoisomerase IV encoded by parC and parE genes), efflux system and changing drug entry [54]. |
| Rifamycin | Interrupting transcription by inhibiting bacterial RNA polymerase [55]. | Mutation of the target (beta subunit of RNA polymerase encoded by rpoB) [56]. |
| Polymyxins | Binding to lipid A of LPS and interfere with outer membrane permeability [57]. | The pmrHFIJKLM (also known as arn operon) and pmrCAB operon—both invove in the biosynthesis of LAra4N and modify the lipid A, thus disrupt lipid A charges [17]; mutations in genes encoding the two-component regulatory systems such as pmrAB [58], phoPQ and plasmid-borne mcr genes confer resistance to colistin—the last line of drug [59,60]. |
| Daptomycin | Binding to anionic phospholipids in the cytoplasmic membrane [61]. | Mutations in gene mprF which encodes the multiple peptide resistance factor [62]. |
| Vancomycin | Binding to the dipeptide terminus d-Ala-d-Ala of peptidoglycan pentapeptide precursors preventing peptidoglycan crosslinking leads to the inhibition of bacterial cell wall synthesis [63]. | Replacing d-Ala-d-Ala with d-Ala-d-lac or d-Ala-d-Ser alternatives to which vancomycin has low affinity [64]. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hasan, C.M.; Dutta, D.; Nguyen, A.N.T. Revisiting Antibiotic Resistance: Mechanistic Foundations to Evolutionary Outlook. Antibiotics 2022, 11, 40. https://doi.org/10.3390/antibiotics11010040
Hasan CM, Dutta D, Nguyen ANT. Revisiting Antibiotic Resistance: Mechanistic Foundations to Evolutionary Outlook. Antibiotics. 2022; 11(1):40. https://doi.org/10.3390/antibiotics11010040
Chicago/Turabian StyleHasan, Chowdhury M., Debprasad Dutta, and An N. T. Nguyen. 2022. "Revisiting Antibiotic Resistance: Mechanistic Foundations to Evolutionary Outlook" Antibiotics 11, no. 1: 40. https://doi.org/10.3390/antibiotics11010040
APA StyleHasan, C. M., Dutta, D., & Nguyen, A. N. T. (2022). Revisiting Antibiotic Resistance: Mechanistic Foundations to Evolutionary Outlook. Antibiotics, 11(1), 40. https://doi.org/10.3390/antibiotics11010040