
Recent Progress in the Discovery and Design of Antimicrobial Peptides Using Traditional Machine Learning and Deep Learning


 , ,
, , 
Abstract
:1. Introduction
1.1. Discovery of Early AMPs
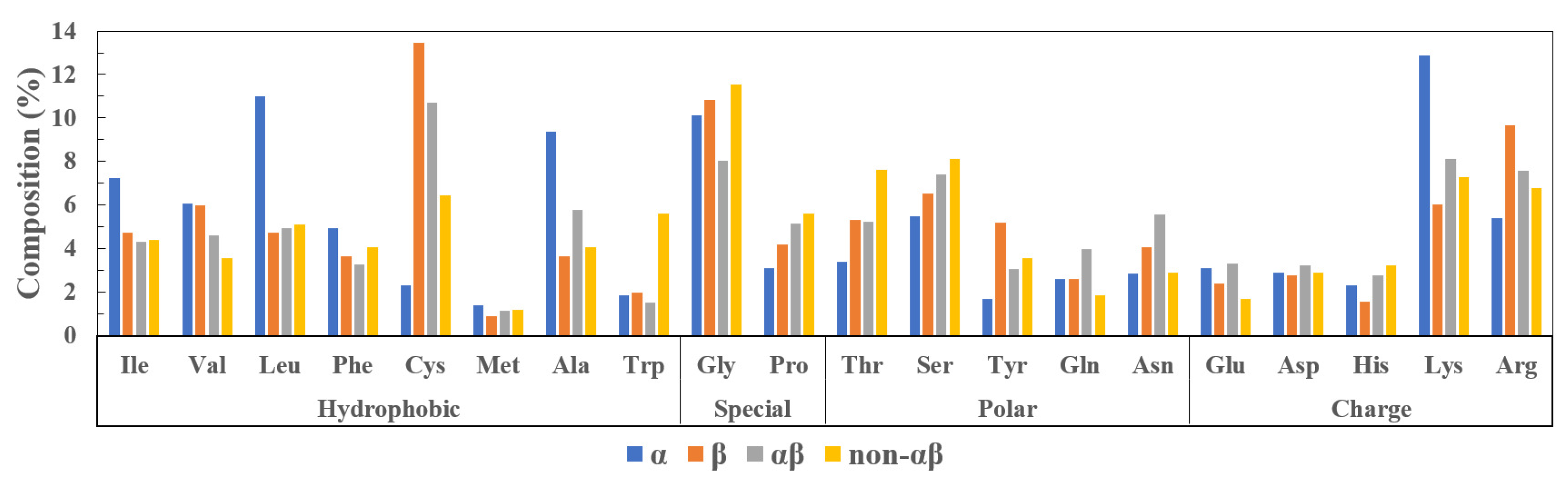
1.2. Classification
1.2.1. -Helical Peptides
1.2.2. -Sheet Peptides
1.2.3. Mixed Peptides
1.2.4. Non- Peptides
1.3. Mechanism of Action
1.4. Therapeutic and Industrial Applications
1.4.1. Biomedicines in Pharmaceutical Industry
1.4.2. Substitutes for Antibiotics and Pesticides in Agriculture and Animal Husbandry
1.4.3. Food Preservatives and Packaging in the Food Industry
1.5. Limitation of AMPs and Bacterial Resistance
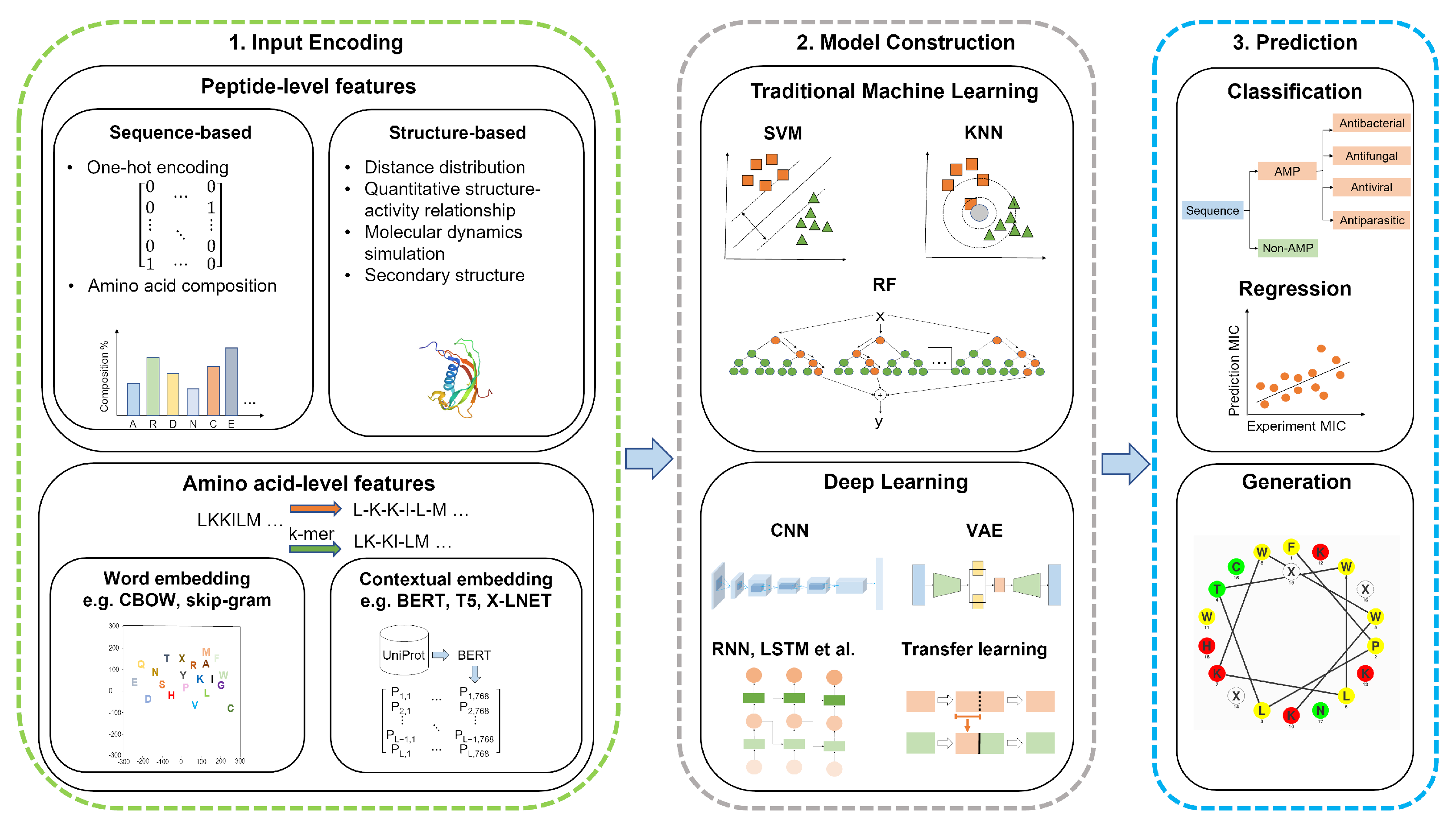
2. AMP Discovery and Design—The Machine Learning Workflow
3. Feature Encoding Methods
3.1. Peptide-Level Features
3.1.1. Sequence-Based Features
3.1.2. Structure-Based Features
3.2. Amino Acid-Level Features
3.2.1. Word Embedding
3.2.2. Contextual Embedding
4. AMP Prediction by Traditional Machine Learning
5. AMP Prediction by Deep Learning
5.1. Deep Neural Networks (DNNs)
5.2. Deep Learning with CNN Layers
5.3. Deep Learning with RNN Layers
5.4. Hybrid Learning
5.4.1. Hybrid of CNN and RNN Layers
5.4.2. Hybrid of DL and Attention Mechanism
5.4.3. Hybrid of Traditional ML and DL
5.5. The Other DL Approaches for Identifying AMPs
5.5.1. Off-the-Shelf DL Architectures
5.5.2. Transfer Learning
5.6. DL for AMP Regression
6. AMP Design by Optimization
7. De Novo AMP Design
8. Limitations and Challenges
8.1. Data Insufficiency
8.2. Limited Modeling beyond Binary Classification of Linear AMPs
8.3. Limited Attempt in Drug-Likeness Prediction of AMPs
8.4. DL Model Optimization and Reproducibility
8.5. Explainable Artificial Intelligence
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mookherjee, N.; Anderson, M.A.; Haagsman, H.P.; Davidson, D.J. Antimicrobial host defence peptides: Functions and clinical potential. Nat. Rev. Drug Discov. 2020, 19, 311–332. [Google Scholar] [CrossRef] [PubMed]
- Diamond, G.; Beckloff, N.; Weinberg, A.; Kisich, O.K. The Roles of Antimicrobial Peptides in Innate Host Defense. Curr. Pharm. Des. 2009, 15, 2377–2392. [Google Scholar] [CrossRef] [Green Version]
- Spohn, R.; Daruka, L.; Lázár, V.; Martins, A.; Vidovics, F.; Grézal, G.; Méhi, O.; Kintses, B.; Számel, M.; Jangir, P.K.; et al. Integrated evolutionary analysis reveals antimicrobial peptides with limited resistance. Nat. Commun. 2019, 10, 4538. [Google Scholar] [CrossRef] [Green Version]
- Ma, R.; Wong, S.W.; Ge, L.; Shaw, C.; Siu, S.W.I.; Kwok, H.F. In-Vitro and MD Simulation Study to Explore Physicochemical Parameters for Antibacterial Peptide to Become Potent Anticancer Peptide. Mol. Ther.-Oncolytics 2020, 16, 7–19. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Li, X.; Wang, Z. APD3: The antimicrobial peptide database as a tool for research and education. Nucleic Acids Res. 2016, 44, D1087–D1093. [Google Scholar] [CrossRef] [Green Version]
- Jhong, J.H.; Yao, L.; Pang, Y.; Li, Z.; Chung, C.R.; Wang, R.; Li, S.; Li, W.; Luo, M.; Ma, R.; et al. dbAMP 2.0: Updated resource for antimicrobial peptides with an enhanced scanning method for genomic and proteomic data. Nucleic Acids Res. 2022, 50, D460–D470. [Google Scholar] [CrossRef]
- Pirtskhalava, M.; Amstrong, A.A.; Grigolava, M.; Chubinidze, M.; Alimbarashvili, E.; Vishnepolsky, B.; Gabrielian, A.; Rosenthal, A.; Hurt, D.E.; Tartakovsky, M. DBAASP v3: Database of antimicrobial/cytotoxic activity and structure of peptides as a resource for development of new therapeutics. Nucleic Acids Res. 2021, 49, D288–D297. [Google Scholar] [CrossRef] [PubMed]
- Gan, B.H.; Gaynord, J.; Rowe, S.M.; Deingruber, T.; Spring, D.R. The multifaceted nature of antimicrobial peptides: Current synthetic chemistry approaches and future directions. Chem. Soc. Rev. 2021, 50, 7820–7880. [Google Scholar] [CrossRef]
- Nakatsuji, T.; Gallo, R.L. Antimicrobial Peptides: Old Molecules with New Ideas. J. Investig. Dermatol. 2012, 132, 887–895. [Google Scholar] [CrossRef] [Green Version]
- Fleming, A.; Wright, A.E. On a remarkable bacteriolytic element found in tissues and secretions. Proc. R. Soc. Lond. Ser. B Contain. Pap. A Biol. Character 1922, 93, 306–317. [Google Scholar] [CrossRef]
- Ragland, S.A.; Criss, A.K. From bacterial killing to immune modulation: Recent insights into the functions of lysozyme. PLoS Pathog. 2017, 13, e1006512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogers, L.A.; Whittier, E.O. Limiting Factors in the Lactic Fermentation. J. Bacteriol. 1928, 16, 211–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, J.; Gwak, J.; Kamarajan, P.; Fenno, J.; Rickard, A.; Kapila, Y. Biomedical applications of nisin. J. Appl. Microbiol. 2016, 120, 1449–1465. [Google Scholar] [CrossRef] [Green Version]
- Severina, E.; Severin, A.; Tomasz, A. Antibacterial efficacy of nisin against multidrug-resistant Gram-positive pathogens. J. Antimicrob. Chemother. 1998, 41, 341–347. [Google Scholar] [CrossRef]
- Dubos, R.J. Studies on a Bactericidal Agent Extracted from a Soil Bacillus: II. Protective Effect of the Bactericidal Agent against Experimental Pneumococcus Infections in Mice. J. Exp. Med. 1939, 70, 11–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simmaco, M.; Kreil, G.; Barra, D. Bombinins, antimicrobial peptides from Bombina species. Biochim. Biophys. Acta (BBA)-Biomembr. 2009, 1788, 1551–1555. [Google Scholar] [CrossRef] [Green Version]
- Zeya, H.I.; Spitznagel, J.K. Antibacterial and Enzymic Basic Proteins from Leukocyte Lysosomes: Separation and Identification. Science 1963, 142, 1085–1087. [Google Scholar] [CrossRef] [PubMed]
- Ganz, T.; Selsted, M.E.; Szklarek, D.; Harwig, S.S.; Daher, K.; Bainton, D.F.; Lehrer, R.I. Defensins. Natural peptide antibiotics of human neutrophils. J. Clin. Investig. 1985, 76, 1427–1435. [Google Scholar] [CrossRef]
- Hultmark, D.; Steiner, H.; Rasmuson, T.; Boman, H.G. Insect Immunity. Purification and Properties of Three Inducible Bactericidal Proteins from Hemolymph of Immunized Pupae of Hyalophora Cecropia. Eur. J. Biochem. 1980, 106, 7–16. [Google Scholar] [CrossRef]
- Zasloff, M. Magainins, a class of antimicrobial peptides from Xenopus skin: Isolation, characterization of two active forms, and partial cDNA sequence of a precursor. Proc. Natl. Acad. Sci. USA 1987, 84, 5449–5453. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Adams, M.E. Lycotoxins, Antimicrobial Peptides from Venom of the Wolf SpiderLycosa carolinensis. J. Biol. Chem. 1998, 273, 2059–2066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nawrot, R.; Barylski, J.; Nowicki, G.; Broniarczyk, J.; Buchwald, W.; Goździcka-Józefiak, A. Plant antimicrobial peptides. Folia Microbiol. 2014, 59, 181–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balls, A.K.; Hale, W.S.; Harris, T.H. A Crystalline Protein Obtained from a Lipoprotein of Wheat Flour. Cereal Chem. 1942, 19, 279–288. [Google Scholar]
- Colilla, F.J.; Rocher, A.; Mendez, E. γ-Purothionins: Amino acid sequence of two polypeptides of a new family of thionins from wheat endosperm. FEBS Lett. 1990, 270, 191–194. [Google Scholar] [CrossRef] [Green Version]
- Broekaert, W.F.; Terras, G.; Cammue, A.; Osborn, R.W. Plant Defensins: Nove1 Antimicrobial Peptides as Components of the Host Defense System. Plant Physiol. 1995, 108, 1353. [Google Scholar] [CrossRef]
- Wang, G. Chapter One—Unifying the classification of antimicrobial peptides in the antimicrobial peptide database. In Methods in Enzymology; Hicks, L.M., Ed.; Academic Press: Cambridge, MA, USA, 2022; Volume 663, pp. 1–18. [Google Scholar] [CrossRef]
- Koehbach, J.; Craik, D.J. The Vast Structural Diversity of Antimicrobial Peptides. Trends Pharmacol. Sci. 2019, 40, 517–528. [Google Scholar] [CrossRef]
- Perumal, P.; Pandey, V.P. Antimicrobial peptides: The role of hydrophobicity in the alpha helical structure. J. Pharm. Pharmacogn. Res. 2013, 1, 39–53. [Google Scholar]
- Xhindoli, D.; Pacor, S.; Benincasa, M.; Scocchi, M.; Gennaro, R.; Tossi, A. The human cathelicidin LL-37—A pore-forming antibacterial peptide and host-cell modulator. Biochim. Biophys. Acta (BBA)-Biomembr. 2016, 1858, 546–566. [Google Scholar] [CrossRef] [PubMed]
- Steiner, H.; Hultmark, D.; Engström, A.; Bennich, H.; Boman, H.G. Sequence and specificity of two antibacterial proteins involved in insect immunity. Nature 1981, 292, 246–248. [Google Scholar] [CrossRef]
- Habermann, E. Bee and Wasp Venoms: The biochemistry and pharmacology of their peptides and enzymes are reviewed. Science 1972, 177, 314–322. [Google Scholar] [CrossRef]
- Shai, Y. Mechanism of the binding, insertion and destabilization of phospholipid bilayer membranes by α-helical antimicrobial and cell non-selective membrane-lytic peptides. Biochim. Biophys. Acta (BBA)-Biomembr. 1999, 1462, 55–70. [Google Scholar] [CrossRef]
- Pino-Angeles, A.; Leveritt, J.M.; Lazaridis, T. Pore Structure and Synergy in Antimicrobial Peptides of the Magainin Family. PLoS Comput. Biol. 2016, 12, e1004570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glaser, R.W.; Sachse, C.; Dürr, U.H.; Wadhwani, P.; Afonin, S.; Strandberg, E.; Ulrich, A.S. Concentration-Dependent Realignment of the Antimicrobial Peptide PGLa in Lipid Membranes Observed by Solid-State 19F-NMR. Biophys. J. 2005, 88, 3392–3397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, I.A.; Elliott, A.G.; Kavanagh, A.M.; Zuegg, J.; Blaskovich, M.A.T.; Cooper, M.A. Contribution of Amphipathicity and Hydrophobicity to the Antimicrobial Activity and Cytotoxicity of β-Hairpin Peptides. ACS Infect. Dis. 2016, 2, 442–450. [Google Scholar] [CrossRef]
- Panteleev, P.V.; Bolosov, I.A.; Balandin, S.V.; Ovchinnikova, T.V. Structure and Biological Functions of β-Hairpin Antimicrobial Peptides. Acta Nat. 2015, 7, 37–47. [Google Scholar] [CrossRef]
- Conibear, A.C.; Craik, D.J. The Chemistry and Biology of Theta Defensins. Angew. Chem. Int. Ed. 2014, 53, 10612–10623. [Google Scholar] [CrossRef]
- Tang, Y.Q.; Yuan, J.; Tran, D.; Miller, C.J.; Ouellette, A.J.; Selsted, M.E. A Cyclic Antimicrobial Peptide Produced in Primate Leukocytes by the Ligation of Two Truncated α-Defensins. Science 1999, 286, 498–502. [Google Scholar] [CrossRef] [Green Version]
- Conibear, A.C.; Bochen, A.; Rosengren, K.J.; Stupar, P.; Wang, C.; Kessler, H.; Craik, D.J. The Cyclic Cystine Ladder of Theta-Defensins as a Stable, Bifunctional Scaffold: A Proof-of-Concept Study Using the Integrin-Binding RGD Motif. ChemBioChem 2014, 15, 451–459. [Google Scholar] [CrossRef]
- Falanga, A.; Nigro, E.; De Biasi, M.; Daniele, A.; Morelli, G.; Galdiero, S.; Scudiero, O. Cyclic Peptides as Novel Therapeutic Microbicides: Engineering of Human Defensin Mimetics. Molecules 2017, 22, 1217. [Google Scholar] [CrossRef] [Green Version]
- Dhople, V.; Krukemeyer, A.; Ramamoorthy, A. The human beta-defensin-3, an antibacterial peptide with multiple biological functions. Biochim. Biophys. Acta (BBA)-Biomembr. 2006, 1758, 1499–1512. [Google Scholar] [CrossRef] [Green Version]
- Cornet, B.; Bonmatin, J.M.; Hetru, C.; Hoffmann, J.A.; Ptak, M.; Vovelle, F. Refined three-dimensional solution structure of insect defensin A. Structure 1995, 3, 435–448. [Google Scholar] [CrossRef]
- Dias, R.d.O.; Franco, O.L. Cysteine-stabilized αβ defensins: From a common fold to antibacterial activity. Peptides 2015, 72, 64–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, D.I.; Prenner, E.J.; Vogel, H.J. Tryptophan- and arginine-rich antimicrobial peptides: Structures and mechanisms of action. Biochim. Biophys. Acta (BBA)-Biomembr. 2006, 1758, 1184–1202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rozek, A.; Friedrich, C.L.; Hancock, R.E.W. Structure of the Bovine Antimicrobial Peptide Indolicidin Bound to Dodecylphosphocholine and Sodium Dodecyl Sulfate Micelles. Biochemistry 2000, 39, 15765–15774. [Google Scholar] [CrossRef]
- Hsu, C.H. Structural and DNA-binding studies on the bovine antimicrobial peptide, indolicidin: Evidence for multiple conformations involved in binding to membranes and DNA. Nucleic Acids Res. 2005, 33, 4053–4064. [Google Scholar] [CrossRef] [Green Version]
- Brogden, K.A. Antimicrobial peptides: Pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 2005, 3, 238–250. [Google Scholar] [CrossRef]
- Liang, W.; Diana, J. The Dual Role of Antimicrobial Peptides in Autoimmunity. Front. Immunol. 2020, 11, 2077. [Google Scholar] [CrossRef]
- Nayab, S.; Aslam, M.A.; Rahman, S.U.; Sindhu, Z.U.D.; Sajid, S.; Zafar, N.; Razaq, M.; Kanwar, R.; Amanullah. A Review of Antimicrobial Peptides: Its Function, Mode of Action and Therapeutic Potential. Int. J. Pept. Res. Ther. 2022, 28, 46. [Google Scholar] [CrossRef]
- Herrell, W.E.; Heilman, D. Experimental and Clinical Studies on Gramicidin 1. J. Clin. Investig. 1941, 20, 583–591. [Google Scholar] [CrossRef] [Green Version]
- Rammelkamp, C.H.; Weinstein, L. Toxic Effects of Tyrothricin, Gramicidin and Tyrocidine. J. Infect. Dis. 1942, 71, 166–173. [Google Scholar] [CrossRef]
- Gharsallaoui, A.; Oulahal, N.; Joly, C.; Degraeve, P. Nisin as a Food Preservative: Part 1: Physicochemical Properties, Antimicrobial Activity, and Main Uses. Crit. Rev. Food Sci. Nutr. 2016, 56, 1262–1274. [Google Scholar] [CrossRef]
- Moretta, A.; Scieuzo, C.; Petrone, A.M.; Salvia, R.; Manniello, M.D.; Franco, A.; Lucchetti, D.; Vassallo, A.; Vogel, H.; Sgambato, A.; et al. Antimicrobial Peptides: A New Hope in Biomedical and Pharmaceutical Fields. Front. Cell. Infect. Microbiol. 2021, 11, 668632. [Google Scholar] [CrossRef] [PubMed]
- Keymanesh, K.; Soltani, S.; Sardari, S. Application of antimicrobial peptides in agriculture and food industry. World J. Microbiol. Biotechnol. 2009, 25, 933–944. [Google Scholar] [CrossRef]
- Peng, Z.; Wang, A.; Xie, L.; Song, W.; Wang, J.; Yin, Z.; Zhou, D.; Li, F. Use of recombinant porcine β-defensin 2 as a medicated feed additive for weaned piglets. Sci. Rep. 2016, 6, 26790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Yang, X.; Wang, A.; Huang, C.; Tang, H.; Zhang, Q.; Fang, Q.; Yu, Z.; Liu, X.; Huang, Q.; et al. Pigs Overexpressing Porcine β-Defensin 2 Display Increased Resilience to Glaesserella parasuis Infection. Antibiotics 2020, 9, 903. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Sameen, D.E.; Ahmed, S.; Dai, J.; Qin, W. Antimicrobial peptides and their application in food packaging. Trends Food Sci. Technol. 2021, 112, 471–483. [Google Scholar] [CrossRef]
- Gruenheid, S.; Le Moual, H. Resistance to antimicrobial peptides in Gram-negative bacteria. FEMS Microbiol. Lett. 2012, 330, 81–89. [Google Scholar] [CrossRef] [Green Version]
- Assoni, L.; Milani, B.; Carvalho, M.R.; Nepomuceno, L.N.; Waz, N.T.; Guerra, M.E.S.; Converso, T.R.; Darrieux, M. Resistance Mechanisms to Antimicrobial Peptides in Gram-Positive Bacteria. Front. Microbiol. 2020, 11, 2362. [Google Scholar] [CrossRef]
- Chen, W.; Ding, H.; Feng, P.; Lin, H.; Chou, K.C. iACP: A sequence-based tool for identifying anticancer peptides. Oncotarget 2016, 7, 16895–16909. [Google Scholar] [CrossRef] [Green Version]
- Manavalan, B.; Shin, T.H.; Kim, M.O.; Lee, G. AIPpred: Sequence-Based Prediction of Anti-inflammatory Peptides Using Random Forest. Front. Pharmacol. 2018, 9, 276. [Google Scholar] [CrossRef] [Green Version]
- Wei, L.; Zhou, C.; Chen, H.; Song, J.; Su, R. ACPred-FL: A sequence-based predictor using effective feature representation to improve the prediction of anti-cancer peptides. Bioinformatics 2018, 34, 4007–4016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torrent, M.; Andreu, D.; Nogués, V.M.; Boix, E. Connecting Peptide Physicochemical and Antimicrobial Properties by a Rational Prediction Model. PLoS ONE 2011, 6, e16968. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; Shrivastava, S.; Kumar Singh, S.; Kumar, A.; Saxena, S. StaBle-ABPpred: A stacked ensemble predictor based on biLSTM and attention mechanism for accelerated discovery of antibacterial peptides. Briefings Bioinform. 2022, 23, bbab439. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Pu, Y.; Tang, J.; Zou, Q.; Guo, F. DeepAVP: A Dual-Channel Deep Neural Network for Identifying Variable-Length Antiviral Peptides. IEEE J. Biomed. Health Inform. 2020, 24, 3012–3019. [Google Scholar] [CrossRef] [PubMed]
- Chou, K.C. Prediction of protein cellular attributes using pseudo-amino acid composition. Proteins Struct. Funct. Bioinform. 2001, 43, 246–255. [Google Scholar] [CrossRef]
- Weathers, E.A.; Paulaitis, M.E.; Woolf, T.B.; Hoh, J.H. Reduced amino acid alphabet is sufficient to accurately recognize intrinsically disordered protein. FEBS Lett. 2004, 576, 348–352. [Google Scholar] [CrossRef] [Green Version]
- Müller, A.T.; Hiss, J.A.; Schneider, G. Recurrent Neural Network Model for Constructive Peptide Design. J. Chem. Inf. Model. 2018, 58, 472–479. [Google Scholar] [CrossRef]
- Van Oort, C.M.; Ferrell, J.B.; Remington, J.M.; Wshah, S.; Li, J. AMPGAN v2: Machine Learning-Guided Design of Antimicrobial Peptides. J. Chem. Inf. Model. 2021, 61, 2198–2207. [Google Scholar] [CrossRef]
- Tyagi, A.; Kapoor, P.; Kumar, R.; Chaudhary, K.; Gautam, A.; Raghava, G.P.S. In silico models for designing and discovering novel anticancer peptides. Sci. Rep. 2013, 3, 2984. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Cheong, H.H.; Siu, S.W.I. xDeep-AcPEP: Deep Learning Method for Anticancer Peptide Activity Prediction Based on Convolutional Neural Network and Multitask Learning. J. Chem. Inf. Model. 2021, 61, 3789–3803. [Google Scholar] [CrossRef]
- Chang, K.Y.; Lin, T.P.; Shih, L.Y.; Wang, C.K. Analysis and prediction of the critical regions of antimicrobial peptides based on conditional random fields. PLoS ONE 2015, 10, e0119490. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ding, Y.; Wen, H.; Lin, Y.; Hu, Y.; Zhang, Y.; Xia, Q.; Lin, Z. QSAR modeling and design of cationic antimicrobial peptides based on structural properties of amino acids. Comb. Chem. High Throughput Screen. 2012, 15, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Sander, O.; Sing, T.; Sommer, I.; Low, A.J.; Cheung, P.K.; Harrigan, P.R.; Lengauer, T.; Domingues, F.S. Structural descriptors of gp120 V3 loop for the prediction of HIV-1 coreceptor usage. PLoS Comput. Biol. 2007, 3, e58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minkiewicz, P.; Iwaniak, A.; Darewicz, M. Annotation of Peptide Structures Using SMILES and Other Chemical Codes–Practical Solutions. Mol. J. Synth. Chem. Nat. Prod. Chem. 2017, 22, 2075. [Google Scholar] [CrossRef] [Green Version]
- Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Model. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- Tyagi, A.; Tuknait, A.; Anand, P.; Gupta, S.; Sharma, M.; Mathur, D.; Joshi, A.; Singh, S.; Gautam, A.; Raghava, G.P. CancerPPD: A database of anticancer peptides and proteins. Nucleic Acids Res. 2015, 43, D837–D843. [Google Scholar] [CrossRef] [Green Version]
- Spänig, S.; Mohsen, S.; Hattab, G.; Hauschild, A.C.; Heider, D. A large-scale comparative study on peptide encodings for biomedical classification. NAR Genom. Bioinform. 2021, 3, lqab039. [Google Scholar] [CrossRef]
- Spänig, S.; Heider, D. Encodings and models for antimicrobial peptide classification for multi-resistant pathogens. BioData Min. 2019, 12, 7. [Google Scholar] [CrossRef] [Green Version]
- Veltri, D.; Kamath, U.; Shehu, A. Deep learning improves antimicrobial peptide recognition. Bioinformatics 2018, 34, 2740–2747. [Google Scholar] [CrossRef] [Green Version]
- Mikolov, T.; Chen, K.; Corrado, G.; Dean, J. Efficient Estimation of Word Representations in Vector Space. In Proceedings of the 1st International Conference on Learning Representations, ICLR 2013, Workshop Track Proceedings, Scottsdale, AZ, USA, 2–4 May 2013; Bengio, Y., LeCun, Y., Eds.; Academic Press: Cambridge, MA, USA, 2013. [Google Scholar]
- Sharma, R.; Shrivastava, S.; Kumar Singh, S.; Kumar, A.; Saxena, S.; Kumar Singh, R. Deep-ABPpred: Identifying antibacterial peptides in protein sequences using bidirectional LSTM with word2vec. Briefings Bioinform. 2021, 22, bbab065. [Google Scholar] [CrossRef]
- Liu, Q.; Kusner, M.J.; Blunsom, P. A Survey on Contextual Embeddings. arXiv 2020, arXiv:2003.07278. [Google Scholar]
- Vaswani, A.; Shazeer, N.; Parmar, N.; Uszkoreit, J.; Jones, L.; Gomez, A.N.; Kaiser, L.; Polosukhin, I. Attention is all you need. In Proceedings of the 31st International Conference on Neural Information Processing Systems (NIPS 2017); Curran Associates Inc.: Red Hook, NY, USA, 2017; pp. 6000–6010. [Google Scholar]
- Ofer, D.; Brandes, N.; Linial, M. The language of proteins: NLP, machine learning & protein sequences. Comput. Struct. Biotechnol. J. 2021, 19, 1750–1758. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lin, J.; Zhao, L.; Zeng, X.; Liu, X. A novel antibacterial peptide recognition algorithm based on BERT. Briefings Bioinform. 2021, 22, bbab200. [Google Scholar] [CrossRef] [PubMed]
- Boutet, E.; Lieberherr, D.; Tognolli, M.; Schneider, M.; Bairoch, A. UniProtKB/Swiss-Prot. Methods Mol. Biol. 2007, 406, 89–112. [Google Scholar]
- Devlin, J.; Chang, M.W.; Lee, K.; Toutanova, K. BERT: Pre-training of Deep Bidirectional Transformers for Language Understanding. In Human Language Technologies, Volume 1 (Long and Short Papers), Proceedings of the 2019 Conference of the North American Chapter of the Association for Computational Linguistics, Minneapolis, MN, USA, 2–7 June 2019; Association for Computational Linguistics: Minneapolis, MN, USA, 2019; pp. 4171–4186. [Google Scholar] [CrossRef]
- Dee, W. LMPred: Predicting antimicrobial peptides using pre-trained language models and deep learning. Bioinform. Adv. 2022, 2, vbac021. [Google Scholar] [CrossRef]
- Raffel, C.; Shazeer, N.; Roberts, A.; Lee, K.; Narang, S.; Matena, M.; Zhou, Y.; Li, W.; Liu, P.J. Exploring the Limits of Transfer Learning with a Unified Text-to-Text Transformer. arXiv 2019, arXiv:1910.10683. [Google Scholar]
- Yang, Z.; Dai, Z.; Yang, Y.; Carbonell, J.G.; Salakhutdinov, R.; Le, Q.V. XLNet: Generalized Autoregressive Pretraining for Language Understanding. arXiv 2019, arXiv:1906.08237. [Google Scholar]
- Porto, W.F.; Pires, A.S.; Franco, O.L. CS-AMPPred: An Updated SVM Model for Antimicrobial Activity Prediction in Cysteine-Stabilized Peptides. PLoS ONE 2012, 7, e51444. [Google Scholar] [CrossRef]
- Porto, W.F.; Fernandes, F.C.; Franco, O.L. An SVM Model Based on Physicochemical Properties to Predict Antimicrobial Activity from Protein Sequences with Cysteine Knot Motifs; Springer: Berlin/Heidelberg, Germany, 2010; pp. 59–62. [Google Scholar]
- Meher, P.K.; Sahu, T.K.; Saini, V.; Rao, A.R. Predicting antimicrobial peptides with improved accuracy by incorporating the compositional, physico-chemical and structural features into Chou’s general PseAAC. Sci. Rep. 2017, 7, 42362. [Google Scholar] [CrossRef] [Green Version]
- Kavousi, K.; Bagheri, M.; Behrouzi, S.; Vafadar, S.; Atanaki, F.F.; Lotfabadi, B.T.; Ariaeenejad, S.; Shockravi, A.; Moosavi-Movahedi, A.A. IAMPE: NMR-Assisted Computational Prediction of Antimicrobial Peptides. J. Chem. Inf. Model. 2020, 60, 4691–4701. [Google Scholar] [CrossRef]
- Xiao, X.; Shao, Y.T.; Cheng, X.; Stamatovic, B. iAMP-CA2L: A new CNN-BiLSTM-SVM classifier based on cellular automata image for identifying antimicrobial peptides and their functional types. Briefings Bioinform. 2021, 22, bbab209. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.; Karnik, S.; Barai, R.S.; Jayaraman, V.K.; Idicula-Thomas, S. CAMP: A useful resource for research on antimicrobial peptides. Nucleic Acids Res. 2010, 38, D774–D780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhadra, P.; Yan, J.; Li, J.; Fong, S.; Siu, S.W.I. AmPEP: Sequence-based prediction of antimicrobial peptides using distribution patterns of amino acid properties and random forest. Sci. Rep. 2018, 8, 1697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, C.R.; Jhong, J.H.; Wang, Z.; Chen, S.; Wan, Y.; Horng, J.T.; Lee, T.Y. Characterization and identification of natural antimicrobial peptides on different organisms. Int. J. Mol. Sci. 2020, 21, 986. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Li, F.; Leier, A.; Xiang, D.; Shen, H.H.; Marquez Lago, T.T.; Li, J.; Yu, D.J.; Song, J. Comprehensive assessment of machine learning-based methods for predicting antimicrobial peptides. Briefings Bioinform. 2021, 22, bbab083. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, V.; Tripathi, P. Detecting antimicrobial peptides by exploring the mutual information of their sequences. J. Biomol. Struct. Dyn. 2020, 38, 5037–5043. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Shrivastava, S.; Kumar Singh, S.; Kumar, A.; Saxena, S.; Kumar Singh, R. AniAMPpred: Artificial intelligence guided discovery of novel antimicrobial peptides in animal kingdom. Briefings Bioinform. 2021, 22, bbab242. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Wang, P.; Lin, W.Z.; Jia, J.H.; Chou, K.C. iAMP-2L: A two-level multi-label classifier for identifying antimicrobial peptides and their functional types. Anal. Biochem. 2013, 436, 168–177. [Google Scholar] [CrossRef]
- Lv, H.; Yan, K.; Guo, Y.; Zou, Q.; Hesham, A.E.L.; Liu, B. AMPpred-EL: An effective antimicrobial peptide prediction model based on ensemble learning. Comput. Biol. Med. 2022, 146, 105577. [Google Scholar] [CrossRef]
- Lertampaiporn, S.; Vorapreeda, T.; Hongsthong, A.; Thammarongtham, C. Ensemble-AMPPred: Robust AMP prediction and recognition using the ensemble learning method with a new hybrid feature for differentiating AMPs. Genes 2021, 12, 137. [Google Scholar] [CrossRef]
- Zarayeneh, N.; Hanifeloo, Z. Antimicrobial peptide prediction using ensemble learning algorithm. arXiv 2020, arXiv:2005.01714. [Google Scholar]
- Caprani, M.C.; Healy, J.; Slattery, O.; O’Keeffe, J. Using an ensemble to identify and classify macroalgae antimicrobial peptides. Interdiscip. Sci. Comput. Life Sci. 2021, 13, 321–333. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Akbar, S.; Tahir, M.; Hayat, M.; Ali, F. iAFPs-EnC-GA: Identifying antifungal peptides using sequential and evolutionary descriptors based multi-information fusion and ensemble learning approach. Chemom. Intell. Lab. Syst. 2022, 222, 104516. [Google Scholar]
- Gunn, S.R. Support vector machines for classification and regression. ISIS Tech. Rep. 1998, 14, 5–16. [Google Scholar]
- Kulkarni, A.; Jayaraman, V.K.; Kulkarni, B.D. Support vector classification with parameter tuning assisted by agent-based technique. Comput. Chem. Eng. 2004, 28, 311–318. [Google Scholar] [CrossRef]
- Ho, T.K. The random subspace method for constructing decision forests. IEEE Trans. Pattern Anal. Mach. Intell. 1998, 20, 832–844. [Google Scholar] [CrossRef] [Green Version]
- Altman, N.S. An Introduction to Kernel and Nearest-Neighbor Nonparametric Regression. Am. Stat. 1992, 46, 175–185. [Google Scholar] [CrossRef]
- Li, Y.; Huang, C.; Ding, L.; Li, Z.; Pan, Y.; Gao, X. Deep Learning in Bioinformatics: Introduction, Application, and Perspective in Big Data Era. Methods 2019, 166, 4–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fjell, C.D.; Jenssen, H.V.; Hilpert, K.; Cheung, W.A.; Panté, N.; Hancock, R.E.W.; Cherkasov, A. Identification of Novel Antibacterial Peptides by Chemoinformatics and Machine Learning. J. Med. Chem. 2009, 52, 2006–2015. [Google Scholar] [CrossRef]
- Yan, J.; Bhadra, P.; Li, A.; Sethiya, P.; Qin, L.; Tai, H.K.; Wong, K.H.; Siu, S.W.I. Deep-AmPEP30: Improve Short Antimicrobial Peptides Prediction with Deep Learning. Mol. Ther.-Nucleic Acids 2020, 20, 882–894. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Guo, Z.; Xia, B.; Zhang, Y.; Liu, X.; Yu, Y.; Tang, N.; Tong, X.; Wang, M.; Ye, X.; et al. Identification of antimicrobial peptides from the human gut microbiome using deep learning. Nat. Biotechnol. 2022, 40, 921–931. [Google Scholar] [CrossRef]
- Li, C.; Sutherland, D.; Hammond, S.A.; Yang, C.; Taho, F.; Bergman, L.; Houston, S.; Warren, R.L.; Wong, T.; Hoang, L.M.N.; et al. AMPlify: Attentive deep learning model for discovery of novel antimicrobial peptides effective against WHO priority pathogens. BMC Genom. 2022, 23, 77. [Google Scholar] [CrossRef] [PubMed]
- Ruiz Puentes, P.; Henao, M.C.; Cifuentes, J.; Muñoz Camargo, C.; Reyes, L.H.; Cruz, J.C.; Arbeláez, P. Rational discovery of antimicrobial peptides by means of artificial intelligence. Membranes 2022, 12, 708. [Google Scholar] [CrossRef]
- Lin, T.T.; Yang, L.Y.; Lu, I.H.; Cheng, W.C.; Hsu, Z.R.; Chen, S.H.; Lin, C.Y. AI4AMP: An Antimicrobial Peptide Predictor Using Physicochemical Property-Based Encoding Method and Deep Learning. mSystems 2021, 6, e00299-21. [Google Scholar] [CrossRef] [PubMed]
- García-Jacas, C.R.; Pinacho-Castellanos, S.A.; García-González, L.A.; Brizuela, C.A. Do deep learning models make a difference in the identification of antimicrobial peptides? Briefings Bioinform. 2022, 23, bbac094. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Akbar, S.; Khan, S.; Hayat, M.; Ali, F.; Ahmed, A.; Tahir, M. Deep-AntiFP: Prediction of antifungal peptides using distanct multi-informative features incorporating with deep neural networks. Chemom. Intell. Lab. Syst. 2021, 208, 104214. [Google Scholar] [CrossRef]
- Timmons, P.B.; Hewage, C.M. ENNAACT is a novel tool which employs neural networks for anticancer activity classification for therapeutic peptides. Biomed. Pharmacother. 2021, 133, 111051. [Google Scholar] [CrossRef]
- Müller, A.T.; Gabernet, G.; Hiss, J.A.; Schneider, G. modlAMP: Python for antimicrobial peptides. Bioinformatics 2017, 33, 2753–2755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timmons, P.B.; Hewage, C.M. ENNAVIA is a novel method which employs neural networks for antiviral and anti-coronavirus activity prediction for therapeutic peptides. Briefings Bioinform. 2021, 22, bbab258. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, S.; Kanehisa, M. AAindex: Amino acid index database. Nucleic Acids Res. 2000, 28, 374. [Google Scholar] [CrossRef] [PubMed]
- Nath, A.; Karthikeyan, S. Enhanced prediction and characterization of CDK inhibitors using optimal class distribution. Interdiscip. Sci. Comput. Life Sci. 2017, 9, 292–303. [Google Scholar] [CrossRef]
- Akbar, S.; Rahman, A.U.; Hayat, M.; Sohail, M. cACP: Classifying anticancer peptides using discriminative intelligent model via Chou’s 5-step rules and general pseudo components. Chemom. Intell. Lab. Syst. 2020, 196, 103912. [Google Scholar] [CrossRef]
- Etchebest, C.; Benros, C.; Bornot, A.; Camproux, A.C.; De Brevern, A. A reduced amino acid alphabet for understanding and designing protein adaptation to mutation. Eur. Biophys. J. 2007, 36, 1059–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, J.; Wang, Z.; Kuen, J.; Ma, L.; Shahroudy, A.; Shuai, B.; Liu, T.; Wang, X.; Wang, G.; Cai, J.; et al. Recent advances in convolutional neural networks. Pattern Recognit. 2018, 77, 354–377. [Google Scholar] [CrossRef] [Green Version]
- Albawi, S.; Mohammed, T.A.; Al-Zawi, S. Understanding of a convolutional neural network. In Proceedings of the 2017 International Conference on Engineering and Technology (ICET), Antalya, Turkey, 21–23 August 2017; pp. 1–6. [Google Scholar] [CrossRef]
- O’Shea, K.; Nash, R. An Introduction to Convolutional Neural Networks. arXiv 2015, arXiv:1511.08458. [Google Scholar]
- Young, T.; Hazarika, D.; Poria, S.; Cambria, E. Recent trends in deep learning based natural language processing. IEEE Comput. Intell. Mag. 2018, 13, 55–75. [Google Scholar] [CrossRef]
- Dua, M.; Barbara, D.; Shehu, A. Exploring Deep Neural Network Architectures: A Case Study on Improving Antimicrobial Peptide Recognition. In Proceedings of the 12th International Conference on Bioinformatics and Computational Biology, San Francisco, CA, USA, 23–25 March 2020; pp. 171–182. [Google Scholar] [CrossRef]
- Su, X.; Xu, J.; Yin, Y.; Quan, X.; Zhang, H. Antimicrobial peptide identification using multi-scale convolutional network. BMC Bioinform. 2019, 20, 730. [Google Scholar] [CrossRef] [Green Version]
- Cao, R.; Wang, M.; Bin, Y.; Zheng, C. DLFF-ACP: Prediction of ACPs based on deep learning and multi-view features fusion. PeerJ 2021, 9, e11906. [Google Scholar] [CrossRef]
- Sun, Y.Y.; Lin, T.T.; Cheng, W.C.; Lu, I.H.; Lin, C.Y.; Chen, S.H. Peptide-Based Drug Predictions for Cancer Therapy Using Deep Learning. Pharmaceuticals 2022, 15, 422. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Shrivastava, S.; Singh, S.K.; Kumar, A.; Singh, A.K.; Saxena, S. Deep-AVPpred: Artificial intelligence driven discovery of peptide drugs for viral infections. IEEE J. Biomed. Health Inform. 2021, 26, 5067–5074. [Google Scholar] [CrossRef] [PubMed]
- Rives, A.; Meier, J.; Sercu, T.; Goyal, S.; Lin, Z.; Liu, J.; Guo, D.; Ott, M.; Zitnick, C.L.; Ma, J.; et al. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. Proc. Natl. Acad. Sci. USA 2021, 118, e2016239118. [Google Scholar] [CrossRef]
- Grønning, A.G.B.; Kacprowski, T.; Schéele, C. MultiPep: A hierarchical deep learning approach for multi-label classification of peptide bioactivities. Biol. Methods Protoc. 2021, 6, bpab021. [Google Scholar] [CrossRef]
- Li, S.; Li, W.; Cook, C.; Zhu, C.; Gao, Y. Independently recurrent neural network (indrnn): Building a longer and deeper rnn. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition, San Juan, PR, USA, 17–19 June 2018; pp. 5457–5466. [Google Scholar]
- Yu, Y.; Si, X.; Hu, C.; Zhang, J. A Review of Recurrent Neural Networks: LSTM Cells and Network Architectures. Neural Comput. 2019, 31, 1235–1270. [Google Scholar] [CrossRef]
- Yi, H.C.; You, Z.H.; Zhou, X.; Cheng, L.; Li, X.; Jiang, T.H.; Chen, Z.H. ACP-DL: A Deep Learning Long Short-Term Memory Model to Predict Anticancer Peptides Using High-Efficiency Feature Representation. Mol. Ther.-Nucleic Acids 2019, 17, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Youmans, M.; Spainhour, J.C.G.; Qiu, P. Classification of Antibacterial Peptides using Long Short-Term Memory Recurrent Neural Networks. IEEE/ACM Trans. Comput. Biol. Bioinform. 2019, 17, 1134–1140. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Jing, R.; Liu, F.; Luo, J.; Li, Y. DeepACP: A Novel Computational Approach for Accurate Identification of Anticancer Peptides by Deep Learning Algorithm. Mol. Ther.-Nucleic Acids 2020, 22, 862–870. [Google Scholar] [CrossRef] [PubMed]
- Hamid, M.N.; Friedberg, I. Identifying antimicrobial peptides using word embedding with deep recurrent neural networks. Bioinformatics 2019, 35, 2009–2016. [Google Scholar] [CrossRef] [Green Version]
- Maier, A.; Köstler, H.; Heisig, M.; Krauss, P.; Yang, S.H. Known operator learning and hybrid machine learning in medical imaging—A review of the past, the present, and the future. Prog. Biomed. Eng. 2022, 4, 022002. [Google Scholar] [CrossRef]
- Fu, H.; Cao, Z.; Li, M.; Wang, S. ACEP: Improving antimicrobial peptides recognition through automatic feature fusion and amino acid embedding. BMC Genom. 2020, 21, 597. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Moriwaki, Y.; Li, C.; Shimizu, K. Prediction of Antifungal Peptides by Deep Learning with Character Embedding. IPSJ Trans. Bioinform. 2019, 12, 21–29. [Google Scholar] [CrossRef] [Green Version]
- Sharma, R.; Shrivastava, S.; Kumar Singh, S.; Kumar, A.; Saxena, S.; Kumar Singh, R. Deep-AFPpred: Identifying novel antifungal peptides using pretrained embeddings from seq2vec with 1DCNN-BiLSTM. Briefings Bioinform. 2022, 23, bbab422. [Google Scholar] [CrossRef] [PubMed]
- Heinzinger, M.; Elnaggar, A.; Wang, Y.; Dallago, C.; Nechaev, D.; Matthes, F.; Rost, B. Modeling aspects of the language of life through transfer-learning protein sequences. BMC Bioinform. 2019, 20, 723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, L.; Lambert, P.; Badia, T. Attention and lexicon regularized LSTM for aspect-based sentiment analysis. In Proceedings of the 57th Annual Meeting of the Association for Computational Linguistics: Student Research Workshop, Florence, Italy, 28 July–2 August 2019; pp. 253–259. [Google Scholar]
- Yang, Z.; Yang, D.; Dyer, C.; He, X.; Smola, A.; Hovy, E. Hierarchical attention networks for document classification. In Proceedings of the 2016 Conference of the North American Chapter of the Association for Computational Linguistics: Human Language Technologies, San Diego, CA, USA, 12–17 June 2016; pp. 1480–1489. [Google Scholar]
- Tossi, A.; Sandri, L.; Giangaspero, A. Amphipathic, α-helical antimicrobial peptides. Pept. Sci. 2000, 55, 4–30. [Google Scholar] [CrossRef]
- Hu, Y.; Wang, Z.; Hu, H.; Wan, F.; Chen, L.; Xiong, Y.; Wang, X.; Zhao, D.; Huang, W.; Zeng, J. ACME: Pan-specific peptide–MHC class I binding prediction through attention-based deep neural networks. Bioinformatics 2019, 35, 4946–4954. [Google Scholar] [CrossRef]
- Xiao, X.; Wang, P.; Chou, K.C. Cellular automata and its applications in protein bioinformatics. Curr. Protein Pept. Sci. 2011, 12, 508–519. [Google Scholar] [CrossRef]
- Hussain, W. sAMP-PFPDeep: Improving accuracy of short antimicrobial peptides prediction using three different sequence encodings and deep neural networks. Briefings Bioinform. 2022, 23, bbab487. [Google Scholar] [CrossRef] [PubMed]
- Simonyan, K.; Zisserman, A. Very Deep Convolutional Networks for Large-Scale Image Recognition. In Proceedings of the 3rd International Conference on Learning Representations, ICLR 2015, Conference Track Proceedings, San Diego, CA, USA, 7–9 May 2015; Bengio, Y., LeCun, Y., Eds.; Academic Press: Cambridge, MA, USA, 2015. [Google Scholar]
- He, K.; Zhang, X.; Ren, S.; Sun, J. Deep residual learning for image recognition. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition, Las Vegas, NV, USA, 27–30 June 2016; pp. 770–778. [Google Scholar]
- Salem, M.; Arshadi, A.K.; Yuan, J.S. AMPDeep: Hemolytic Activity Prediction of Antimicrobial Peptides using Transfer Learning. BMC Bioinform. 2022, 23, 389. [Google Scholar] [CrossRef] [PubMed]
- Elnaggar, A.; Heinzinger, M.; Dallago, C.; Rihawi, G.; Wang, Y.; Jones, L.; Gibbs, T.; Feher, T.; Angerer, C.; Steinegger, M.; et al. ProtTrans: Towards cracking the language of Life’s code through self-supervised deep learning and high performance computing. arXiv 2020, arXiv:2007.06225. [Google Scholar] [CrossRef]
- Witten, J.; Witten, Z. Deep learning regression model for antimicrobial peptide design. bioRxiv 2019, 692681. [Google Scholar] [CrossRef] [Green Version]
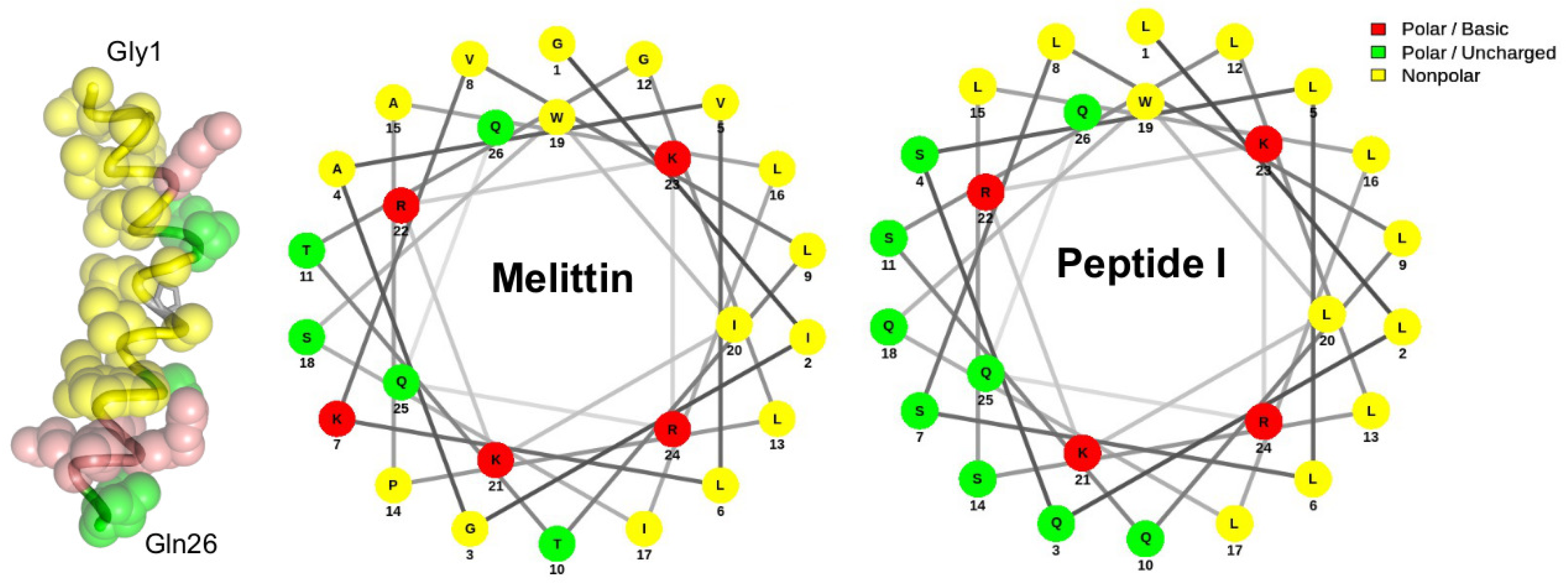
- DeGrado, W.F.; Kezdy, F.J.; Kaiser, E.T. Design, synthesis, and characterization of a cytotoxic peptide with melittin-like activity. J. Am. Chem. Soc. 1981, 103, 679–681. [Google Scholar] [CrossRef]
- Mól, A.; Castro, M.; Fontes, W. NetWheels: A web application to create high quality peptide helical wheel and net projections. bioRxiv 2018, 416347. [Google Scholar] [CrossRef] [Green Version]
- Karami, Y.; Khakzad, H.; Shirazi, H.; Arab, S. Protein structure prediction using bio-inspired algorithm: A review. In Proceedings of the 16th CSI International Symposium on Artificial Intelligence and Signal Processing (AISP 2012), Shiraz, Fars, Iran, 2–3 May 2012; pp. 201–206. [Google Scholar]
- Korichi, M.; Gerbaud, V.; Talou, T.; Floquet, P.; Meniai, A.H.; Nacef, S. Computer-aided aroma design. II. Quantitative structure–odour relationship. Chem. Eng. Process. Process Intensif. 2008, 47, 1912–1925. [Google Scholar] [CrossRef] [Green Version]
- Maccari, G.; Di Luca, M.; Nifosí, R.; Cardarelli, F.; Signore, G.; Boccardi, C.; Bifone, A. Antimicrobial Peptides Design by Evolutionary Multiobjective Optimization. PLoS Comput. Biol. 2013, 9, e1003212. [Google Scholar] [CrossRef] [Green Version]
- Dathe, M.; Nikolenko, H.; Meyer, J.; Beyermann, M.; Bienert, M. Optimization of the antimicrobial activity of magainin peptides by modification of charge. FEBS Lett. 2001, 501, 146–150. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, M.; Hinkley, T.; Tsuda, S.; Abul-Haija, Y.M.; McBurney, R.T.; Kulikov, V.; Mathieson, J.S.; Galiñanes Reyes, S.; Castro, M.D.; Cronin, L. Using Evolutionary Algorithms and Machine Learning to Explore Sequence Space for the Discovery of Antimicrobial Peptides. Chem 2018, 4, 533–543. [Google Scholar] [CrossRef] [Green Version]
- Boone, K.; Wisdom, C.; Camarda, K.; Spencer, P.; Tamerler, C. Combining genetic algorithm with machine learning strategies for designing potent antimicrobial peptides. BMC Bioinform. 2021, 22, 239. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, Z. Rough Set Theory and Its Applications to Data Analysis. Cybern. Syst. 1998, 29, 661–688. [Google Scholar] [CrossRef]
- Blondelle, S.E.; Houghten, R.A. Design of model amphipathic peptides having potent antimicrobial activities. Biochemistry 1992, 31, 12688–12694. [Google Scholar] [CrossRef] [PubMed]
- Chowdhary, K.R. Natural Language Processing. In Fundamentals of Artificial Intelligence; Springer: New Delhi, India, 2020; pp. 603–649. [Google Scholar] [CrossRef]
- Singh, S.P.; Kumar, A.; Darbari, H.; Singh, L.; Rastogi, A.; Jain, S. Machine translation using deep learning: An overview. In Proceedings of the 2017 International Conference on Computer, Communications and Electronics (Comptelix), Jaipur, India, 1–2 July 2017; pp. 162–167. [Google Scholar]
- Popel, M.; Tomkova, M.; Tomek, J.; Kaiser, L.; Uszkoreit, J.; Bojar, O.; Žabokrtskỳ, Z. Transforming machine translation: A deep learning system reaches news translation quality comparable to human professionals. Nat. Commun. 2020, 11, 4381. [Google Scholar] [CrossRef] [PubMed]
- Mutabazi, E.; Ni, J.; Tang, G.; Cao, W. A Review on Medical Textual Question Answering Systems Based on Deep Learning Approaches. Appl. Sci. 2021, 11, 5456. [Google Scholar] [CrossRef]
- Sharma, Y.; Gupta, S. Deep Learning Approaches for Question Answering System. Procedia Comput. Sci. 2018, 132, 785–794. [Google Scholar] [CrossRef]
- Liu, S.; Zhang, X.; Zhang, S.; Wang, H.; Zhang, W. Neural Machine Reading Comprehension: Methods and Trends. Appl. Sci. 2019, 9, 3698. [Google Scholar] [CrossRef] [Green Version]
- Yin, W.; Kann, K.; Yu, M.; Schütze, H. Comparative Study of CNN and RNN for Natural Language Processing. arXiv 2017, arXiv:1702.01923. [Google Scholar]
- Zulqarnain, M.; Ghazali, R.; Ghouse, M.G.; Mushtaq, M.F. Efficient processing of GRU based on word embedding for text classification. JOIV Int. J. Inform. Vis. 2019, 3, 377–383. [Google Scholar] [CrossRef] [Green Version]
- Strubell, E.; Ganesh, A.; McCallum, A. Energy and policy considerations for deep learning in NLP. arXiv 2019, arXiv:1906.02243. [Google Scholar]
- Radford, A.; Narasimhan, K.; Salimans, T.; Sutskever, I. Improving Language Understanding by Generative Pre-Training; OpenAI: San Francisco, CA, USA, 2018. [Google Scholar]
- Kamath, U.; Graham, K.L.; Emara, W. Bidirectional encoder representations from transformers (BERT). In Transformers for Machine Learning; Chapman and Hall/CRC: New York, NY, USA, 2022; pp. 43–70. [Google Scholar]
- Loose, C.; Jensen, K.; Rigoutsos, I.; Stephanopoulos, G. A linguistic model for the rational design of antimicrobial peptides. Nature 2006, 443, 867–869. [Google Scholar] [CrossRef]
- Bolatchiev, A.; Baturin, V.; Shchetinin, E.; Bolatchieva, E. Novel Antimicrobial Peptides Designed Using a Recurrent Neural Network Reduce Mortality in Experimental Sepsis. Antibiotics 2022, 11, 411. [Google Scholar] [CrossRef]
- Sherstinsky, A. Fundamentals of Recurrent Neural Network (RNN) and Long Short-Term Memory (LSTM) network. Phys. D Nonlinear Phenom. 2020, 404, 132306. [Google Scholar] [CrossRef] [Green Version]
- Waghu, F.H.; Barai, R.S.; Gurung, P.; Idicula-Thomas, S. CAMPR3: A database on sequences, structures and signatures of antimicrobial peptides. Nucleic Acids Res. 2016, 44, D1094–D1097. [Google Scholar] [CrossRef] [Green Version]
- Das, P.; Wadhawan, K.; Chang, O.; Sercu, T.; Santos, C.D.; Riemer, M.; Chenthamarakshan, V.; Padhi, I.; Mojsilovic, A. PepCVAE: Semi-Supervised Targeted Design of Antimicrobial Peptide Sequences. arXiv 2018, arXiv:1810.07743. [Google Scholar]
- Gómez-Bombarelli, R.; Wei, J.N.; Duvenaud, D.; Hernández-Lobato, J.M.; Sánchez-Lengeling, B.; Sheberla, D.; Aguilera-Iparraguirre, J.; Hirzel, T.D.; Adams, R.P.; Aspuru-Guzik, A. Automatic Chemical Design Using a Data-Driven Continuous Representation of Molecules. ACS Cent. Sci. 2018, 4, 268–276. [Google Scholar] [CrossRef]
- Blaschke, T.; Olivecrona, M.; Engkvist, O.; Bajorath, J.; Chen, H. Application of generative autoencoder in de novo molecular design. Mol. Inform. 2018, 37, 1700123. [Google Scholar] [CrossRef] [Green Version]
- Dean, S.N.; Walper, S.A. Variational Autoencoder for Generation of Antimicrobial Peptides. ACS Omega 2020, 5, 20746–20754. [Google Scholar] [CrossRef] [PubMed]
- Das, P.; Sercu, T.; Wadhawan, K.; Padhi, I.; Gehrmann, S.; Cipcigan, F.; Chenthamarakshan, V.; Strobelt, H.; dos Santos, C.; Chen, P.Y.; et al. Accelerated antimicrobial discovery via deep generative models and molecular dynamics simulations. Nat. Biomed. Eng. 2021, 5, 613–623. [Google Scholar] [CrossRef] [PubMed]
- Creswell, A.; White, T.; Dumoulin, V.; Arulkumaran, K.; Sengupta, B.; Bharath, A.A. Generative Adversarial Networks: An Overview. IEEE Signal Process. Mag. 2018, 35, 53–65. [Google Scholar] [CrossRef] [Green Version]
- Goodfellow, I.; Pouget-Abadie, J.; Mirza, M.; Xu, B.; Warde-Farley, D.; Ozair, S.; Courville, A.; Bengio, Y. Generative Adversarial Nets. Advances in Neural Information Processing Systems 27 (NIPS 2014); Curran Associates, Inc.: Red Hook, NY, USA, 2014; Volume 27. [Google Scholar]
- Bhagyashree; Kushwaha, V.; Nandi, G.C. Study of Prevention of Mode Collapse in Generative Adversarial Network (GAN). In Proceedings of the 2020 IEEE 4th Conference on Information & Communication Technology (CICT), Chennai, India, 3–5 December 2020; pp. 1–6. [Google Scholar] [CrossRef]
- Tucs, A.; Tran, D.P.; Yumoto, A.; Ito, Y.; Uzawa, T.; Tsuda, K. Generating Ampicillin-Level Antimicrobial Peptides with Activity-Aware Generative Adversarial Networks. ACS Omega 2020, 5, 22847–22851. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Lin, Y.; Liu, J.; Chen, X.; Ma, C.; Xi, X.; Zhou, M.; Chen, T.; Burrows, J.F.; Wang, L. Targeted Modification and Structure-Activity Study of GL-29, an Analogue of the Antimicrobial Peptide Palustrin-2ISb. Antibiotics 2022, 11, 1048. [Google Scholar] [CrossRef]
- Shorten, C.; Khoshgoftaar, T.M. A survey on Image Data Augmentation for Deep Learning. J. Big Data 2019, 6, 60. [Google Scholar] [CrossRef]
- Chen, X.G.; Zhang, W.; Yang, X.; Li, C.; Chen, H. ACP-DA: Improving the Prediction of Anticancer Peptides Using Data Augmentation. Front. Genet. 2021, 12, 698477. [Google Scholar] [CrossRef]
- Lee, B.; Shin, M.K.; Hwang, I.W.; Jung, J.; Shim, Y.J.; Kim, G.W.; Kim, S.T.; Jang, W.; Sung, J.S. A Deep Learning Approach with Data Augmentation to Predict Novel Spider Neurotoxic Peptides. Int. J. Mol. Sci. 2021, 22, 12291. [Google Scholar] [CrossRef]
- Han, X.; Zhang, L.; Zhou, K.; Wang, X. ProGAN: Protein solubility generative adversarial nets for data augmentation in DNN framework. Comput. Chem. Eng. 2019, 131, 106533. [Google Scholar] [CrossRef]
- Ramazi, S.; Mohammadi, N.; Allahverdi, A.; Khalili, E.; Abdolmaleki, P. A review on antimicrobial peptides databases and the computational tools. Database 2022, 2022, baac011. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; You, Z.B. Predicting minimum inhibitory concentration of antimicrobial peptides by the pseudo-amino acid composition and Gaussian kernel regression. In Proceedings of the 2015 8th International Conference on Biomedical Engineering and Informatics (BMEI), Shenyang, China, 14–16 October 2015; pp. 301–305. [Google Scholar]
- Pane, K.; Durante, L.; Crescenzi, O.; Cafaro, V.; Pizzo, E.; Varcamonti, M.; Zanfardino, A.; Izzo, V.; Di Donato, A.; Notomista, E. Antimicrobial potency of cationic antimicrobial peptides can be predicted from their amino acid composition: Application to the detection of “cryptic” antimicrobial peptides. J. Theor. Biol. 2017, 419, 254–265. [Google Scholar] [CrossRef]
- Li, W.; Separovic, F.; O’Brien-Simpson, N.M.; Wade, J.D. Chemically modified and conjugated antimicrobial peptides against superbugs. Chem. Soc. Rev. 2021, 50, 4932–4973. [Google Scholar] [CrossRef] [PubMed]
- Di Natale, C.; De Benedictis, I.; De Benedictis, A.; Marasco, D. Metal–peptide complexes as promising antibiotics to fight emerging drug resistance: New perspectives in tuberculosis. Antibiotics 2020, 9, 337. [Google Scholar] [CrossRef] [PubMed]
- La Manna, S.; Di Natale, C.; Onesto, V.; Marasco, D. Self-Assembling Peptides: From Design to Biomedical Applications. Int. J. Mol. Sci. 2021, 22, 12662. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.H.; Lu, T.K. Development and Challenges of Antimicrobial Peptides for Therapeutic Applications. Antibiotics 2020, 9, 24. [Google Scholar] [CrossRef]
- Mathur, D.; Singh, S.; Mehta, A.; Agrawal, P.; Raghava, G.P.S. In silico approaches for predicting the half-life of natural and modified peptides in blood. PLoS ONE 2018, 13, e0196829. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Singla, D.; Rashid, M.; Raghava, G.P.S. Designing of peptides with desired half-life in intestine-like environment. BMC Bioinform. 2014, 15, 282. [Google Scholar] [CrossRef] [Green Version]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, P.; Eckert, A.O.; Schrey, A.K.; Preissner, R. ProTox-II: A webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018, 46, W257–W263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, S.; Kapoor, P.; Chaudhary, K.; Gautam, A.; Kumar, R.; Consortium, O.S.D.D.; Raghava, G.P.S. In Silico Approach for Predicting Toxicity of Peptides and Proteins. PLoS ONE 2013, 8, e73957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taho, F. Antimicrobial Peptide Host Toxicity Prediction with Transfer Learning for Proteins. Ph.D. Thesis, University of British Columbia, Vancouver, BC, Canada, 2020. [Google Scholar] [CrossRef]
- Hicks, A.L.; Wheeler, N.; Sánchez-Busó, L.; Rakeman, J.L.; Harris, S.R.; Grad, Y.H. Evaluation of parameters affecting performance and reliability of machine learning-based antibiotic susceptibility testing from whole genome sequencing data. PLoS Comput. Biol. 2019, 15, e1007349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartley, M.; Olsson, T.S. dtoolai: Reproducibility for deep learning. Patterns 2020, 1, 100073. [Google Scholar] [CrossRef]
- Alahmari, S.S.; Goldgof, D.B.; Mouton, P.R.; Hall, L.O. Challenges for the repeatability of deep learning models. IEEE Access 2020, 8, 211860–211868. [Google Scholar] [CrossRef]
- Pham, H.V.; Qian, S.; Wang, J.; Lutellier, T.; Rosenthal, J.; Tan, L.; Yu, Y.; Nagappan, N. Problems and opportunities in training deep learning software systems: An analysis of variance. In Proceedings of the 35th IEEE/ACM International Conference on Automated Software Engineering, Melbourne, Australia, 21–25 September 2020; pp. 771–783. [Google Scholar]
- Gundersen, O.E.; Coakley, K.; Kirkpatrick, C. Sources of Irreproducibility in Machine Learning: A Review. arXiv 2022, arXiv:2204.07610. [Google Scholar]
- Gunning, D.; Stefik, M.; Choi, J.; Miller, T.; Stumpf, S.; Yang, G.Z. XAI—Explainable artificial intelligence. Sci. Robot. 2019, 4, eaay7120. [Google Scholar] [CrossRef]
- Lundberg, S.M.; Erion, G.; Chen, H.; DeGrave, A.; Prutkin, J.M.; Nair, B.; Katz, R.; Himmelfarb, J.; Bansal, N.; Lee, S.I. From local explanations to global understanding with explainable AI for trees. Nat. Mach. Intell. 2020, 2, 56–67. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Non- | Unknown | ||||
|---|---|---|---|---|---|
| Total no. of peptides | 494 | 89 | 120 | 22 | 2710 |
| Average length | 29.66 | 35.08 | 58.92 | 26.82 | -- |
| Average net charge | +3.63 | +3.65 | +5.37 | +2.55 | -- |
| Technique | De Novo Sequence | Length | Target Species | Activity MIC (g/mL) | Reference |
|---|---|---|---|---|---|
| Manually designed | Ac-LKLLKKLLKKLKKLLKKL-NH2 | 18 | S. areus E. coli P. aeruginosa | 64 64 128 | [171] |
| Ensemble learning, ANN | ALFGILKKAFGKILTIFAGLPGVV | MCF7 A549 | 9.8 (EC50) 8.6 | 24 | [196] |
| GLGDFIKAIAKHLGPLIGILPSKLKVAA | 28 | MCF7 A549 | 4.5 11.3 | ||
| FLGPTIGKIAKFILKHIVGLGDAALV | 26 | MCF7 A549 | 2.6 10.7 | ||
| GLFAILKKLVNLVG | 15 | MCF7 A549 | 2.3 4.6 | ||
| GLFKIISKLAKKA | 13 | MCF7 A549 | 27.7 36.3 | ||
| VAE | KKIKRFLRKIG | 11 | E. coli A. baumannii S. aureus | 11 36 0.4 | [190] |
| KLFRIIKRIFKG | 12 | E. coli A. baumannii S. aureus | 0.2 0.8 >400 | ||
| VAE, LSTM | YLRLIRYMAKMI-CONH2 | 12 | S. aureus E. coli P. aeruginosa A. baumannii MDR K. pneumoniae polyR K. pneumoniae | 7.8 31.25 125 15.6 31.25 31 | [191] |
| FPLTWLKWWKWKK-CONH2 | 13 | S. aureus E. coli P. aeruginosa A. baumannii MDR K. pneumoniae polyR K. pneumoniae | 15.6 31.25 62.5 31.25 15.6 16 | ||
| GAN | ILPLLKKFGKKFGKKVWKAL IKALLALPKLAKKIAKKFLK GLRSSVKTLLRGLLGIIKKF GLKKLFSKIKIIGSALKNLA FLPAFKNVISKILKALKKKV FLGPIIKTVRAVLCAIKKL | 20 20 20 20 20 20 | E. coli E. coli E. coli E. coli E. coli E. coli | 25 50 >100 2.1 12.5 25 | [195] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, J.; Cai, J.; Zhang, B.; Wang, Y.; Wong, D.F.; Siu, S.W.I. Recent Progress in the Discovery and Design of Antimicrobial Peptides Using Traditional Machine Learning and Deep Learning. Antibiotics 2022, 11, 1451. https://doi.org/10.3390/antibiotics11101451
Yan J, Cai J, Zhang B, Wang Y, Wong DF, Siu SWI. Recent Progress in the Discovery and Design of Antimicrobial Peptides Using Traditional Machine Learning and Deep Learning. Antibiotics. 2022; 11(10):1451. https://doi.org/10.3390/antibiotics11101451
Chicago/Turabian StyleYan, Jielu, Jianxiu Cai, Bob Zhang, Yapeng Wang, Derek F. Wong, and Shirley W. I. Siu. 2022. "Recent Progress in the Discovery and Design of Antimicrobial Peptides Using Traditional Machine Learning and Deep Learning" Antibiotics 11, no. 10: 1451. https://doi.org/10.3390/antibiotics11101451
APA StyleYan, J., Cai, J., Zhang, B., Wang, Y., Wong, D. F., & Siu, S. W. I. (2022). Recent Progress in the Discovery and Design of Antimicrobial Peptides Using Traditional Machine Learning and Deep Learning. Antibiotics, 11(10), 1451. https://doi.org/10.3390/antibiotics11101451