
Evolution of Bacterial Persistence to Antibiotics during a 50,000-Generation Experiment in an Antibiotic-Free Environment


Abstract
:1. Introduction
2. Results
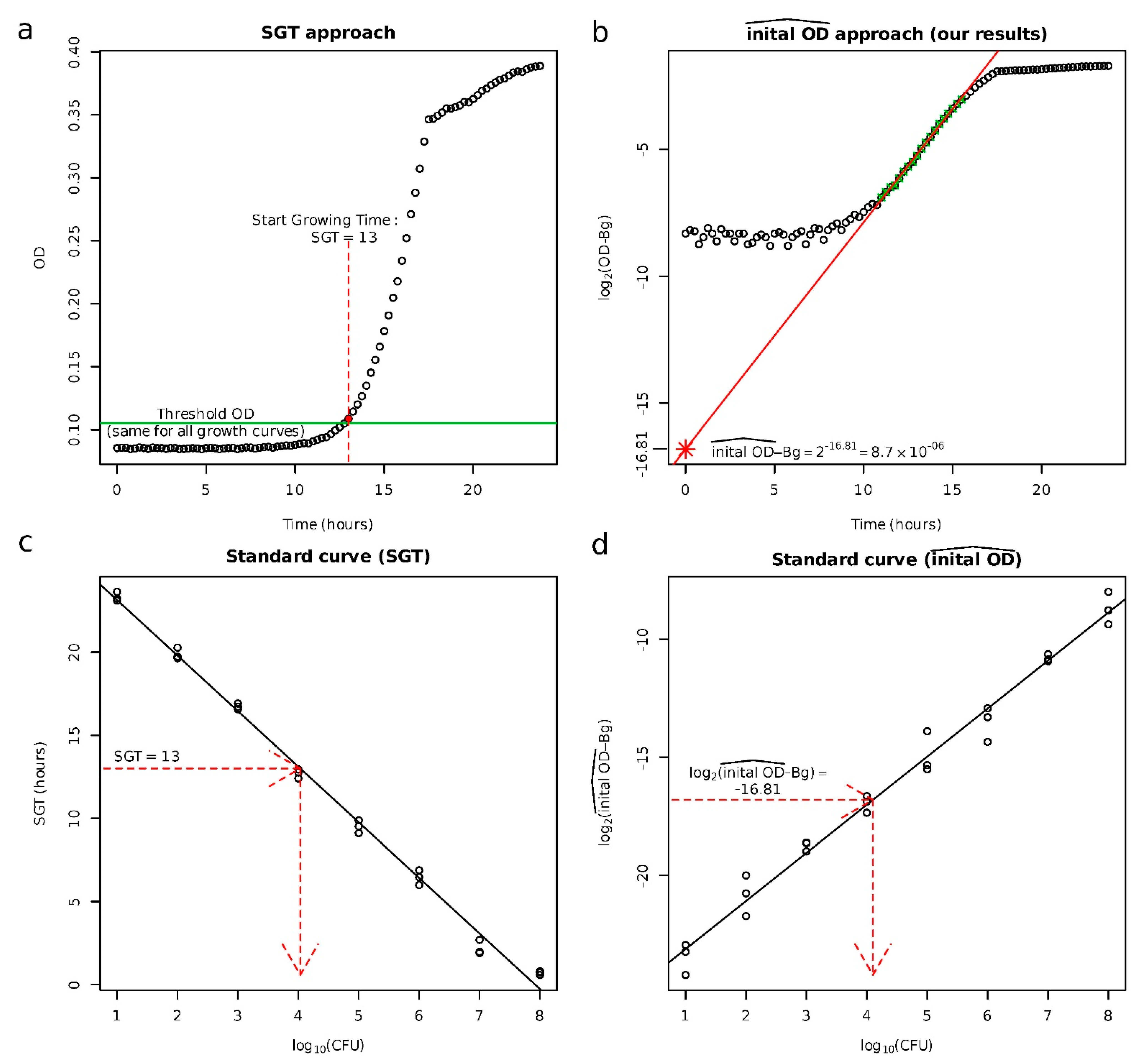
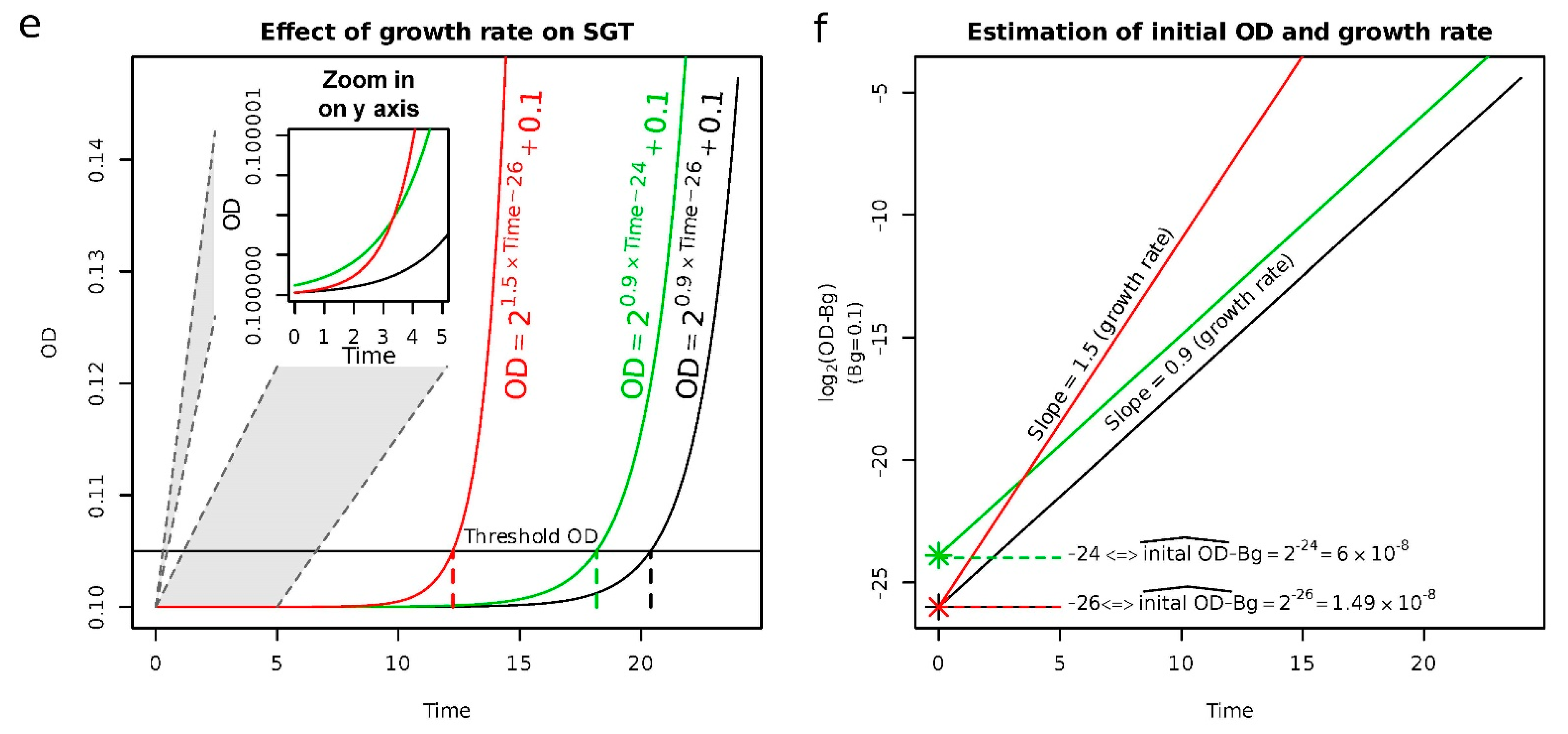
2.1. Validation of the Approach to Quantify Persister Cells
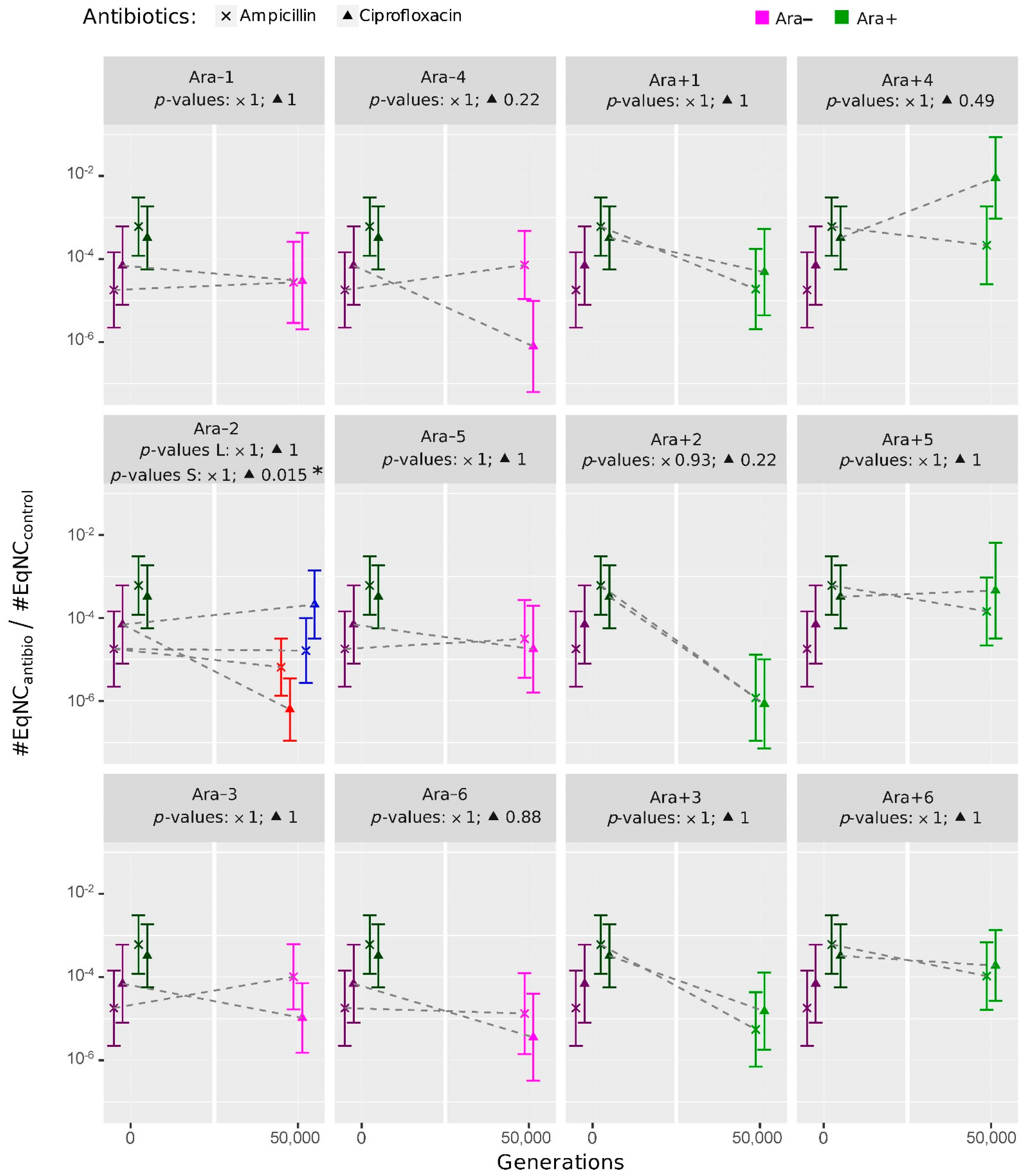
2.2. Evolution of Persistence in the ‘LTEE-50K’ Analysis
2.2.1. Overall Trends in the Persistence Level to Ampicillin and Ciprofloxacin
2.2.2. Evolution of Persistence after 50,000 Generations of Evolution
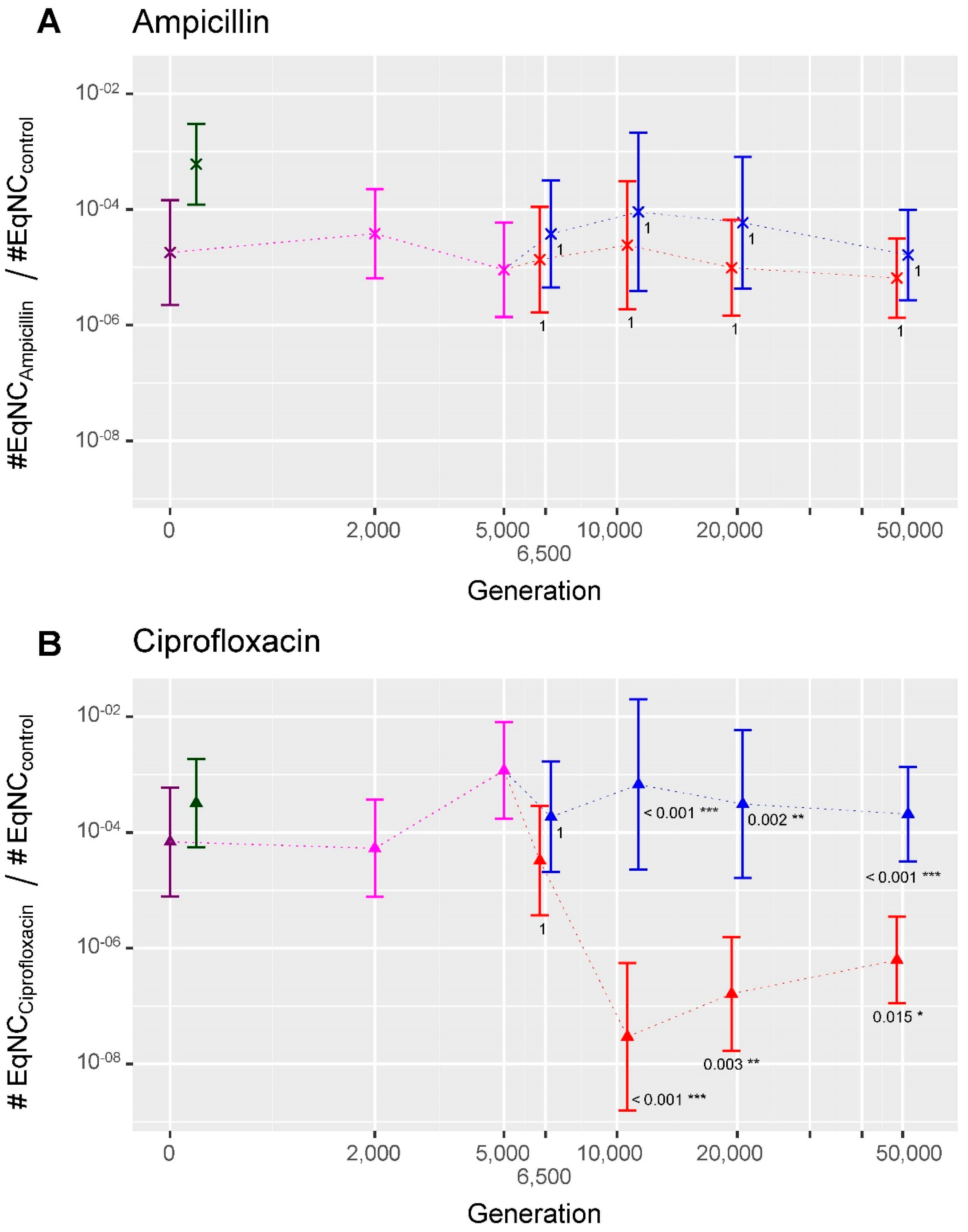
2.3. Evolution of Persistence in the ‘Ara−2_S_L’ Analysis
3. Discussion
4. Materials and Methods
4.1. Strains
4.2. Measuring the Proportion of Persister Cells
4.3. Rationale of Data Analyses
4.4. Quantification of the Population Size of Persister Cells
4.5. Estimating and Comparing Persistence
4.6. Relationship between Persistence and Mutator Phenotype
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A. Estimation of the Equivalent Number of Normal Cells (#EqNC)


References
- Interagency Coordination Group on Antimicrobial Resistance. No Time to Wait: Securing the Future from Drug-Resistant Infections. In Report to the Secretary-General of the United Nations; WHO: Geneva, Switzerland, 2019; Available online: https://www.who.int/publications/i/item/no-time-to-wait-securing-the-future-from-drug-resistant-infections (accessed on 20 February 2022).
- Kraker, M.E.A.D.; Stewardson, A.J.; Harbarth, S. Will 10 Million People Die a Year Due to Antimicrobial Resistance by 2050? PLoS Med. 2016, 13, e1002184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassini, A.; Högberg, L.D.; Plachouras, D.; Quattrocchi, A.; Hoxha, A.; Simonsen, G.S.; Colomb-Cotinat, M.; Kretzschmar, M.E.; Devleesschauwer, B.; Cecchini, M.; et al. Attributable Deaths and Disability-Adjusted Life-Years Caused by Infections with Antibiotic-Resistant Bacteria in the EU and the European Economic Area in 2015: A Population-Level Modelling Analysis. Lancet Infect. Dis. 2019, 19, 56–66. [Google Scholar] [CrossRef] [Green Version]
- Brauner, A.; Fridman, O.; Gefen, O.; Balaban, N.Q. Distinguishing between Resistance, Tolerance and Persistence to Antibiotic Treatment. Nat. Rev. Microbiol. 2016, 14, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Balaban, N.Q.; Helaine, S.; Lewis, K.; Ackermann, M.; Aldridge, B.; Andersson, D.I.; Brynildsen, M.P.; Bumann, D.; Camilli, A.; Collins, J.J.; et al. Definitions and Guidelines for Research on Antibiotic Persistence. Nat. Rev. Microbiol. 2019, 17, 441–448. [Google Scholar] [CrossRef] [Green Version]
- Bakkeren, E.; Diard, M.; Hardt, W.-D. Evolutionary Causes and Consequences of Bacterial Antibiotic Persistence. Nat. Rev. Microbiol. 2020, 18, 479–490. [Google Scholar] [CrossRef]
- Van den Bergh, B.; Fauvart, M.; Michiels, J. Formation, Physiology, Ecology, Evolution and Clinical Importance of Bacterial Persisters. FEMS Microbiol. Rev. 2017, 41, 219–251. [Google Scholar] [CrossRef]
- Jung, S.-H.; Ryu, C.-M.; Kim, J.-S. Bacterial Persistence: Fundamentals and Clinical Importance. J. Microbiol. 2019, 57, 829–835. [Google Scholar] [CrossRef]
- Cohen, N.R.; Lobritz, M.A.; Collins, J.J. Microbial Persistence and the Road to Drug Resistance. Cell Host Microbe 2013, 13, 632–642. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.V.; Lee, D.Y.; Li, B.; Quinlan, M.P.; Takahashi, F.; Maheswaran, S.; McDermott, U.; Azizian, N.; Zou, L.; Fischbach, M.A.; et al. A Chromatin-Mediated Reversible Drug-Tolerant State in Cancer Cell Subpopulations. Cell 2010, 141, 69–80. [Google Scholar] [CrossRef] [Green Version]
- Swayden, M.; Chhouri, H.; Anouar, Y.; Grumolato, L. Tolerant/Persister Cancer Cells and the Path to Resistance to Targeted Therapy. Cells 2020, 9, 2601. [Google Scholar] [CrossRef]
- Vallette, F.M.; Olivier, C.; Lézot, F.; Oliver, L.; Cochonneau, D.; Lalier, L.; Cartron, P.-F.; Heymann, D. Dormant, Quiescent, Tolerant and Persister Cells: Four Synonyms for the Same Target in Cancer. Biochem. Pharmacol. 2019, 162, 169–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehman, J.; Clune, J.; Misevic, D.; Adami, C.; Altenberg, L.; Beaulieu, J.; Bentley, P.J.; Bernard, S.; Beslon, G.; Bryson, D.M.; et al. The Surprising Creativity of Digital Evolution: A Collection of Anecdotes from the Evolutionary Computation and Artificial Life Research Communities. Artif. Life 2020, 26, 274–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, R.A.; Gollan, B.; Helaine, S. Persistent Bacterial Infections and Persister Cells. Nat. Rev. Microbiol. 2017, 15, 453–464. [Google Scholar] [CrossRef] [PubMed]
- Gollan, B.; Grabe, G.; Michaux, C.; Helaine, S. Bacterial Persisters and Infection: Past, Present, and Progressing. Annu. Rev. Microbiol. 2019, 73, 359–385. [Google Scholar] [CrossRef]
- Fauvart, M.; De Groote, V.N.; Michiels, J. Role of Persister Cells in Chronic Infections: Clinical Relevance and Perspectives on Anti-Persister Therapies. J. Med. Microbiol. 2011, 60, 699–709. [Google Scholar] [CrossRef]
- Li, L.; Mendis, N.; Trigui, H.; Oliver, J.D.; Faucher, S.P. The Importance of the Viable but Non-Culturable State in Human Bacterial Pathogens. Front. Microbiol. 2014, 5, 258. [Google Scholar] [CrossRef] [Green Version]
- Lin, P.L.; Flynn, J.L. Understanding Latent Tuberculosis: A Moving Target. J. Immunol. 2010, 185, 15–22. [Google Scholar] [CrossRef] [Green Version]
- Windels, E.M.; Michiels, J.E.; Fauvart, M.; Wenseleers, T.; Van den Bergh, B.; Michiels, J. Bacterial Persistence Promotes the Evolution of Antibiotic Resistance by Increasing Survival and Mutation Rates. ISME J. 2019, 13, 1239–1251. [Google Scholar] [CrossRef]
- Hobby, G.L.; Meyer, K.; Chaffee, E. Observations on the Mechanism of Action of Penicillin. Exp. Biol. Med. 1942, 50, 281–285. [Google Scholar] [CrossRef]
- Balaban, N.Q.; Merrin, J.; Chait, R.; Kowalik, L.; Leibler, S. Bacterial Persistence as a Phenotypic Switch. Science 2004, 305, 5. [Google Scholar] [CrossRef] [Green Version]
- Suter, L.; Widmer, A. Phenotypic Effects of Salt and Heat Stress over Three Generations in Arabidopsis Thaliana. PLoS ONE 2013, 8, e80819. [Google Scholar] [CrossRef] [PubMed]
- Badyaev, A.V. Environmental Stress and Developmental Stability in Dentition of the Yellowstone Grizzly Bears. Behav. Ecol. 1998, 9, 339–344. [Google Scholar] [CrossRef] [Green Version]
- Vøllestad, L.A.; Hindar, K. Developmental Stability and Environmental Stress in Salmo salar (Atlantic Salmon). Heredity 1997, 78, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Hurtado, L.; Castrezana, S.; Mateos, M.; Mclaurin, D.; Tello, M.K.; Campoy, J.; Markow, T. Developmental Stability and Environmental Stress in Natural Populations of Drosophila Pachea. Ecotoxicology 1997, 6, 233–238. [Google Scholar] [CrossRef]
- Badyaev, A.V. Stress-Induced Variation in Evolution: From Behavioural Plasticity to Genetic Assimilation. Proc. Biol. Sci. R. Soc. 2005, 272, 877–886. [Google Scholar] [CrossRef] [Green Version]
- Vogt, G. Disentangling the Environmentally Induced and Stochastic Developmental Components of Phenotypic Variation. In Phenotypic Switching: Implications in Biology and Medicine; Elsevier: Amsterdam, The Netherlands, 2020; pp. 207–251. ISBN 978-0-12-817996-3. [Google Scholar]
- Vogt, G. Stochastic Developmental Variation, an Epigenetic Source of Phenotypic Diversity with Far-Reaching Biological Consequences. J. Biosci. 2015, 40, 159–204. [Google Scholar] [CrossRef]
- Krishna, S.; Laxman, S. Emergence of Metabolic Heterogeneity in Cell Populations: Lessons from Budding Yeast. In Phenotypic Switching: Implications in Biology and Medicine; Elsevier: Amsterdam, The Netherlands, 2020; pp. 335–360. ISBN 978-0-12-817996-3. [Google Scholar]
- Schwartz, M.H.; Waldbauer, J.R.; Zhang, L.; Pan, T. Global TRNA Misacylation Induced by Anaerobiosis and Antibiotic Exposure Broadly Increases Stress Resistance in Escherichia coli. Nucleic Acids Res. 2016, 44, gkw856. [Google Scholar] [CrossRef] [Green Version]
- Rocabert, C.; Beslon, G.; Knibbe, C.; Bernard, S. Phenotypic Noise and the Cost of Complexity. Evol. Biol. 2020, 74, 2221–2237. [Google Scholar] [CrossRef]
- Wang, Q.; Huang, L.; Wen, K.; Yu, J. China The Mean and Noise of Stochastic Gene Transcription with Cell Division. Math. Biosci. Eng. 2018, 15, 1255–1270. [Google Scholar] [CrossRef] [Green Version]
- Prado Casanova, M. Noise and Synthetic Biology: How to Deal with Stochasticity? Nanoethics 2020, 14, 113–122. [Google Scholar] [CrossRef]
- Kaneko, K.; Furusawa, C. Relevance of Phenotypic Noise to Adaptation and Evolution. IET Syst. Biol. 2008, 2, 234–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hallgrimsson, B.; Willmore, K.; Hall, B.K. Canalization, Developmental Stability, and Morphological Integration in Primate Limbs. Am. J. Phys. Anthropol. 2002, 119, 131–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortbauer, M. Abiotic Stress Adaptation: Protein Folding Stability and Dynamics. In Abiotic Stress–Plant Responses and Applications in Agriculture; Vahdati, K., Ed.; InTech: London, UK, 2013; ISBN 978-953-51-1024-8. [Google Scholar]
- Jorgen, R.; Helen, O.; Niclas, J. What Is Bet-Hedging, Really? Proc. R. Soc. B 2010, 277, 1153–1154. [Google Scholar] [CrossRef]
- Levin, B.R.; Concepción-Acevedo, J.; Udekwu, K.I. Persistence: A Copacetic and Parsimonious Hypothesis for the Existence of Non-Inherited Resistance to Antibiotics. Curr. Opin. Microbiol. 2014, 21, 18–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girgis, H.S.; Harris, K.; Tavazoie, S. Large Mutational Target Size for Rapid Emergence of Bacterial Persistence. Proc. Natl. Acad. Sci. USA 2012, 109, 12740–12745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nichol, D.; Robertson, M.; Jeavons, P.; Anderson, A.R.A. Stochasticity in the Genotype-Phenotype Map: Implications for the Robustness and Persistence of Bet-Hedging. Genetics 2016, 204, 1523–1539. [Google Scholar] [CrossRef]
- Pu, Y.; Ke, Y.; Bai, F. Active Efflux in Dormant Bacterial Cells–New Insights into Antibiotic Persistence. Drug Resist. Updates 2017, 30, 7–14. [Google Scholar] [CrossRef]
- Pu, Y.; Zhao, Z.; Li, Y.; Zou, J.; Ma, Q.; Zhao, Y.; Ke, Y.; Zhu, Y.; Chen, H.; Baker, M.A.B.; et al. Enhanced Efflux Activity Facilitates Drug Tolerance in Dormant Bacterial Cells. Mol. Cell 2016, 62, 284–294. [Google Scholar] [CrossRef] [Green Version]
- LaFleur, M.D.; Qi, Q.; Lewis, K. Patients with Long-Term Oral Carriage Harbor High-Persister Mutants of Candida Albicans. AAC 2010, 54, 39–44. [Google Scholar] [CrossRef] [Green Version]
- Stepanyan, K.; Wenseleers, T.; Duéñez-Guzmán, E.A.; Muratori, F.; Van den Bergh, B.; Verstraeten, N.; De Meester, L.; Verstrepen, K.J.; Fauvart, M.; Michiels, J. Fitness Trade-Offs Explain Low Levels of Persister Cells in the Opportunistic Pathogen Pseudomonas aeruginosa. Mol. Ecol. 2015, 24, 1572–1583. [Google Scholar] [CrossRef] [Green Version]
- Goneau, L.W.; Yeoh, N.S.; MacDonald, K.W.; Cadieux, P.A.; Burton, J.P.; Razvi, H.; Reid, G. Selective Target Inactivation Rather than Global Metabolic Dormancy Causes Antibiotic Tolerance in Uropathogens. Antimicrob. Agents Chemother. 2014, 58, 2089–2097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vulin, C.; Leimer, N.; Huemer, M.; Ackermann, M.; Zinkernagel, A.S. Prolonged Bacterial Lag Time Results in Small Colony Variants That Represent a Sub-Population of Persisters. Nat. Commun. 2018, 9, 4074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van den Bergh, B.; Michiels, J.E.; Wenseleers, T.; Windels, E.M.; Boer, P.V.; Kestemont, D.; De Meester, L.; Verstrepen, K.J.; Verstraeten, N.; Fauvart, M.; et al. Frequency of Antibiotic Application Drives Rapid Evolutionary Adaptation of Escherichia coli Persistence. Nat. Microbiol. 2016, 1, 16020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Good, B.H.; Mcdonald, M.J.; Barrick, J.E.; Lenski, R.E.; Desai, M.M. The Dynamics of Molecular Evolution over 60,000 Generations. Nature 2017, 551, 45–50. [Google Scholar] [CrossRef]
- Lamrabet, O.; Martin, M.; Lenski, R.E.; Schneider, D. Changes in Intrinsic Antibiotic Susceptibility during a Long-Term Evolution Experiment with Escherichia coli. MBio 2019, 10, e00189-19. [Google Scholar] [CrossRef] [Green Version]
- Hazan, R.; Que, Y.-A.; Maura, D.; Rahme, L.G. A Method for High Throughput Determination of Viable Bacteria Cell Counts in 96-Well Plates. BMC Microbiol. 2012, 12, 259. [Google Scholar] [CrossRef] [Green Version]
- Plucain, J.; Hindre, T.; Le Gac, M.; Tenaillon, O.; Cruveiller, S.; Medigue, C.; Leiby, N.; Harcombe, W.R.; Marx, C.J.; Lenski, R.E.; et al. Epistasis and Allele Specificity in the Emergence of a Stable Polymorphism in Escherichia coli. Science 2014, 343, 1366–1369. [Google Scholar] [CrossRef]
- Wiser, M.J.; Ribeck, N.; Lenski, R.E. Long-Term Dynamics of Adaptation in Asexual Populations. Science 2013, 342, 1364–1367. [Google Scholar] [CrossRef] [Green Version]
- Lenski, R.E.; Rose, M.R.; Simpson, S.C.; Tadler, S.C. Long-Term Experimental Evolution in Escherichia coli. I. Adaptation and Divergence During 2000 Generations. Am. Nat. 1991, 138, 1315–1341. [Google Scholar] [CrossRef]
- Consuegra, J.; Plucain, J.; Gaffé, J.; Hindré, T.; Schneider, D. Genetic Basis of Exploiting Ecological Opportunity During the Long-Term Diversification of a Bacterial Population. J. Mol. Evol. 2017, 85, 26–36. [Google Scholar] [CrossRef]
- Vogwill, T.; Comfort, A.C.; Furió, V.; MacLean, R.C. Persistence and Resistance as Complementary Bacterial Adaptations to Antibiotics. J. Evol. Biol. 2016, 29, 1223–1233. [Google Scholar] [CrossRef] [PubMed]
- Hofsteenge, N.; van Nimwegen, E.; Silander, O.K. Quantitative Analysis of Persister Fractions Suggests Different Mechanisms of Formation among Environmental Isolates of E. Coli. BMC Microbiol. 2013, 13, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, B.; Rozen, D.E. Genetic Variation for Antibiotic Persistence in Escherichia coli. Evolution 2012, 66, 933–939. [Google Scholar] [CrossRef] [PubMed]
- Roemhild, R.; Gokhale, C.S.; Dirksen, P.; Blake, C.; Rosenstiel, P.; Traulsen, A.; Andersson, D.I.; Schulenburg, H. Cellular Hysteresis as a Principle to Maximize the Efficacy of Antibiotic Therapy. Proc. Natl. Acad. Sci. USA 2018, 115, 9767–9772. [Google Scholar] [CrossRef] [Green Version]
- Roemhild, R.; Schulenburg, H. Evolutionary Ecology Meets the Antibiotic Crisis. Evol. Med. Public Health 2019, 2019, 37–45. [Google Scholar] [CrossRef]
- Mitchell, A.; Romano, G.H.; Groisman, B.; Yona, A.; Dekel, E.; Kupiec, M.; Dahan, O.; Pilpel, Y. Adaptive Prediction of Environmental Changes by Microorganisms. Nature 2009, 460, 220–224. [Google Scholar] [CrossRef]
- López García de Lomana, A.; Kaur, A.; Turkarslan, S.; Beer, K.D.; Mast, F.D.; Smith, J.J.; Aitchison, J.D.; Baliga, N.S. Adaptive Prediction Emerges over Short Evolutionary Time Scales. Genome Biol. Evol. 2017, 9, 1616–1623. [Google Scholar] [CrossRef] [Green Version]
- Rozen, D.E.; Lenski, R.E. Long-Term Experimental Evolution in Escherichia coli. VIII. Dynamics of a Balanced Polymorphism. Am. Nat. 2000, 155, 24–35. [Google Scholar] [CrossRef]
- Rozen, D.E.; Philippe, N.; Arjan de Visser, J.; Lenski, R.E.; Schneider, D. Death and Cannibalism in a Seasonal Environment Facilitate Bacterial Coexistence. Ecol. Lett. 2009, 12, 34–44. [Google Scholar] [CrossRef]
- Großkopf, T.; Consuegra, J.; Gaffé, J.; Willison, J.C.; Lenski, R.E.; Soyer, O.S.; Schneider, D. Metabolic Modelling in a Dynamic Evolutionary Framework Predicts Adaptive Diversification of Bacteria in a Long-Term Evolution Experiment. BMC Evol. Biol. 2016, 16, 163. [Google Scholar] [CrossRef] [Green Version]
- Keren, I.; Kaldalu, N.; Spoering, A.; Wang, Y.; Lewis, K. Persister Cells and Tolerance to Antimicrobials. FEMS Microbiol. Lett. 2004, 230, 13–18. [Google Scholar] [CrossRef] [Green Version]
- Verstraete, L.; Van den Bergh, B.; Verstraeten, N.; Michiels, J. Ecology and Evolution of Antibiotic Persistence. Trends Microbiol. 2022. [Google Scholar] [CrossRef] [PubMed]
- Žiemytė, M.; Carda-Diéguez, M.; Rodríguez-Díaz, J.C.; Ventero, M.P.; Mira, A.; Ferrer, M.D. Real-Time Monitoring of Pseudomonas aeruginosa Biofilm Growth Dynamics and Persister Cells’ Eradication. Emerg. Microbes Infect. 2021, 10, 2062–2075. [Google Scholar] [CrossRef]
- Jeong, H.; Barbe, V.; Lee, C.H.; Vallenet, D.; Yu, D.S.; Choi, S.-H.; Couloux, A.; Lee, S.-W.; Yoon, S.H.; Cattolico, L.; et al. Genome Sequences of Escherichia coli B Strains REL606 and BL21(DE3). J. Mol. Biol. 2009, 394, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Rozen, D.E.; Schneider, D.; Lenski, R.E. Long-Term Experimental Evolution in Escherichia coli. XIII. Phylogenetic History of a Balanced Polymorphism. J. Mol. Evol. 2005, 61, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Bates, D.; Mächler, M.; Bolker, B.; Walker, S. Fitting Linear Mixed-Effects Models Using Lme4. J. Stat. Softw. 2015, 67, 1–48. [Google Scholar] [CrossRef]
- Luke, S.G. Evaluating Significance in Linear Mixed-Effects Models in R. Behav. Res. 2017, 49, 1494–1502. [Google Scholar] [CrossRef]
- Li, P.; Redden, D.T. Comparing Denominator Degrees of Freedom Approximations for the Generalized Linear Mixed Model in Analyzing Binary Outcome in Small Sample Cluster-Randomized Trials. BMC Med. Res. Methodol. 2015, 15, 38. [Google Scholar] [CrossRef] [Green Version]
- Hothorn, T.; Bretz, F.; Westfall, P. Simultaneous Inference in General Parametric Models. Biom. J. 2008, 50, 346–363. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clone | LTEE Population | Generation | Mutator State * | Analyses ** | |
|---|---|---|---|---|---|
| REL606 | Ancestor (Ara−) | 0 | N | LTEE-50K | Ara−2_S_L |
| REL607 | Ancestor (Ara+) | 0 | N | LTEE-50K | Ara−2_S_L |
| 11330 | Ara−1 | 50,000 | M | LTEE-50K | |
| 1165A | Ara−2 (BC ***) | 2000 | N | Ara−2_S_L | |
| 2180A | Ara−2 (BC ***) | 5000 | M | Ara−2_S_L | |
| 6.5KS1 | Ara−2 (S) | 6500 | M | Ara−2_S_L | |
| 6.5KL4 | Ara−2 (L) | 6500 | M | Ara−2_S_L | |
| 11KS1 | Ara−2 (S) | 11,000 | M | Ara−2_S_L | |
| 11KL1 | Ara−2 (L) | 11,000 | M | Ara−2_S_L | |
| 20KS1 | Ara−2 (S) | 20,000 | M | Ara−2_S_L | |
| 20KL1 | Ara−2 (L) | 20,000 | M | Ara−2_S_L | |
| 13335 | Ara−2 (S) | 50,000 | N | LTEE-50K | Ara−2_S_L |
| 11333 | Ara−2 (L) | 50,000 | M | LTEE-50K | Ara−2_S_L |
| 11364 | Ara−3 | 50,000 | M | LTEE-50K | |
| 11336 | Ara−4 | 50,000 | M | LTEE-50K | |
| 11339 | Ara−5 | 50,000 | N | LTEE-50K | |
| 11389 | Ara−6 | 50,000 | N | LTEE-50K | |
| 11392 | Ara+1 | 50,000 | N | LTEE-50K | |
| 11342 | Ara+2 | 50,000 | N | LTEE-50K | |
| 11345 | Ara+3 | 50,000 | M | LTEE-50K | |
| 11348 | Ara+4 | 50,000 | N | LTEE-50K | |
| 11367 | Ara+5 | 50,000 | N | LTEE-50K | |
| 11370 | Ara+6 | 50,000 | M | LTEE-50K | |
| Variable | df | F-Value | p-Value |
|---|---|---|---|
| (dilution) | 1, 100.58 | 595.11 | <0.001 |
| Clone ID | 23, 59.92 | 48.86 | <0.001 |
| Antibiotic | 2, 1342.65 | 144.94 | <0.001 |
| (dilution) | 2, 131.47 | 7.63 | <0.001 |
| Antibiotic × clone ID | 44, 399.89 | 9.45 | <0.001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mathé-Hubert, H.; Amia, R.; Martin, M.; Gaffé, J.; Schneider, D. Evolution of Bacterial Persistence to Antibiotics during a 50,000-Generation Experiment in an Antibiotic-Free Environment. Antibiotics 2022, 11, 451. https://doi.org/10.3390/antibiotics11040451
Mathé-Hubert H, Amia R, Martin M, Gaffé J, Schneider D. Evolution of Bacterial Persistence to Antibiotics during a 50,000-Generation Experiment in an Antibiotic-Free Environment. Antibiotics. 2022; 11(4):451. https://doi.org/10.3390/antibiotics11040451
Chicago/Turabian StyleMathé-Hubert, Hugo, Rafika Amia, Mikaël Martin, Joël Gaffé, and Dominique Schneider. 2022. "Evolution of Bacterial Persistence to Antibiotics during a 50,000-Generation Experiment in an Antibiotic-Free Environment" Antibiotics 11, no. 4: 451. https://doi.org/10.3390/antibiotics11040451
APA StyleMathé-Hubert, H., Amia, R., Martin, M., Gaffé, J., & Schneider, D. (2022). Evolution of Bacterial Persistence to Antibiotics during a 50,000-Generation Experiment in an Antibiotic-Free Environment. Antibiotics, 11(4), 451. https://doi.org/10.3390/antibiotics11040451