
Evaluation of a Conformationally Constrained Indole Carboxamide as a Potential Efflux Pump Inhibitor in Pseudomonas aeruginosa

Abstract
:1. Introduction
2. Results and Discussion
2.1. Identification and In-Depth Potentiation Evaluation of a Potent First-Generation Indole 2-Carboxamide EPI
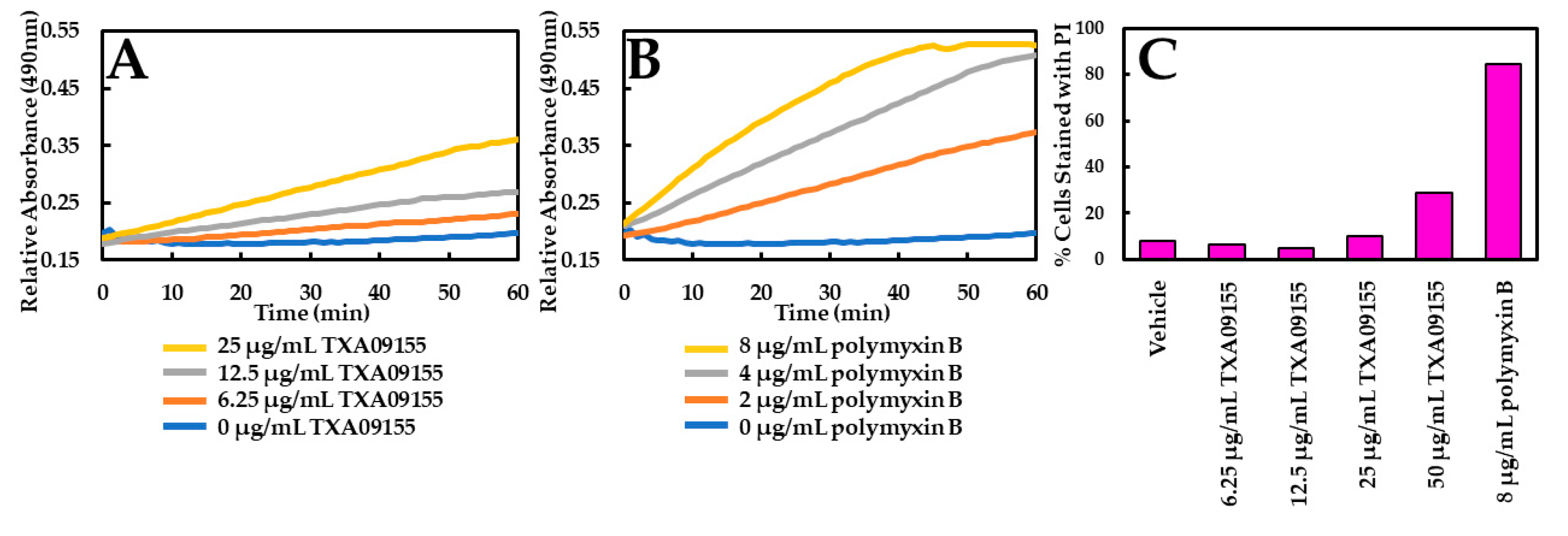
2.2. TXA09155 Plays a Minimal Role in Membrane Disruption
2.3. TXA09155 Inhibits the Efflux of Ethidium Bromide and Levofloxacin
2.4. TXA09155 Does Not Affect the Proton Gradient across P. aeruginosa Inner Membrane
2.5. TXA09155 Does Not Deplete ATP Levels in P. aeruginosa
2.6. TXA09155 Is Active in P. aeruginosa Mutants Overexpressing MexAB-OprM and MexXY-OprM Efflux Pumps
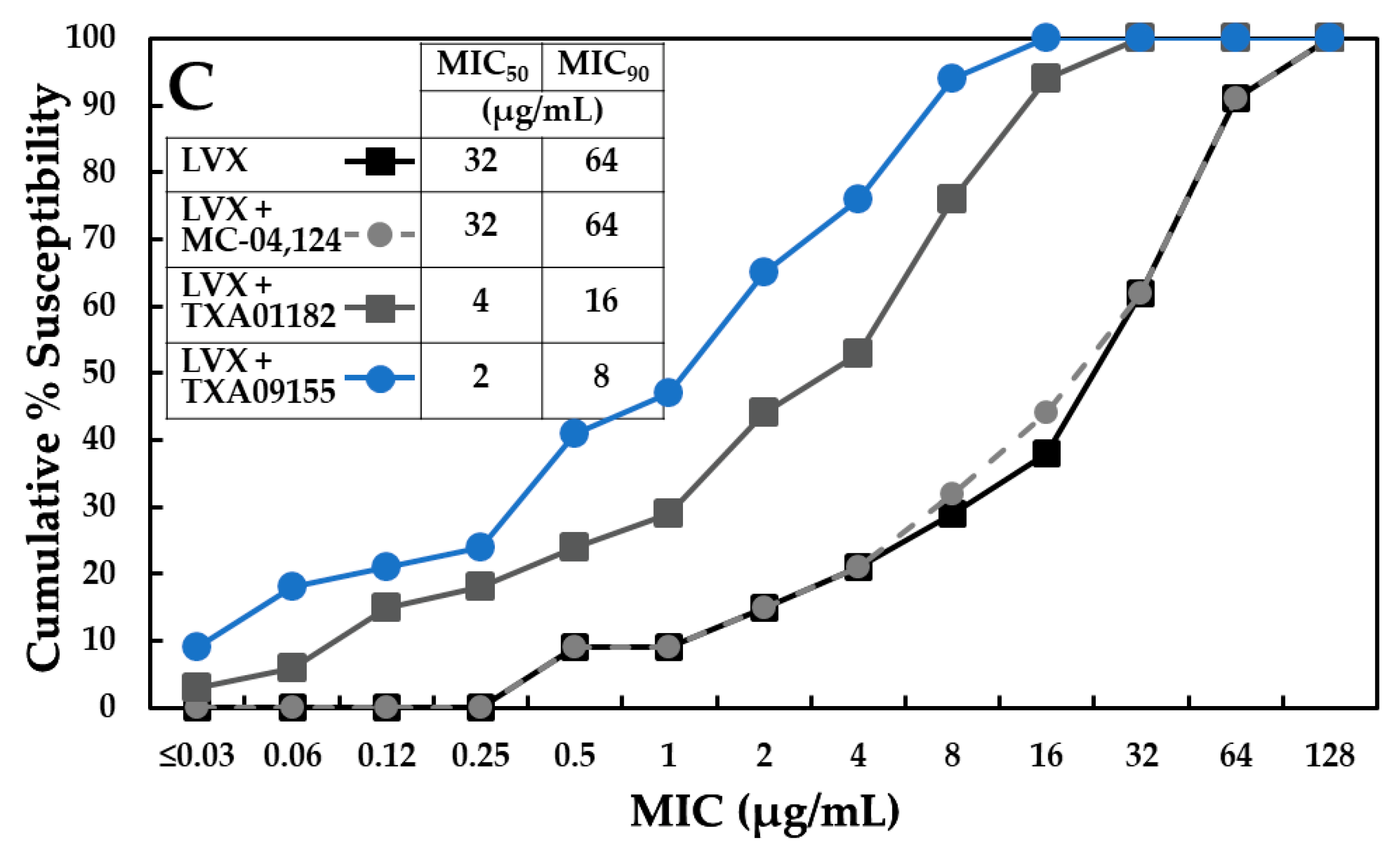
2.7. TXA09155 Potentiates Levofloxacin in P. aeruginosa Clinical Isolates from the United States and Other Countries
2.8. TXA09155 Shows Superior Potency against Multidrug-Resistant P. aeruginosa Clinical Isolates from the CDC-FDA
2.9. TXA09155 Lowers the Frequency of Resistance to Levofloxacin
2.10. Genes Involved in Resistance to TXA09155 Alone or Combined with Levofloxacin
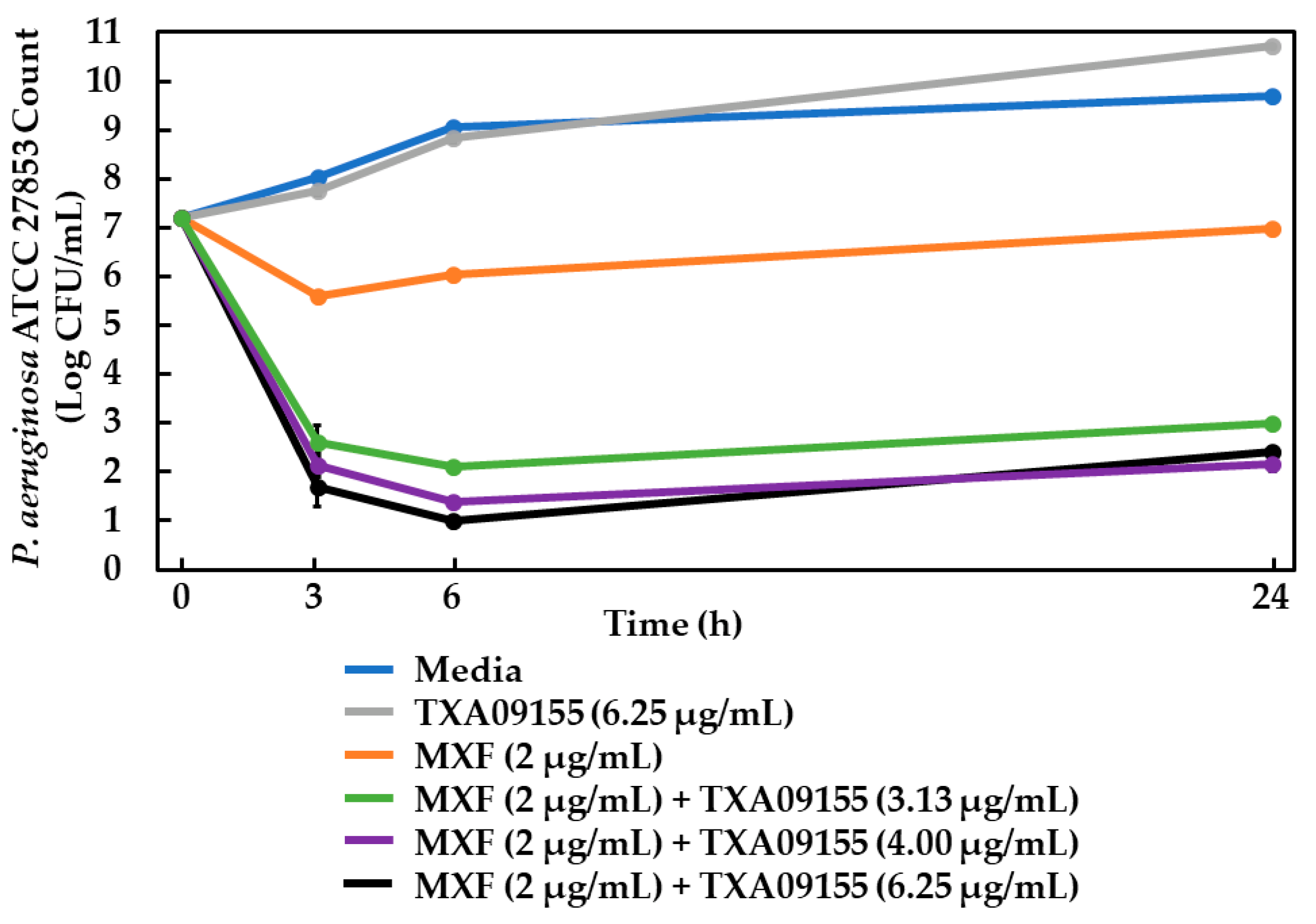
2.11. Time-Kill Assay
2.12. TXA09155 Has a Favorable Physiochemical and ADME Profile
3. Materials and methods
3.1. Synthesis Reagents, Analytical Instruments and Reaction Conditions
3.2. Bacterial Strains, Media, and Reagents
3.3. Micromyx and IHMA Clinical Isolates for Susceptibility Studies
3.4. Minimum Inhibitory Concentration (MIC) Assay for Potentiation of Antimicrobial Activity against P. aeruginosa
3.5. Flow Cytometry Assay for Permeabilization of Inner Cell Membranes to Propidium Iodide (PI) in P. aeruginosa
3.6. Nitrocefin Cellular Assay for Outer Cell Membrane Permeabilization Assessment in P. aeruginosa
3.7. Fluorescence-Based Cellular Assay for Inhibition of Pump-Mediated Efflux of Ethidium Bromide (EtBr)
3.8. Levofloxacin Accumulation Assay
3.9. Membrane Polarization Assay
3.10. Determination of Intracellular ATP Levels
3.11. Frequency of Resistance (FoR) Study
3.12. Whole-Genome Sequencing
3.13. Time-Kill Studies
3.14. Metabolic Stability Studies and hERG Assay
4. Conclusions
5. Patents
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sadikot, R.T.; Blackwell, T.S.; Christman, J.W.; Prince, A.S. Pathogen-host interactions in Pseudomonas aeruginosa pneumonia. Am. J. Respir. Crit. Care Med. 2005, 171, 1209–1223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reynolds, D.; Kollef, M. The Epidemiology and Pathogenesis and Treatment of Pseudomonas aeruginosa Infections: An Update. Drugs 2021, 81, 2117–2131. [Google Scholar] [CrossRef] [PubMed]
- Ehlenbach, W.J.; Curtis, J.R. Noninvasive ventilation for patients near the end of life: What do we know and what do we need to know? Crit. Care Med. 2008, 36, 1003–1004. [Google Scholar] [CrossRef] [PubMed]
- Micek, S.T.; Wunderink, R.G.; Kollef, M.H.; Chen, C.; Rello, J.; Chastre, J.; Antonelli, M.; Welte, T.; Clair, B.; Ostermann, H.; et al. An international multicenter retrospective study of Pseudomonas aeruginosa nosocomial pneumonia: Impact of multidrug resistance. Crit. Care 2015, 19, 219. [Google Scholar] [CrossRef] [Green Version]
- Lister, P.D.; Wolter, D.J.; Hanson, N.D. Antibacterial-resistant Pseudomonas aeruginosa: Clinical impact and complex regulation of chromosomally encoded resistance mechanisms. Clin. Microbiol. Rev. 2009, 22, 582–610. [Google Scholar] [CrossRef] [Green Version]
- Amaral, L.; Martins, A.; Spengler, G.; Molnar, J. Efflux pumps of Gram-negative bacteria: What they do, how they do it, with what and how to deal with them. Front. Pharmacol. 2014, 4, 168. [Google Scholar] [CrossRef] [Green Version]
- Li, X.Z.; Nikaido, H. Efflux-mediated drug resistance in bacteria. Drugs 2004, 64, 159–204. [Google Scholar] [CrossRef]
- Nikaido, H.; Pages, J.M. Broad-specificity efflux pumps and their role in multidrug resistance of Gram-negative bacteria. FEMS Microbiol. Rev. 2012, 36, 340–363. [Google Scholar] [CrossRef] [Green Version]
- Colclough, A.L.; Alav, I.; Whittle, E.E.; Pugh, H.L.; Darby, E.M.; Legood, S.W.; McNeil, H.E.; Blair, J.M. RND efflux pumps in Gram-negative bacteria; regulation, structure and role in antibiotic resistance. Future Microbiol. 2020, 15, 143–157. [Google Scholar] [CrossRef]
- Daury, L.; Orange, F.; Taveau, J.C.; Verchere, A.; Monlezun, L.; Gounou, C.; Marreddy, R.K.; Picard, M.; Broutin, I.; Pos, K.M.; et al. Tripartite assembly of RND multidrug efflux pumps. Nat. Commun. 2016, 7, 10731. [Google Scholar] [CrossRef] [Green Version]
- Poole, K. Multidrug efflux pumps and antimicrobial resistance in Pseudomonas aeruginosa and related organisms. J. Mol. Microbiol. Biotechnol. 2001, 3, 255–264. [Google Scholar]
- Poole, K.; Srikumar, R. Multidrug efflux in Pseudomonas aeruginosa: Components, mechanisms and clinical significance. Curr. Top. Med. Chem. 2001, 1, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Serra, C.; Bouharkat, B.; Tir Touil-Meddah, A.; Guenin, S.; Mullie, C. MexXY Multidrug Efflux System Is More Frequently Overexpressed in Ciprofloxacin Resistant French Clinical Isolates Compared to Hospital Environment Ones. Front. Microbiol. 2019, 10, 366. [Google Scholar] [CrossRef]
- Blanco, P.; Sanz-Garcia, F.; Hernando-Amado, S.; Martinez, J.L.; Alcalde-Rico, M. The development of efflux pump inhibitors to treat Gram-negative infections. Expert Opin. Drug Discov. 2018, 13, 919–931. [Google Scholar] [CrossRef]
- Wang, Y.; Venter, H.; Ma, S. Efflux Pump Inhibitors: A Novel Approach to Combat Efflux-Mediated Drug Resistance in Bacteria. Curr. Drug Targets 2016, 17, 702–719. [Google Scholar] [CrossRef]
- Pages, J.M.; Masi, M.; Barbe, J. Inhibitors of efflux pumps in Gram-negative bacteria. Trends Mol. Med. 2005, 11, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Lomovskaya, O.; Warren, M.S.; Lee, A.; Galazzo, J.; Fronko, R.; Lee, M.; Blais, J.; Cho, D.; Chamberland, S.; Renau, T.; et al. Identification and characterization of inhibitors of multidrug resistance efflux pumps in Pseudomonas aeruginosa: Novel agents for combination therapy. Antimicrob. Agents Chemother. 2001, 45, 105–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renau, T.E.; Leger, R.; Filonova, L.; Flamme, E.M.; Wang, M.; Yen, R.; Madsen, D.; Griffith, D.; Chamberland, S.; Dudley, M.N.; et al. Conformationally-restricted analogues of efflux pump inhibitors that potentiate the activity of levofloxacin in Pseudomonas aeruginosa. Bioorg. Med. Chem. Lett. 2003, 13, 2755–2758. [Google Scholar] [CrossRef]
- Yoshida, K.; Nakayama, K.; Ohtsuka, M.; Kuru, N.; Yokomizo, Y.; Sakamoto, A.; Takemura, M.; Hoshino, K.; Kanda, H.; Nitanai, H.; et al. MexAB-OprM specific efflux pump inhibitors in Pseudomonas aeruginosa. Part 7: Highly soluble and in vivo active quaternary ammonium analogue D13-9001, a potential preclinical candidate. Bioorg. Med. Chem. 2007, 15, 7087–7097. [Google Scholar] [CrossRef]
- Opperman, T.J.; Kwasny, S.M.; Kim, H.S.; Nguyen, S.T.; Houseweart, C.; D’Souza, S.; Walker, G.C.; Peet, N.P.; Nikaido, H.; Bowlin, T.L. Characterization of a novel pyranopyridine inhibitor of the AcrAB efflux pump of Escherichia coli. Antimicrob. Agents Chemother. 2014, 58, 722–733. [Google Scholar] [CrossRef] [Green Version]
- Lomovskaya, O.; Bostian, K.A. Practical applications and feasibility of efflux pump inhibitors in the clinic—A vision for applied use. Biochem. Pharmacol. 2006, 71, 910–918. [Google Scholar] [CrossRef]
- Farrell, L.J.; Lo, R.; Wanford, J.J.; Jenkins, A.; Maxwell, A.; Piddock, L.J.V. Revitalizing the drug pipeline: AntibioticDB, an open access database to aid antibacterial research and development. J. Antimicrob. Chemother. 2018, 73, 2284–2297. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Z.; Plesiat, P.; Nikaido, H. The challenge of efflux-mediated antibiotic resistance in Gram-negative bacteria. Clin. Microbiol. Rev. 2015, 28, 337–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahmood, H.Y.; Jamshidi, S.; Sutton, J.M.; Rahman, K.M. Current Advances in Developing Inhibitors of Bacterial Multidrug Efflux Pumps. Curr. Med. Chem. 2016, 23, 1062–1081. [Google Scholar] [CrossRef]
- Schweizer, H.P. Understanding efflux in Gram-negative bacteria: Opportunities for drug discovery. Expert Opin. Drug Discov. 2012, 7, 633–642. [Google Scholar] [CrossRef] [PubMed]
- TAXIS Pharmaceuticals. Tackling resistance in multidrug-resistant bacterial infections. Biopharma Deal. 2020, 14, B23. [Google Scholar]
- Blankson, G.; Parhi, A.K.; Kaul, M.; Pilch, D.S.; LaVoie, E.J. Structure-activity relationships of potentiators of the antibiotic activity of clarithromycin against Escherichia coli. Eur. J. Med. Chem. 2019, 178, 30–38. [Google Scholar] [CrossRef]
- Blankson, G.A.; Parhi, A.K.; Kaul, M.; Pilch, D.S.; LaVoie, E.J. Advances in the structural studies of antibiotic potentiators against Escherichia coli. Bioorg. Med. Chem. 2019, 27, 3254–3278. [Google Scholar] [CrossRef]
- Yuan, Y.; Rosado-Lugo, J.D.; Zhang, Y.; Datta, P.; Sun, Y.; Cao, Y.; Banerjee, A.; Parhi, A.K. Evaluation of Heterocyclic Carboxamides as Potential Efflux Pump Inhibitors in Pseudomonas aeruginosa. Antibiotics 2021, 11, 30. [Google Scholar] [CrossRef]
- Pierschbacher, M.D.; Ruoslahti, E. Influence of stereochemistry of the sequence Arg-Gly-Asp-Xaa on binding specificity in cell adhesion. J. Biol. Chem. 1987, 262, 17294–17298. [Google Scholar] [CrossRef]
- Wang, J.X.; Dipasquale, A.J.; Bray, A.M.; Maeji, N.J.; Spellmeyer, D.C.; Geysen, H.M. Systematic study of substance P analogs. II. Rapid screening of 512 substance P stereoisomers for binding to NK1 receptor. Int. J. Pept. Protein Res. 1993, 42, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Horwell, D.C.; Howson, W.; Higginbottom, M.; Naylor, D.; Ratcliffe, G.S.; Williams, S. Quantitative structure-activity relationships (QSARs) of N-terminus fragments of NK1 tachykinin antagonists: A comparison of classical QSARs and three-dimensional QSARs from similarity matrices. J. Med. Chem. 1995, 38, 4454–4462. [Google Scholar] [CrossRef] [PubMed]
- Hancock, R.E.; Wong, P.G. Compounds which increase the permeability of the Pseudomonas aeruginosa outer membrane. Antimicrob. Agents Chemother. 1984, 26, 48–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kugelberg, E.; Lofmark, S.; Wretlind, B.; Andersson, D.I. Reduction of the fitness burden of quinolone resistance in Pseudomonas aeruginosa. J. Antimicrob. Chemother. 2005, 55, 22–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konai, M.M.; Haldar, J. Lysine-Based Small Molecule Sensitizes Rifampicin and Tetracycline against Multidrug-Resistant Acinetobacter baumannii and Pseudomonas aeruginosa. ACS Infect. Dis. 2020, 6, 91–99. [Google Scholar] [CrossRef]
- Mahey, N.; Tambat, R.; Chandal, N.; Verma, D.K.; Thakur, K.G.; Nandanwar, H. Repurposing Approved Drugs as Fluoroquinolone Potentiators to Overcome Efflux Pump Resistance in Staphylococcus aureus. Microbiol. Spectr. 2021, 9, e0095121. [Google Scholar] [CrossRef]
- Nikaido, H.; Takatsuka, Y. Mechanisms of RND multidrug efflux pumps. Biochim. Biophys. Acta 2009, 1794, 769–781. [Google Scholar] [CrossRef] [Green Version]
- Hudson, M.A.; Siegele, D.A.; Lockless, S.W. Use of a Fluorescence-Based Assay To Measure Escherichia coli Membrane Potential Changes in High Throughput. Antimicrob. Agents Chemother. 2020, 64, e00910-20. [Google Scholar] [CrossRef]
- Novo, D.; Perlmutter, N.G.; Hunt, R.H.; Shapiro, H.M. Accurate flow cytometric membrane potential measurement in bacteria using diethyloxacarbocyanine and a ratiometric technique. Cytometry 1999, 35, 55–63. [Google Scholar] [CrossRef]
- Kashket, E.R. The proton motive force in bacteria: A critical assessment of methods. Annu. Rev. Microbiol. 1985, 39, 219–242. [Google Scholar] [CrossRef]
- Llanes, C.; Hocquet, D.; Vogne, C.; Benali-Baitich, D.; Neuwirth, C.; Plesiat, P. Clinical strains of Pseudomonas aeruginosa overproducing MexAB-OprM and MexXY efflux pumps simultaneously. Antimicrob. Agents Chemother. 2004, 48, 1797–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moosavi, S.M.; Pouresmaeil, O.; Zandi, H.; Emadi, S.; Akhavan, F.; Torki, A.; Astani, A. The Evaluation of Antibiotic Resistance and nalB Mutants in Pseudomonas eruginosa Isolated from Burnt Patients of Shohada Mehrab Yazd Hospital Burn Ward. Rep. Biochem. Mol. Biol. 2020, 9, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Boutoille, D.; Corvec, S.; Caroff, N.; Giraudeau, C.; Espaze, E.; Caillon, J.; Plesiat, P.; Reynaud, A. Detection of an IS21 insertion sequence in the mexR gene of Pseudomonas aeruginosa increasing beta-lactam resistance. FEMS Microbiol. Lett. 2004, 230, 143–146. [Google Scholar] [CrossRef]
- Jalal, S.; Wretlind, B. Mechanisms of quinolone resistance in clinical strains of Pseudomonas aeruginosa. Microb. Drug. Resist. 1998, 4, 257–261. [Google Scholar] [CrossRef]
- Aghazadeh, M.; Hojabri, Z.; Mahdian, R.; Nahaei, M.R.; Rahmati, M.; Hojabri, T.; Pirzadeh, T.; Pajand, O. Role of efflux pumps: MexAB-OprM and MexXY(-OprA), AmpC cephalosporinase and OprD porin in non-metallo-beta-lactamase producing Pseudomonas aeruginosa isolated from cystic fibrosis and burn patients. Infect. Genet. Evol. 2014, 24, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Castanheira, M.; Mills, J.C.; Farrell, D.J.; Jones, R.N. Mutation-driven beta-lactam resistance mechanisms among contemporary ceftazidime-nonsusceptible Pseudomonas aeruginosa isolates from U.S. hospitals. Antimicrob. Agents Chemother. 2014, 58, 6844–6850. [Google Scholar] [CrossRef] [Green Version]
- Suresh, M.; Nithya, N.; Jayasree, P.R.; Vimal, K.P.; Manish Kumar, P.R. Mutational analyses of regulatory genes, mexR, nalC, nalD and mexZ of mexAB-oprM and mexXY operons, in efflux pump hyperexpressing multidrug-resistant clinical isolates of Pseudomonas aeruginosa. World J. Microbiol. Biotechnol. 2018, 34, 83. [Google Scholar] [CrossRef]
- Chopra, I.; Roberts, M. Tetracycline antibiotics: Mode of action, applications, molecular biology, and epidemiology of bacterial resistance. Microbiol. Mol. Biol. Rev. 2001, 65, 232–260. [Google Scholar] [CrossRef] [Green Version]
- Dinos, G.P.; Athanassopoulos, C.M.; Missiri, D.A.; Giannopoulou, P.C.; Vlachogiannis, I.A.; Papadopoulos, G.E.; Papaioannou, D.; Kalpaxis, D.L. Chloramphenicol Derivatives as Antibacterial and Anticancer Agents: Historic Problems and Current Solutions. Antibiotics 2016, 5, 20. [Google Scholar] [CrossRef]
- Kalkut, G. Sulfonamides and trimethoprim. Cancer Investig. 1998, 16, 612–615. [Google Scholar] [CrossRef]
- Gleckman, R.; Blagg, N.; Joubert, D.W. Trimethoprim: Mechanisms of action, antimicrobial activity, bacterial resistance, pharmacokinetics, adverse reactions, and therapeutic indications. Pharmacotherapy 1981, 1, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Elowe, N.H.; Blanchard, J.E.; Cechetto, J.D.; Brown, E.D. Experimental screening of dihydrofolate reductase yields a “test set” of 50,000 small molecules for a computational data-mining and docking competition. J. Biomol. Screen. 2005, 10, 653–657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wrobel, A.; Baradyn, M.; Ratkiewicz, A.; Drozdowska, D. Synthesis, Biological Activity, and Molecular Dynamics Study of Novel Series of a Trimethoprim Analogs as Multi-Targeted Compounds: Dihydrofolate Reductase (DHFR) Inhibitors and DNA-Binding Agents. Int. J. Mol. Sci. 2021, 22, 3685. [Google Scholar] [CrossRef]
- CDC and FDA Antibiotic Resistance Isolate Bank. Available online: https://wwwn.cdc.gov/ARIsolateBank/Panel/PanelDetail?ID=12 (accessed on 6 April 2022).
- Braz, V.S.; Furlan, J.P.; Fernandes, A.F.; Stehling, E.G. Mutations in NalC induce MexAB-OprM overexpression resulting in high level of aztreonam resistance in environmental isolates of Pseudomonas aeruginosa. FEMS Microbiol. Lett. 2016, 363, fnw166. [Google Scholar] [CrossRef] [Green Version]
- Maeda, T.; Garcia-Contreras, R.; Pu, M.; Sheng, L.; Garcia, L.R.; Tomas, M.; Wood, T.K. Quorum quenching quandary: Resistance to antivirulence compounds. ISME J. 2012, 6, 493–501. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.P.; Xu, Y.H.; Wang, Z.X.; Fang, Y.P.; Shen, J.L. Overexpression of MexAB-OprM efflux pump in carbapenem-resistant Pseudomonas aeruginosa. Arch. Microbiol. 2016, 198, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, D.; Ghose, A.; Dhar Chanda, D.; Das Talukdar, A.; Dutta Choudhury, M.; Paul, D.; Maurya, A.P.; Chakravarty, A.; Bhattacharjee, A. Premature Termination of MexR Leads to Overexpression of MexAB-OprM Efflux Pump in Pseudomonas aeruginosa in a Tertiary Referral Hospital in India. PLoS ONE 2016, 11, e0149156. [Google Scholar] [CrossRef]
- Pai, H.; Kim, J.; Kim, J.; Lee, J.H.; Choe, K.W.; Gotoh, N. Carbapenem resistance mechanisms in Pseudomonas aeruginosa clinical isolates. Antimicrob. Agents Chemother. 2001, 45, 480–484. [Google Scholar] [CrossRef] [Green Version]
- Bruchmann, S.; Dotsch, A.; Nouri, B.; Chaberny, I.F.; Haussler, S. Quantitative contributions of target alteration and decreased drug accumulation to Pseudomonas aeruginosa fluoroquinolone resistance. Antimicrob. Agents Chemother. 2013, 57, 1361–1368. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, K.V.; Nguyen, T.V.; Nguyen, H.T.T.; Le, D.V. Mutations in the gyrA, parC, and mexR genes provide functional insights into the fluoroquinolone-resistant Pseudomonas aeruginosa isolated in Vietnam. Infect. Drug Resist. 2018, 11, 275–282. [Google Scholar] [CrossRef] [Green Version]
- Kriengkauykiat, J.; Porter, E.; Lomovskaya, O.; Wong-Beringer, A. Use of an efflux pump inhibitor to determine the prevalence of efflux pump-mediated fluoroquinolone resistance and multidrug resistance in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2005, 49, 565–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliver, A.; Canton, R.; Campo, P.; Baquero, F.; Blazquez, J. High frequency of hypermutable Pseudomonas aeruginosa in cystic fibrosis lung infection. Science 2000, 288, 1251–1254. [Google Scholar] [CrossRef] [PubMed]
- Macfarlane, E.L.A.; Kwasnicka, A.; Hancock, R.E.W. Role of Pseudomonas aeruginosa PhoP-phoQ in resistance to antimicrobial cationic peptides and aminoglycosides. Microbiology 2000, 146, 2543–2554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gooderham, W.J.; Gellatly, S.L.; Sanschagrin, F.; McPhee, J.B.; Bains, M.; Cosseau, C.; Levesque, R.C.; Hancock, R.E.W. The sensor kinase PhoQ mediates virulence in Pseudomonas aeruginosa. Microbiology 2009, 155, 699–711. [Google Scholar] [CrossRef] [Green Version]
- Hirvas, L.; Coleman, J.; Koski, P.; Vaara, M. Bacterial ‘histone-like protein I’ (HLP-I) is an outer membrane constituent? FEBS Lett. 1990, 262, 123–126. [Google Scholar] [CrossRef] [Green Version]
- Peng, X.; Xu, C.; Ren, H.; Lin, X.; Wu, L.; Wang, S. Proteomic analysis of the sarcosine-insoluble outer membrane fraction of Pseudomonas aeruginosa responding to ampicilin, kanamycin, and tetracycline resistance. J. Proteome Res. 2005, 4, 2257–2265. [Google Scholar] [CrossRef]
- Dotsch, A.; Becker, T.; Pommerenke, C.; Magnowska, Z.; Jansch, L.; Haussler, S. Genomewide identification of genetic determinants of antimicrobial drug resistance in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2009, 53, 2522–2531. [Google Scholar] [CrossRef] [Green Version]
- Goemans, C.; Denoncin, K.; Collet, J.F. Folding mechanisms of periplasmic proteins. Biochim. Biophys. Acta 2014, 1843, 1517–1528. [Google Scholar] [CrossRef] [Green Version]
- Jarchow, S.; Luck, C.; Gorg, A.; Skerra, A. Identification of potential substrate proteins for the periplasmic Escherichia coli chaperone Skp. Proteomics 2008, 8, 4987–4994. [Google Scholar] [CrossRef]
- Babu, M.; Diaz-Mejia, J.J.; Vlasblom, J.; Gagarinova, A.; Phanse, S.; Graham, C.; Yousif, F.; Ding, H.; Xiong, X.; Nazarians-Armavil, A.; et al. Genetic interaction maps in Escherichia coli reveal functional crosstalk among cell envelope biogenesis pathways. PLoS Genet. 2011, 7, e1002377. [Google Scholar] [CrossRef]
- Rosenberg, E.Y.; Ma, D.; Nikaido, H. AcrD of Escherichia coli is an aminoglycoside efflux pump. J. Bacteriol. 2000, 182, 1754–1756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srikumar, R.; Paul, C.J.; Poole, K. Influence of mutations in the mexR repressor gene on expression of the MexA-MexB-oprM multidrug efflux system of Pseudomonas aeruginosa. J. Bacteriol. 2000, 182, 1410–1414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morita, Y.; Sobel, M.L.; Poole, K. Antibiotic inducibility of the MexXY multidrug efflux system of Pseudomonas aeruginosa: Involvement of the antibiotic-inducible PA5471 gene product. J. Bacteriol. 2006, 188, 1847–1855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afgan, E.; Baker, D.; Batut, B.; van den Beek, M.; Bouvier, D.; Cech, M.; Chilton, J.; Clements, D.; Coraor, N.; Gruning, B.A.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res 2018, 46, W537–W544. [Google Scholar] [CrossRef] [Green Version]
- Cohen, S.P.; McMurry, L.M.; Hooper, D.C.; Wolfson, J.S.; Levy, S.B. Cross-resistance to fluoroquinolones in multiple-antibiotic-resistant (Mar) Escherichia coli selected by tetracycline or chloramphenicol: Decreased drug accumulation associated with membrane changes in addition to OmpF reduction. Antimicrob. Agents Chemother. 1989, 33, 1318–1325. [Google Scholar] [CrossRef] [Green Version]
- Ma, D.; Cook, D.N.; Alberti, M.; Pon, N.G.; Nikaido, H.; Hearst, J.E. Molecular cloning and characterization of acrA and acrE genes of Escherichia coli. J. Bacteriol. 1993, 175, 6299–6313. [Google Scholar] [CrossRef] [Green Version]
- Gajdacs, M.; Barath, Z.; Karpati, K.; Szabo, D.; Usai, D.; Zanetti, S.; Donadu, M.G. No Correlation between Biofilm Formation, Virulence Factors, and Antibiotic Resistance in Pseudomonas aeruginosa: Results from a Laboratory-Based In Vitro Study. Antibiotics 2021, 10, 1134. [Google Scholar] [CrossRef]
- Fang, Y.; Baloch, Z.; Zhang, W.; Hu, Y.; Zheng, R.; Song, Y.; Tai, W.; Xia, X. Emergence of Carbapenem-Resistant ST244, ST292, and ST2446 Pseudomonas aeruginosa Clones in Burn Patients in Yunnan Province. Infect. Drug Resist. 2022, 15, 1103–1114. [Google Scholar] [CrossRef]
- Tjandra, K.C.; Ram-Mohan, N.; Abe, R.; Hashemi, M.M.; Lee, J.H.; Chin, S.M.; Roshardt, M.A.; Liao, J.C.; Wong, P.K.; Yang, S. Diagnosis of Bloodstream Infections: An Evolution of Technologies towards Accurate and Rapid Identification and Antibiotic Susceptibility Testing. Antibiotics 2022, 11, 511. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antimicrobial | MICs (µg/mL) | ||
|---|---|---|---|
| Lack of EPI | +6.25 µg/mL of TXA01182 (Fold Change) | +6.25 µg/mL of TXA09155 (Fold Change) | |
| Moxifloxacin | 2 | 0.063 (32) | 0.031 (16) |
| Levofloxacin | 1 | 0.125 (8) | 0.063 (16) |
| Doxycycline | 32 | 2 (16) | 1 (32) |
| Minocycline | 32 | 1 (32) | 0.5 (64) |
| Chloramphenicol | >256 | 32 (>8) | 16 (>16) |
| Antibiotics | Antibiotic MIC (μg/mL) in Presence of TXA09155 ‡ at a Concentration (μg/mL) of: | Emax * | EC50 ** | FICindex | ||||
|---|---|---|---|---|---|---|---|---|
| 0 | 3.13 | 6.25 | 12.5 | 25 | ||||
| Levofloxacin | 1 | 1 | 0.063 | 0.063 | 0.063 | 16 | 6.25 | 0.188 |
| Moxifloxacin | 2 | 2 | 0.031 | 0.031 | 0.016 | 125 | 6.25 | 0.14 |
| Doxycycline | 32 | 16 | 1 | 1 | 1 | 32 | 6.25 | 0.156 |
| Minocycline | 32 | 16 | 0.5 | 0.25 | 0.125 | 256 | 6.25 | 0.14 |
| Cefpirome | 8 | 4 | 1 | 0.5 | 0.5 | 16 | 6.25 | 0.25 |
| Aztreonam | 8 | 4 | 2 | 0.25 | 0.25 | 32 | 6.25 | 0.125 |
| Chloramphenicol | >256 | >256 | 16 | 4 | 2 | >128 | 6.25 | 0.187 |
| Cotrimoxazole | 256 | 256 | 8 | 4 | 2 | 128 | 6.25 | 0.156 |
| Imipenem # | 2 | 2 | 2 | 2 | 2 | 1 | ND | ND |
| Antibiotics | MIC Ratios | |||
|---|---|---|---|---|
| K1455 (↑mexAB-oprM)/K3698 (∆oprM) | K1455 (↑mexAB-oprM) ± TXA09155 # | K2415 (↑mexXY-oprM) ± TXA09155 # | K3698 (∆oprM) ± TXA09155 # | |
| Cefpirome | 4 | 8 | 8 | 2 |
| Levofloxacin | 8 | 16 | 16 | 2 |
| Cotrimoxazole | 8 | 32 | 16 | 4 |
| Doxycycline | >8 | ≥64 | 32 | 8 |
| Minocycline | >4 | >128 | 64 | 16 |
| Chloramphenicol | 4 | 32 | 16 | 8 |
| Imipenem * | 1 | 1 | 1 | 1 |
| Strain | Levofloxacin (4 μg/mL) | Levofloxacin (4 μg/mL) + TXA01182 (12.5 μg/mL) |
|---|---|---|
| P. aeruginosa ATCC 27853 | 4.97 × 10−7 | <3.33 × 10−9 |
| Resistance to: | Parent Strain | FoR | Strain Name | Mutation | Gene Role |
|---|---|---|---|---|---|
| TXA09155 alone (4×-MIC) | P. aeruginosa ATCC 27853 | 1.40 × 10−6 | EPIR1S | phoQ-L175R | Two-component regulatory system |
| EPIR9S | phoQ-L175R | ||||
| EPIR20L | phoQ-H248D | ||||
| TXA09155 alone (1×-MIC) | 1.90 × 10−3 | EPIR43 | ompH-Q127X * | Skp-like periplasmic chaperone | |
| TXA09155 (⅛×-MIC) + LVX (1×-MIC) | P. aeruginosa DA7232 | 2.48 × 10−8 | EPIR24L | trpS-R171S | Tryptophan-tRNA ligase |
| MW | Solubility at pH 7.4 (μM) | cLogP | HLM and RLM Metabolic Stability | CYP Inhibition IC50 (μM) | ||||
|---|---|---|---|---|---|---|---|---|
| CYP 1A2 | CYP 2C19 | CYP 2C9 | CYP 2D6 | CYP 3A4 | ||||
| 366 | 155 ± 3 | 2.6 | >60% | >100 | >100 | >100 | 67.2 | 28.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Rosado-Lugo, J.D.; Datta, P.; Sun, Y.; Cao, Y.; Banerjee, A.; Yuan, Y.; Parhi, A.K. Evaluation of a Conformationally Constrained Indole Carboxamide as a Potential Efflux Pump Inhibitor in Pseudomonas aeruginosa. Antibiotics 2022, 11, 716. https://doi.org/10.3390/antibiotics11060716
Zhang Y, Rosado-Lugo JD, Datta P, Sun Y, Cao Y, Banerjee A, Yuan Y, Parhi AK. Evaluation of a Conformationally Constrained Indole Carboxamide as a Potential Efflux Pump Inhibitor in Pseudomonas aeruginosa. Antibiotics. 2022; 11(6):716. https://doi.org/10.3390/antibiotics11060716
Chicago/Turabian StyleZhang, Yongzheng, Jesus D. Rosado-Lugo, Pratik Datta, Yangsheng Sun, Yanlu Cao, Anamika Banerjee, Yi Yuan, and Ajit K. Parhi. 2022. "Evaluation of a Conformationally Constrained Indole Carboxamide as a Potential Efflux Pump Inhibitor in Pseudomonas aeruginosa" Antibiotics 11, no. 6: 716. https://doi.org/10.3390/antibiotics11060716
APA StyleZhang, Y., Rosado-Lugo, J. D., Datta, P., Sun, Y., Cao, Y., Banerjee, A., Yuan, Y., & Parhi, A. K. (2022). Evaluation of a Conformationally Constrained Indole Carboxamide as a Potential Efflux Pump Inhibitor in Pseudomonas aeruginosa. Antibiotics, 11(6), 716. https://doi.org/10.3390/antibiotics11060716