
The Mechanism of Bacterial Resistance and Potential Bacteriostatic Strategies

Abstract
:1. Introduction
2. Mechanism of Antibiotics Resistance
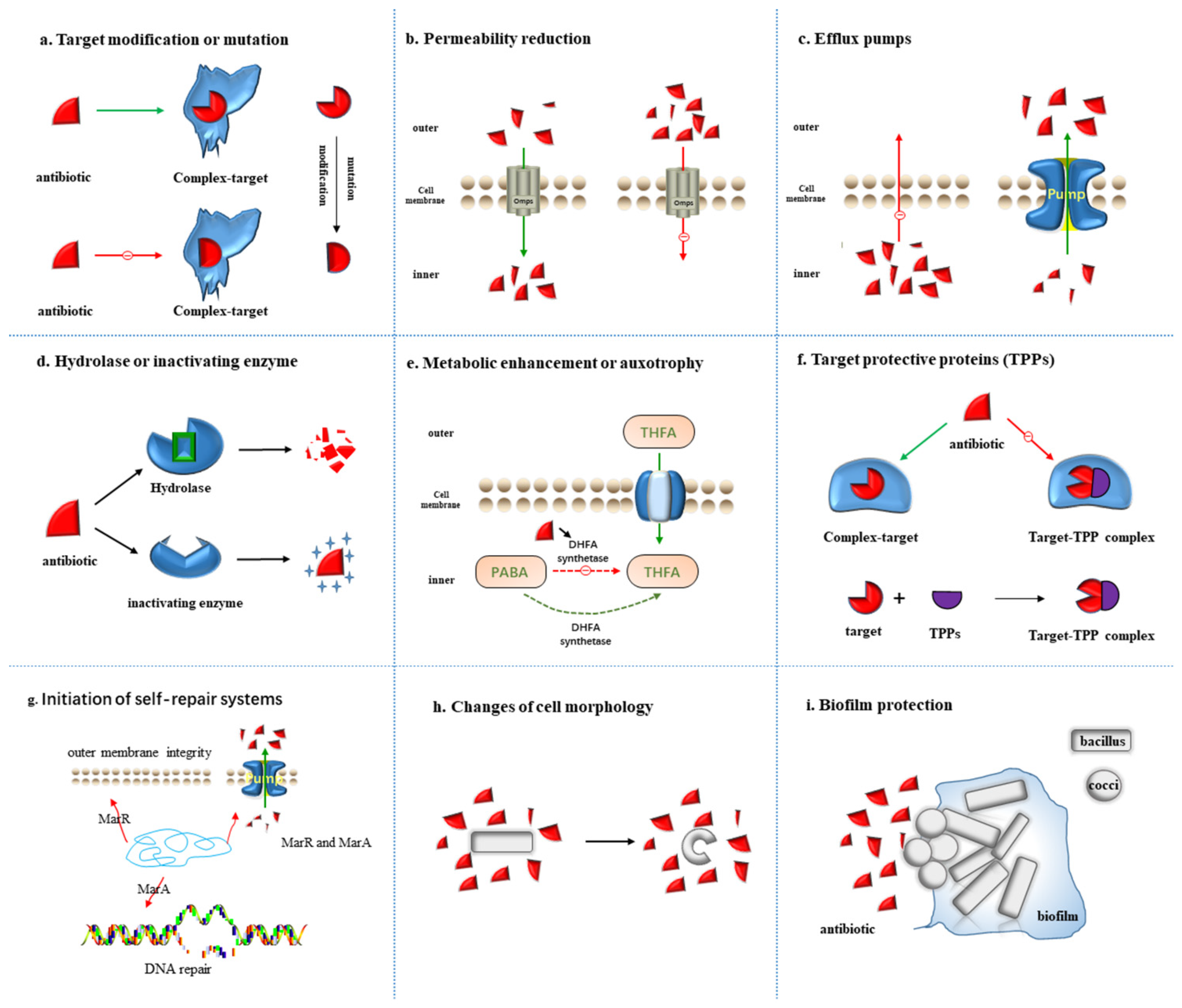
2.1. Target Modification or Mutation
2.2. Permeability Reduction
2.3. Efflux Pumps
2.4. Hydrolase or Inactivating Enzyme
2.5. Metabolic Alteration or Auxotrophy
2.6. Target Protective Proteins (TPPs)
2.7. Initiation of Self-Repair Systems
2.8. Changes of Cell Morphology
2.9. Biofilm Protection
3. Antibacterial Methods
3.1. Newly Potential Bacteriostatic Compound Molecule
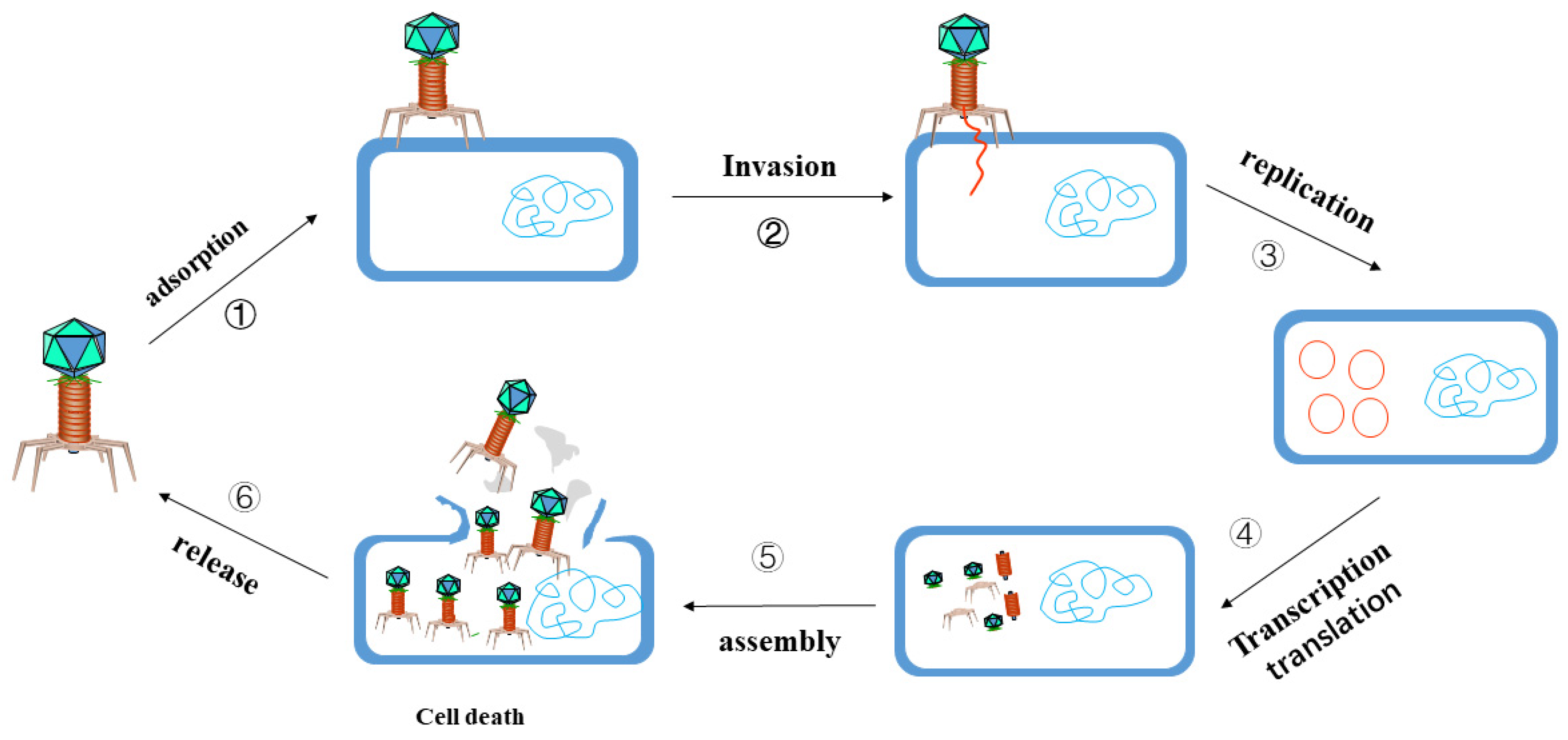
3.2. Phage Targeted Elimination of MDR Bacteria
3.2.1. The Function of Phage Endolysin
3.2.2. Phage and Antibiotic Combination Therapy
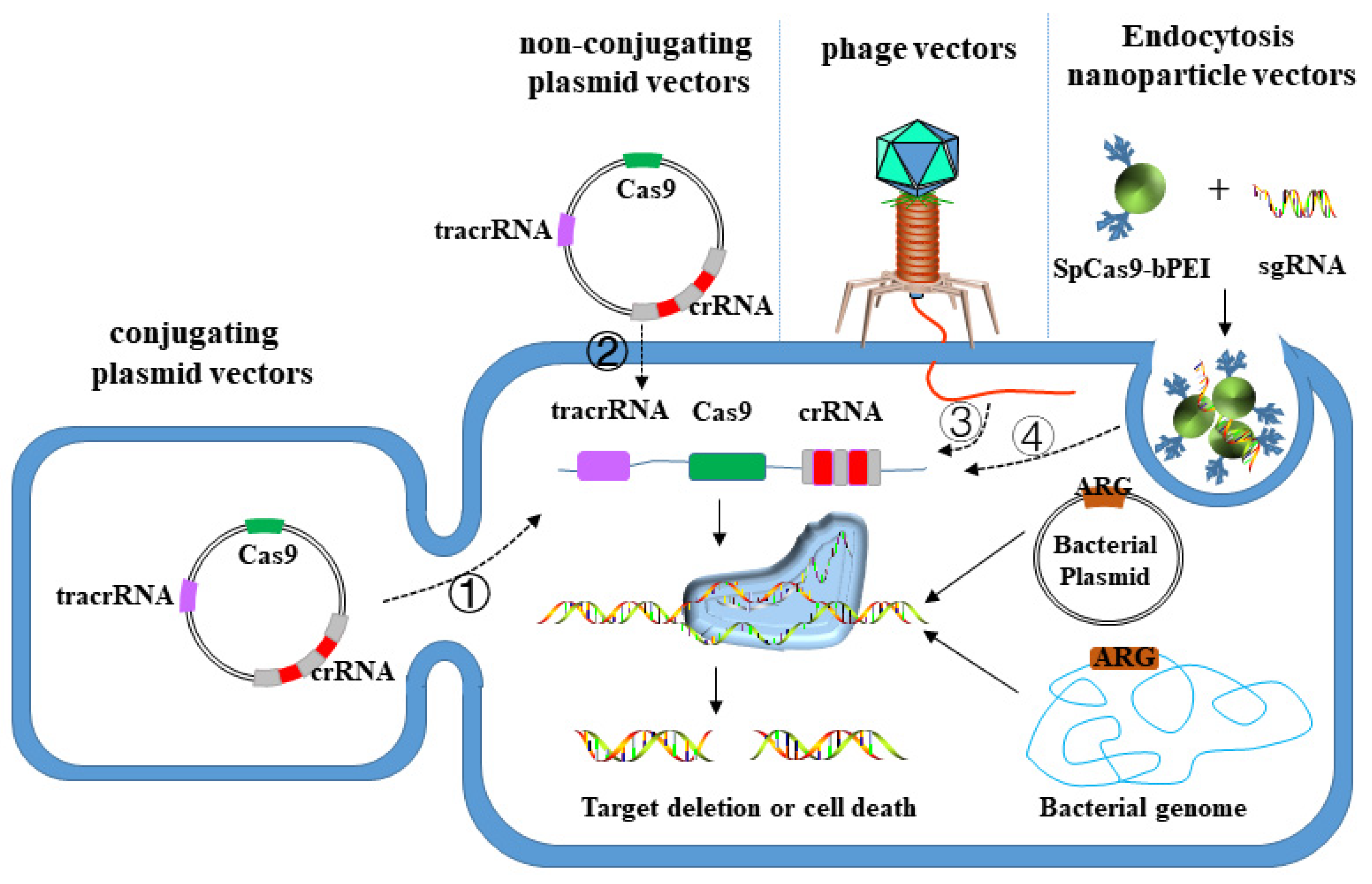
3.3. CRISPR-Cas System Targeted Elimination of MDR Bacteria
3.3.1. Plasmid Vector
3.3.2. Phage Vector
3.3.3. Nanoparticle Vector
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Roope, L.S.J.; Smith, R.D.; Pouwels, K.B.; Buchanan, J.; Abel, L.; Eibich, P.; Butler, C.C.; Tan, P.S.; Walker, A.S.; Robotham, J.V.; et al. The challenge of antimicrobial resistance: What economics can contribute. Science 2019, 364, eaau4679. [Google Scholar] [CrossRef] [PubMed]
- Theuretzbacher, U.; Gottwalt, S.; Beyer, P.; Butler, M.; Czaplewski, L.; Lienhardt, C.; Moja, L.; Paul, M.; Paulin, S.; Rex, J.H.; et al. Analysis of the clinical antibacterial and antituberculosis pipeline. Lancet Infect. Dis. 2019, 19, e40–e50. [Google Scholar] [CrossRef]
- Zhang, Q.Q.; Ying, G.G.; Pan, C.G.; Liu, Y.S.; Zhao, J.L. Comprehensive evaluation of antibiotics emission and fate in the river basins of China: Source analysis, multimedia modeling, and linkage to bacterial resistance. Env. Sci. Technol. 2015, 49, 6772–6782. [Google Scholar] [CrossRef] [PubMed]
- Antimicrobial Resistance, C. Global burden of bacterial antimicrobial resistance in 2019: A systematic analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef]
- Diseases, G.B.D.; Injuries, C. Global burden of 369 diseases and injuries in 204 countries and territories, 1990-2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet 2020, 396, 1204–1222. [Google Scholar] [CrossRef]
- Fleming, A. On the antibacterial action of cultures of a penicillium, with special reference to their use in the isolation of B. influenzae. 1929. Bull. World Health Organ. 2001, 79, 780–790. [Google Scholar]
- Chain, E.; Florey, H.W.; Gardner, A.D.; Heatley, N.G.; Jennings, M.A.; Orr-Ewing, J.; Sanders, A.G. Penicillin as a chemotherapeutic agent. Lancet 1940, 236, 226–228. [Google Scholar] [CrossRef]
- Lee, A.S.; de Lencastre, H.; Garau, J.; Kluytmans, J.; Malhotra-Kumar, S.; Peschel, A.; Harbarth, S. Methicillin-resistant Staphylococcus aureus. Nat. Reviews. Dis. Primers 2018, 4, 18033. [Google Scholar] [CrossRef]
- Yong, D.; Toleman, M.A.; Giske, C.G.; Cho, H.S.; Sundman, K.; Lee, K.; Walsh, T.R. Characterization of a new metallo-beta-lactamase gene, bla(NDM-1), and a novel erythromycin esterase gene carried on a unique genetic structure in Klebsiella pneumoniae sequence type 14 from India. Antimicrob. Agents Chemother. 2009, 53, 5046–5054. [Google Scholar] [CrossRef]
- Liu, Y.Y.; Wang, Y.; Walsh, T.R.; Yi, L.X.; Zhang, R.; Spencer, J.; Doi, Y.; Tian, G.; Dong, B.; Huang, X.; et al. Emergence of plasmid-mediated colistin resistance mechanism MCR-1 in animals and human beings in China: A microbiological and molecular biological study. Lancet. Infect. Dis. 2016, 16, 161–168. [Google Scholar] [CrossRef]
- Who Publishes List Of Bacteria for Which New Antibiotics Are Urgently Needed. Saudi Med. J. 2017, 38, 444–445.
- Mohr, K.I. History of Antibiotics Research. Curr. Top Microbiol. Immunol. 2016, 398, 237–272. [Google Scholar] [CrossRef]
- Lewis, K. The Science of Antibiotic Discovery. Cell 2020, 181, 29–45. [Google Scholar] [CrossRef] [PubMed]
- D’Costa, V.M.; King, C.E.; Kalan, L.; Morar, M.; Sung, W.W.; Schwarz, C.; Froese, D.; Zazula, G.; Calmels, F.; Debruyne, R.; et al. Antibiotic resistance is ancient. Nature 2011, 477, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Lebreton, F.; Manson, A.L.; Saavedra, J.T.; Straub, T.J.; Earl, A.M.; Gilmore, M.S. Tracing the Enterococci from Paleozoic Origins to the Hospital. Cell 2017, 169, 849–861.e813. [Google Scholar] [CrossRef] [PubMed]
- Shukla, R.; Medeiros-Silva, J.; Parmar, A.; Vermeulen, B.J.A.; Das, S.; Paioni, A.L.; Jekhmane, S.; Lorent, J.; Bonvin, A.; Baldus, M.; et al. Mode of action of teixobactins in cellular membranes. Nat. Commun. 2020, 11, 2848. [Google Scholar] [CrossRef]
- Luther, A.; Urfer, M.; Zahn, M.; Muller, M.; Wang, S.Y.; Mondal, M.; Vitale, A.; Hartmann, J.B.; Sharpe, T.; Monte, F.L.; et al. Chimeric peptidomimetic antibiotics against Gram-negative bacteria. Nature 2019, 576, 452–458. [Google Scholar] [CrossRef]
- Smith, P.A.; Koehler, M.F.T.; Girgis, H.S.; Yan, D.; Chen, Y.; Chen, Y.; Crawford, J.J.; Durk, M.R.; Higuchi, R.I.; Kang, J.; et al. Optimized arylomycins are a new class of Gram-negative antibiotics. Nature 2018, 561, 189–194. [Google Scholar] [CrossRef]
- Culp, E.J.; Waglechner, N.; Wang, W.; Fiebig-Comyn, A.A.; Hsu, Y.P.; Koteva, K.; Sychantha, D.; Coombes, B.K.; Van Nieuwenhze, M.S.; Brun, Y.V.; et al. Evolution-guided discovery of antibiotics that inhibit peptidoglycan remodelling. Nature 2020, 578, 582–587. [Google Scholar] [CrossRef]
- Mitcheltree, M.J.; Pisipati, A.; Syroegin, E.A.; Silvestre, K.J.; Klepacki, D.; Mason, J.D.; Terwilliger, D.W.; Testolin, G.; Pote, A.R.; Wu, K.J.Y.; et al. A synthetic antibiotic class overcoming bacterial multidrug resistance. Nature 2021, 599, 507–512. [Google Scholar] [CrossRef]
- Dethlefsen, L.; Huse, S.; Sogin, M.L.; Relman, D.A. The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol. 2008, 6, e280. [Google Scholar] [CrossRef] [PubMed]
- Trudil, D. Phage lytic enzymes: A history. Virol. Sin. 2015, 30, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Duzgunes, N.; Sessevmez, M.; Yildirim, M. Bacteriophage Therapy of Bacterial Infections: The Rediscovered Frontier. Pharmaceuticals 2021, 14, 34. [Google Scholar] [CrossRef]
- Schooley, R.T.; Biswas, B.; Gill, J.J.; Hernandez-Morales, A.; Lancaster, J.; Lessor, L.; Barr, J.J.; Reed, S.L.; Rohwer, F.; Benler, S.; et al. Development and Use of Personalized Bacteriophage-Based Therapeutic Cocktails To Treat a Patient with a Disseminated Resistant Acinetobacter baumannii Infection. Antimicrob. Agents Chemother. 2017, 61, e00954-17. [Google Scholar] [CrossRef] [PubMed]
- Nir-Paz, R.; Gelman, D.; Khouri, A.; Sisson, B.M.; Fackler, J.; Alkalay-Oren, S.; Khalifa, L.; Rimon, A.; Yerushalmy, O.; Bader, R.; et al. Successful Treatment of Antibiotic-resistant, Poly-microbial Bone Infection With Bacteriophages and Antibiotics Combination. Clin. Infect Dis. 2019, 69, 2015–2018. [Google Scholar] [CrossRef]
- Eskenazi, A.; Lood, C.; Wubbolts, J.; Hites, M.; Balarjishvili, N.; Leshkasheli, L.; Askilashvili, L.; Kvachadze, L.; van Noort, V.; Wagemans, J.; et al. Combination of pre-adapted bacteriophage therapy and antibiotics for treatment of fracture-related infection due to pandrug-resistant Klebsiella pneumoniae. Nat. Commun. 2022, 13, 302. [Google Scholar] [CrossRef]
- Du, D.; Wang-Kan, X.; Neuberger, A.; van Veen, H.W.; Pos, K.M.; Piddock, L.J.V.; Luisi, B.F. Multidrug efflux pumps: Structure, function and regulation. Nat. Rev. Microbiol. 2018, 16, 523–539. [Google Scholar] [CrossRef]
- Davies, J. Origins and evolution of antibiotic resistance. Microbiologia 1996, 12, 9–16. [Google Scholar] [CrossRef]
- Goldstein, B.P. Resistance to rifampicin: A review. J. Antibiot. 2014, 67, 625–630. [Google Scholar] [CrossRef]
- Ahmad, S.; Mokaddas, E.; Fares, E. Characterization of rpoB mutations in rifampin-resistant clinical Mycobacterium tuberculosis isolates from Kuwait and Dubai. Diagn. Microbiol. Infect. Dis. 2002, 44, 245–252. [Google Scholar] [CrossRef]
- Chen, L.; Gan, X.; Li, N.; Wang, J.; Li, K.; Zhang, H. rpoB gene mutation profile in rifampicin-resistant Mycobacterium tuberculosis clinical isolates from Guizhou, one of the highest incidence rate regions in China. J. Antimicrob. Chemother. 2010, 65, 1299–1301. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Hu, Z.; Zhao, Y.; Cai, X.; Luo, C.; Zou, C.; Liu, X. The beginning of the rpoB gene in addition to the rifampin resistance determination region might be needed for identifying rifampin/rifabutin cross-resistance in multidrug-resistant Mycobacterium tuberculosis isolates from Southern China. J. Clin. Microbiol. 2012, 50, 81–85. [Google Scholar] [CrossRef] [Green Version]
- Siu, G.K.; Zhang, Y.; Lau, T.C.; Lau, R.W.; Ho, P.L.; Yew, W.W.; Tsui, S.K.; Cheng, V.C.; Yuen, K.Y.; Yam, W.C. Mutations outside the rifampicin resistance-determining region associated with rifampicin resistance in Mycobacterium tuberculosis. J. Antimicrob. Chemother. 2011, 66, 730–733. [Google Scholar] [CrossRef]
- Miyachiro, M.M.; Contreras-Martel, C.; Dessen, A. Penicillin-Binding Proteins (PBPs) and Bacterial Cell Wall Elongation Complexes. Subcell. Biochem. 2019, 93, 273–289. [Google Scholar] [CrossRef] [PubMed]
- Miragaia, M. Factors Contributing to the Evolution of mecA-Mediated β-lactam Resistance in Staphylococci: Update and New Insights From Whole Genome Sequencing (WGS). Front. Microbiol. 2018, 9, 2723. [Google Scholar] [CrossRef]
- Nikaido, H. Molecular basis of bacterial outer membrane permeability revisited. Microbiol. Mol. Biol. Rev. 2003, 67, 593–656. [Google Scholar] [CrossRef] [PubMed]
- Chevalier, S.; Bouffartigues, E.; Bodilis, J.; Maillot, O.; Lesouhaitier, O.; Feuilloley, M.G.J.; Orange, N.; Dufour, A.; Cornelis, P. Structure, function and regulation of Pseudomonas aeruginosa porins. FEMS Microbiol. Rev. 2017, 41, 698–722. [Google Scholar] [CrossRef]
- McPhee, J.B.; Tamber, S.; Brazas, M.D.; Lewenza, S.; Hancock, R.E.W. Antibiotic Resistance Due to Reduced Uptake. In Antimicrobial Drug Resistance: Mechanisms of Drug Resistance; Mayers, D.L., Ed.; Humana Press: Totowa, NJ, USA, 2009; pp. 97–110. [Google Scholar]
- Nadeem, S.F.; Gohar, U.F.; Tahir, S.F.; Mukhtar, H.; Pornpukdeewattana, S.; Nukthamna, P.; Moula Ali, A.M.; Bavisetty, S.C.B.; Massa, S. Antimicrobial resistance: More than 70 years of war between humans and bacteria. Crit. Rev. Microbiol. 2020, 46, 578–599. [Google Scholar] [CrossRef]
- Moya-Torres, A.; Mulvey, M.R.; Kumar, A.; Oresnik, I.J.; Brassinga, A.K.C. The lack of OmpF, but not OmpC, contributes to increased antibiotic resistance in Serratia marcescens. Microbiol. (Read. Engl.) 2014, 160, 1882–1892. [Google Scholar] [CrossRef]
- Ziervogel, B.K.; Roux, B. The Binding of Antibiotics in OmpF Porin. Structure 2013, 21, 76–87. [Google Scholar] [CrossRef]
- Bafna, J.A.; Sans-Serramitjana, E.; Acosta-Gutiérrez, S.; Bodrenko, I.V.; Hörömpöli, D.; Berscheid, A.; Brötz-Oesterhelt, H.; Winterhalter, M.; Ceccarelli, M. Kanamycin Uptake into Escherichia coli Is Facilitated by OmpF and OmpC Porin Channels Located in the Outer Membrane. ACS Infect. Dis. 2020, 6, 1855–1865. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Chang, S.; Qiao, W.; Luo, Q.; Chen, Y.; Jia, Z.; Coleman, J.; Zhang, K.; Wang, T.; Zhang, Z.; et al. Structural insights into outer membrane asymmetry maintenance in Gram-negative bacteria by MlaFEDB. Nat. Struct. Mol. Biol. 2021, 28, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Rahman, T.; Yarnall, B.; Doyle, D.A. Efflux drug transporters at the forefront of antimicrobial resistance. Eur. Biophys. J. EBJ 2017, 46, 647–653. [Google Scholar] [CrossRef] [PubMed]
- Amaral, L.; Martins, A.; Spengler, G.; Molnar, J. Efflux pumps of Gram-negative bacteria: What they do, how they do it, with what and how to deal with them. Front. Pharm. 2014, 4, 168. [Google Scholar] [CrossRef]
- Blanco, P.; Hernando-Amado, S.; Reales-Calderon, J.A.; Corona, F.; Lira, F.; Alcalde-Rico, M.; Bernardini, A.; Sanchez, M.B.; Martinez, J.L. Bacterial Multidrug Efflux Pumps: Much More Than Antibiotic Resistance Determinants. Microorganisms 2016, 4, 14. [Google Scholar] [CrossRef]
- Hernando-Amado, S.; Blanco, P.; Alcalde-Rico, M.; Corona, F.; Reales-Calderón, J.A.; Sánchez, M.B.; Martínez, J.L. Multidrug efflux pumps as main players in intrinsic and acquired resistance to antimicrobials. Drug Resist. Updates Rev. Comment. Antimicrob. Anticancer. Chemother. 2016, 28, 13–27. [Google Scholar] [CrossRef]
- Li, X.Z.; Plesiat, P.; Nikaido, H. The challenge of efflux-mediated antibiotic resistance in Gram-negative bacteria. Clin. Microbiol. Rev. 2015, 28, 337–418. [Google Scholar] [CrossRef] [PubMed]
- Kourtesi, C.; Ball, A.R.; Huang, Y.Y.; Jachak, S.M.; Vera, D.M.; Khondkar, P.; Gibbons, S.; Hamblin, M.R.; Tegos, G.P. Microbial efflux systems and inhibitors: Approaches to drug discovery and the challenge of clinical implementation. Open Microbiol. J. 2013, 7, 34–52. [Google Scholar] [CrossRef]
- Costa, S.S.; Viveiros, M.; Amaral, L.; Couto, I. Multidrug Efflux Pumps in Staphylococcus aureus: An Update. Open Microbiol. J. 2013, 7, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Reygaert, W.C. An overview of the antimicrobial resistance mechanisms of bacteria. AIMS Microbiol. 2018, 4, 482–501. [Google Scholar] [CrossRef] [PubMed]
- Swick, M.C.; Morgan-Linnell, S.K.; Carlson, K.M.; Zechiedrich, L. Expression of multidrug efflux pump genes acrAB-tolC, mdfA, and norE in Escherichia coli clinical isolates as a function of fluoroquinolone and multidrug resistance. Antimicrob. Agents Chemother. 2011, 55, 921–924. [Google Scholar] [CrossRef] [PubMed]
- Hansen, L.H.; Jensen, L.B.; Sorensen, H.I.; Sorensen, S.J. Substrate specificity of the OqxAB multidrug resistance pump in Escherichia coli and selected enteric bacteria. J. Antimicrob. Chemother. 2007, 60, 145–147. [Google Scholar] [CrossRef] [PubMed]
- Peterson, E.; Kaur, P. Antibiotic Resistance Mechanisms in Bacteria: Relationships Between Resistance Determinants of Antibiotic Producers, Environmental Bacteria, and Clinical Pathogens. Front. Microbiol. 2018, 9, 2928. [Google Scholar] [CrossRef] [PubMed]
- Tooke, C.L.; Hinchliffe, P.; Bragginton, E.C.; Colenso, C.K.; Hirvonen, V.H.A.; Takebayashi, Y.; Spencer, J. β-Lactamases and β-Lactamase Inhibitors in the 21st Century. J. Mol. Biol. 2019, 431, 3472–3500. [Google Scholar] [CrossRef]
- Ghafourian, S.; Sadeghifard, N.; Soheili, S.; Sekawi, Z. Extended Spectrum Beta-lactamases: Definition, Classification and Epidemiology. Curr. Issues Mol. Biol. 2015, 17, 11–21. [Google Scholar]
- Sawa, T.; Kooguchi, K.; Moriyama, K. Molecular diversity of extended-spectrum β-lactamases and carbapenemases, and antimicrobial resistance. J. Intensive Care 2020, 8, 13. [Google Scholar] [CrossRef]
- Sköld, O. Aminoglycosides. In Antibiotics and Antibiotic Resistance; John Wiley & Sons: Hoboken, NJ, USA, 2011; Volume 6, pp. 103–113. [Google Scholar]
- Schwarz, S.; Shen, J.; Kadlec, K.; Wang, Y.; Brenner Michael, G.; Fessler, A.T.; Vester, B. Lincosamides, Streptogramins, Phenicols, and Pleuromutilins: Mode of Action and Mechanisms of Resistance. Cold Spring Harb. Perspect Med. 2016, 6, a027037. [Google Scholar] [CrossRef]
- Lopatkin, A.J.; Bening, S.C.; Manson, A.L.; Stokes, J.M.; Kohanski, M.A.; Badran, A.H.; Earl, A.M.; Cheney, N.J.; Yang, J.H.; Collins, J.J. Clinically relevant mutations in core metabolic genes confer antibiotic resistance. Science 2021, 371, eaba0862. [Google Scholar] [CrossRef]
- Zampieri, M. The genetic underground of antibiotic resistance. Science 2021, 371, 783–784. [Google Scholar] [CrossRef]
- Mee, M.T.; Collins, J.J.; Church, G.M.; Wang, H.H. Syntrophic exchange in synthetic microbial communities. Proc. Natl. Acad. Sci. USA 2014, 111, E2149–E2156. [Google Scholar] [CrossRef]
- Croft, M.T.; Lawrence, A.D.; Raux-Deery, E.; Warren, M.J.; Smith, A.G. Algae acquire vitamin B12 through a symbiotic relationship with bacteria. Nature 2005, 438, 90–93. [Google Scholar] [CrossRef] [PubMed]
- Sieuwerts, S.; Molenaar, D.; van Hijum, S.A.; Beerthuyzen, M.; Stevens, M.J.; Janssen, P.W.; Ingham, C.J.; de Bok, F.A.; de Vos, W.M.; van Hylckama Vlieg, J.E. Mixed-culture transcriptome analysis reveals the molecular basis of mixed-culture growth in Streptococcus thermophilus and Lactobacillus bulgaricus. Appl. Env. Microbiol. 2010, 76, 7775–7784. [Google Scholar] [CrossRef] [PubMed]
- Zelezniak, A.; Andrejev, S.; Ponomarova, O.; Mende, D.R.; Bork, P.; Patil, K.R. Metabolic dependencies drive species co-occurrence in diverse microbial communities. Proc. Natl. Acad. Sci. USA 2015, 112, 6449–6454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seif, Y.; Choudhary, K.S.; Hefner, Y.; Anand, A.; Yang, L.; Palsson, B.O. Metabolic and genetic basis for auxotrophies in Gram-negative species. Proc. Natl. Acad. Sci. USA 2020, 117, 6264–6273. [Google Scholar] [CrossRef]
- Sun, H.; Yao, Z.; Wang, D.; Wu, X.; Lin, Z.; Liu, Y. A deep insight into the toxic mechanism for sulfonamides based on bacterial cell-cell communication. Environ. Int. 2019, 129, 185–193. [Google Scholar] [CrossRef]
- Yu, J.S.L.; Correia-Melo, C.; Zorrilla, F.; Herrera-Dominguez, L.; Wu, M.Y.; Hartl, J.; Campbell, K.; Blasche, S.; Kreidl, M.; Egger, A.S.; et al. Microbial communities form rich extracellular metabolomes that foster metabolic interactions and promote drug tolerance. Nat. Microbiol. 2022, 7, 542–555. [Google Scholar] [CrossRef]
- Wilson, D.N.; Hauryliuk, V.; Atkinson, G.C.; O’Neill, A.J. Target protection as a key antibiotic resistance mechanism. Nat. Rev. Microbiol. 2020, 18, 637–648. [Google Scholar] [CrossRef]
- Sharkey, L.K.; Edwards, T.A.; O’Neill, A.J. ABC-F Proteins Mediate Antibiotic Resistance through Ribosomal Protection. mBio 2016, 7, e01975. [Google Scholar] [CrossRef]
- Sharkey, L.K.R.; O’Neill, A.J. Antibiotic Resistance ABC-F Proteins: Bringing Target Protection into the Limelight. ACS Infect. Dis. 2018, 4, 239–246. [Google Scholar] [CrossRef]
- Su, W.; Kumar, V.; Ding, Y.; Ero, R.; Serra, A.; Lee, B.S.T.; Wong, A.S.W.; Shi, J.; Sze, S.K.; Yang, L.; et al. Ribosome protection by antibiotic resistance ATP-binding cassette protein. Proc. Natl. Acad. Sci. USA 2018, 115, 5157–5162. [Google Scholar] [CrossRef]
- Seo, H.S.; Abedin, S.; Kamp, D.; Wilson, D.N.; Nierhaus, K.H.; Cooperman, B.S. EF-G-dependent GTPase on the ribosome. conformational change and fusidic acid inhibition. Biochemistry 2006, 45, 2504–2514. [Google Scholar] [CrossRef] [PubMed]
- Castanheira, M.; Watters, A.A.; Mendes, R.E.; Farrell, D.J.; Jones, R.N. Occurrence and molecular characterization of fusidic acid resistance mechanisms among Staphylococcus spp. from European countries (2008). J. Antimicrob. Chemother. 2010, 65, 1353–1358. [Google Scholar] [CrossRef]
- McLaws, F.; Chopra, I.; O’Neill, A.J. High prevalence of resistance to fusidic acid in clinical isolates of Staphylococcus epidermidis. J. Antimicrob. Chemother. 2008, 61, 1040–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- George, A.M.; Levy, S.B. Amplifiable resistance to tetracycline, chloramphenicol, and other antibiotics in Escherichia coli: Involvement of a non-plasmid-determined efflux of tetracycline. J. Bacteriol. 1983, 155, 531–540. [Google Scholar] [CrossRef] [PubMed]
- Hao, Z.; Lou, H.; Zhu, R.; Zhu, J.; Zhang, D.; Zhao, B.S.; Zeng, S.; Chen, X.; Chan, J.; He, C.; et al. The multiple antibiotic resistance regulator MarR is a copper sensor in Escherichia coli. Nat. Chem. Biol. 2014, 10, 21–28. [Google Scholar] [CrossRef]
- Ariza, R.R.; Cohen, S.P.; Bachhawat, N.; Levy, S.B.; Demple, B. Repressor mutations in the marRAB operon that activate oxidative stress genes and multiple antibiotic resistance in Escherichia coli. J. Bacteriol. 1994, 176, 143–148. [Google Scholar] [CrossRef]
- McMurry, L.M.; George, A.M.; Levy, S.B. Active efflux of chloramphenicol in susceptible Escherichia coli strains and in multiple-antibiotic-resistant (Mar) mutants. Antimicrob. Agents Chemother. 1994, 38, 542–546. [Google Scholar] [CrossRef]
- Zhang, A.; Rosner, J.L.; Martin, R.G. Transcriptional activation by MarA, SoxS and Rob of two tolC promoters using one binding site: A complex promoter configuration for tolC in Escherichia coli. Mol. Microbiol. 2008, 69, 1450–1455. [Google Scholar] [CrossRef]
- Sharma, P.; Haycocks, J.R.J.; Middlemiss, A.D.; Kettles, R.A.; Sellars, L.E.; Ricci, V.; Piddock, L.J.V.; Grainger, D.C. The multiple antibiotic resistance operon of enteric bacteria controls DNA repair and outer membrane integrity. Nat. Commun. 2017, 8, 1444. [Google Scholar] [CrossRef]
- Banerjee, S.; Lo, K.; Ojkic, N.; Stephens, R.; Scherer, N.F.; Dinner, A.R. Mechanical feedback promotes bacterial adaptation to antibiotics. Nat. Phys. 2021, 17, 403–409. [Google Scholar] [CrossRef]
- Mickiewicz, K.M.; Kawai, Y.; Drage, L.; Gomes, M.C.; Davison, F.; Pickard, R.; Hall, J.; Mostowy, S.; Aldridge, P.D.; Errington, J. Possible role of L-form switching in recurrent urinary tract infection. Nat. Commun. 2019, 10, 4379. [Google Scholar] [CrossRef] [PubMed]
- Tolker-Nielsen, T. Biofilm Development. Microbiol. Spectr. 2015, 3, MB-0001-2014. [Google Scholar] [CrossRef]
- Høiby, N.; Bjarnsholt, T.; Givskov, M.; Molin, S.; Ciofu, O. Antibiotic resistance of bacterial biofilms. Int. J. Antimicrob. Agents 2010, 35, 322–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bottery, M.J.; Pitchford, J.W.; Friman, V.P. Ecology and evolution of antimicrobial resistance in bacterial communities. ISME J. 2021, 15, 939–948. [Google Scholar] [CrossRef] [PubMed]
- Vega, N.M.; Gore, J. Collective antibiotic resistance: Mechanisms and implications. Curr. Opin. Microbiol. 2014, 21, 28–34. [Google Scholar] [CrossRef]
- Orazi, G.; O’Toole, G.A. Pseudomonas aeruginosa Alters Staphylococcus aureus Sensitivity to Vancomycin in a Biofilm Model of Cystic Fibrosis Infection. mBio 2017, 8, e00873-17. [Google Scholar] [CrossRef]
- Beaudoin, T.; Yau, Y.C.W.; Stapleton, P.J.; Gong, Y.; Wang, P.W.; Guttman, D.S.; Waters, V. Staphylococcus aureus interaction with Pseudomonas aeruginosa biofilm enhances tobramycin resistance. NPJ Biofilms Microbiomes 2017, 3, 25. [Google Scholar] [CrossRef]
- Molina-Santiago, C.; Daddaoua, A.; Fillet, S.; Duque, E.; Ramos, J.L. Interspecies signalling: Pseudomonas putida efflux pump TtgGHI is activated by indole to increase antibiotic resistance. Environ. Microbiol. 2014, 16, 1267–1281. [Google Scholar] [CrossRef]
- Ryan, R.P.; Fouhy, Y.; Garcia, B.F.; Watt, S.A.; Niehaus, K.; Yang, L.; Tolker-Nielsen, T.; Dow, J.M. Interspecies signalling via the Stenotrophomonas maltophilia diffusible signal factor influences biofilm formation and polymyxin tolerance in Pseudomonas aeruginosa. Mol. Microbiol. 2008, 68, 75–86. [Google Scholar] [CrossRef]
- Ling, L.L.; Schneider, T.; Peoples, A.J.; Spoering, A.L.; Engels, I.; Conlon, B.P.; Mueller, A.; Schäberle, T.F.; Hughes, D.E.; Epstein, S.; et al. A new antibiotic kills pathogens without detectable resistance. Nature 2015, 517, 455–459. [Google Scholar] [CrossRef]
- Maffioli, S.I.; Sosio, M.; Ebright, R.H.; Donadio, S. Discovery, properties, and biosynthesis of pseudouridimycin, an antibacterial nucleoside-analog inhibitor of bacterial RNA polymerase. J. Ind. Microbiol. Biotechnol. 2019, 46, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Smitha Rao, C.V.; Anné, J. Bacterial type I signal peptidases as antibiotic targets. Future Microbiol. 2011, 6, 1279–1296. [Google Scholar] [CrossRef]
- Imai, Y.; Meyer, K.J.; Iinishi, A.; Favre-Godal, Q.; Green, R.; Manuse, S.; Caboni, M.; Mori, M.; Niles, S.; Ghiglieri, M.; et al. A new antibiotic selectively kills Gram-negative pathogens. Nature 2019, 576, 459–464. [Google Scholar] [CrossRef]
- Stokes, J.M.; Yang, K.; Swanson, K.; Jin, W.; Cubillos-Ruiz, A.; Donghia, N.M.; MacNair, C.R.; French, S.; Carfrae, L.A.; Bloom-Ackermann, Z.; et al. A Deep Learning Approach to Antibiotic Discovery. Cell 2020, 180, 688–702.e13. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.K., 2nd; Sheehan, J.P.; Bratton, B.P.; Moore, G.M.; Mateus, A.; Li, S.H.; Kim, H.; Rabinowitz, J.D.; Typas, A.; Savitski, M.M.; et al. A Dual-Mechanism Antibiotic Kills Gram-Negative Bacteria and Avoids Drug Resistance. Cell 2020, 181, 1518–1532.e14. [Google Scholar] [CrossRef] [PubMed]
- Shatalin, K.; Nuthanakanti, A.; Kaushik, A.; Shishov, D.; Peselis, A.; Shamovsky, I.; Pani, B.; Lechpammer, M.; Vasilyev, N.; Shatalina, E.; et al. Inhibitors of bacterial H2S biogenesis targeting antibiotic resistance and tolerance. Science 2021, 372, 1169–1175. [Google Scholar] [CrossRef]
- Li, L.; Koirala, B.; Hernandez, Y.; MacIntyre, L.W.; Ternei, M.A.; Russo, R.; Brady, S.F. Identification of structurally diverse menaquinone-binding antibiotics with in vivo activity against multidrug-resistant pathogens. Nat. Microbiol. 2022, 7, 120–131. [Google Scholar] [CrossRef]
- Wang, Z.; Koirala, B.; Hernandez, Y.; Zimmerman, M.; Park, S.; Perlin, D.S.; Brady, S.F. A naturally inspired antibiotic to target multidrug-resistant pathogens. Nature 2022, 601, 606–611. [Google Scholar] [CrossRef]
- Wang, Z.; Koirala, B.; Hernandez, Y.; Zimmerman, M.; Brady, S.F. Bioinformatic prospecting and synthesis of a bifunctional lipopeptide antibiotic that evades resistance. Science 2022, 376, 991–996. [Google Scholar] [CrossRef]
- Clokie, M.R.; Millard, A.D.; Letarov, A.V.; Heaphy, S. Phages in nature. Bacteriophage 2011, 1, 31–45. [Google Scholar] [CrossRef]
- Koshland, D.; Sauer, R.T.; Botstein, D. Diverse effects of mutations in the signal sequence on the secretion of beta-lactamase in Salmonella typhimurium. Cell 1982, 30, 903–914. [Google Scholar] [CrossRef]
- Ho, H.; Miu, A.; Alexander, M.K.; Garcia, N.K.; Oh, A.; Zilberleyb, I.; Reichelt, M.; Austin, C.D.; Tam, C.; Shriver, S.; et al. Structural basis for dual-mode inhibition of the ABC transporter MsbA. Nature 2018, 557, 196–201. [Google Scholar] [CrossRef] [PubMed]
- Nayfach, S.; Páez-Espino, D.; Call, L.; Low, S.J.; Sberro, H.; Ivanova, N.N.; Proal, A.D.; Fischbach, M.A.; Bhatt, A.S.; Hugenholtz, P.; et al. Metagenomic compendium of 189,680 DNA viruses from the human gut microbiome. Nat. Microbiol. 2021, 6, 960–970. [Google Scholar] [CrossRef]
- Little, J.S.; Dedrick, R.M.; Freeman, K.G.; Cristinziano, M.; Smith, B.E.; Benson, C.A.; Jhaveri, T.A.; Baden, L.R.; Solomon, D.A.; Hatfull, G.F. Bacteriophage treatment of disseminated cutaneous Mycobacterium chelonae infection. Nat. Commun. 2022, 13, 2313. [Google Scholar] [CrossRef] [PubMed]
- Taylor, N.M.; Prokhorov, N.S.; Guerrero-Ferreira, R.C.; Shneider, M.M.; Browning, C.; Goldie, K.N.; Stahlberg, H.; Leiman, P.G. Structure of the T4 baseplate and its function in triggering sheath contraction. Nature 2016, 533, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Kostyuchenko, V.A.; Chipman, P.R.; Leiman, P.G.; Arisaka, F.; Mesyanzhinov, V.V.; Rossmann, M.G. The tail structure of bacteriophage T4 and its mechanism of contraction. Nat. Struct. Mol. Biol. 2005, 12, 810–813. [Google Scholar] [CrossRef]
- Fischetti, V.A. Bacteriophage lytic enzymes: Novel anti-infectives. Trends Microbiol. 2005, 13, 491–496. [Google Scholar] [CrossRef]
- Briers, Y.; Walmagh, M.; Van Puyenbroeck, V.; Cornelissen, A.; Cenens, W.; Aertsen, A.; Oliveira, H.; Azeredo, J.; Verween, G.; Pirnay, J.P.; et al. Engineered endolysin-based “Artilysins” to combat multidrug-resistant gram-negative pathogens. mBio 2014, 5, e01379-14. [Google Scholar] [CrossRef]
- Vollmer, W.; Bertsche, U. Murein (peptidoglycan) structure, architecture and biosynthesis in Escherichia coli. Biochim. Biophys. Acta 2008, 1778, 1714–1734. [Google Scholar] [CrossRef]
- Low, L.Y.; Yang, C.; Perego, M.; Osterman, A.; Liddington, R. Role of net charge on catalytic domain and influence of cell wall binding domain on bactericidal activity, specificity, and host range of phage lysins. J. Biol. Chem. 2011, 286, 34391–34403. [Google Scholar] [CrossRef]
- Yang, H.; Linden, S.B.; Wang, J.; Yu, J.; Nelson, D.C.; Wei, H. A chimeolysin with extended-spectrum streptococcal host range found by an induced lysis-based rapid screening method. Sci. Rep. 2015, 5, 17257. [Google Scholar] [CrossRef] [PubMed]
- Domenech, M.; Garcia, E.; Moscoso, M. In vitro destruction of Streptococcus pneumoniae biofilms with bacterial and phage peptidoglycan hydrolases. Antimicrob. Agents Chemother. 2011, 55, 4144–4148. [Google Scholar] [CrossRef] [PubMed]
- Schuch, R.; Nelson, D.; Fischetti, V.A. A bacteriolytic agent that detects and kills Bacillus anthracis. Nature 2002, 418, 884–889. [Google Scholar] [CrossRef]
- Abdelrahman, F.; Easwaran, M.; Daramola, O.I.; Ragab, S.; Lynch, S.; Oduselu, T.J.; Khan, F.M.; Ayobami, A.; Adnan, F.; Torrents, E.; et al. Phage-Encoded Endolysins. Antibiotics 2021, 10, 124. [Google Scholar] [CrossRef]
- Broendum, S.S.; Buckle, A.M.; McGowan, S. Catalytic diversity and cell wall binding repeats in the phage-encoded endolysins. Mol. Microbiol. 2018, 110, 879–896. [Google Scholar] [CrossRef] [PubMed]
- Navarre, W.W.; Ton-That, H.; Faull, K.F.; Schneewind, O. Multiple enzymatic activities of the murein hydrolase from staphylococcal phage phi11. Identification of a D-alanyl-glycine endopeptidase activity. J. Biol. Chem. 1999, 274, 15847–15856. [Google Scholar] [CrossRef]
- Loessner, M.J. Bacteriophage endolysins--current state of research and applications. Curr. Opin. Microbiol. 2005, 8, 480–487. [Google Scholar] [CrossRef]
- Sass, P.; Bierbaum, G. Lytic activity of recombinant bacteriophage phi11 and phi12 endolysins on whole cells and biofilms of Staphylococcus aureus. Appl. Env. Microbiol. 2007, 73, 347–352. [Google Scholar] [CrossRef]
- Son, J.S.; Lee, S.J.; Jun, S.Y.; Yoon, S.J.; Kang, S.H.; Paik, H.R.; Kang, J.O.; Choi, Y.J. Antibacterial and biofilm removal activity of a podoviridae Staphylococcus aureus bacteriophage SAP-2 and a derived recombinant cell-wall-degrading enzyme. Appl. Microbiol. Biotechnol. 2010, 86, 1439–1449. [Google Scholar] [CrossRef]
- Gutierrez, D.; Ruas-Madiedo, P.; Martinez, B.; Rodriguez, A.; Garcia, P. Effective removal of staphylococcal biofilms by the endolysin LysH5. PLoS ONE 2014, 9, e107307. [Google Scholar] [CrossRef]
- Linden, S.B.; Zhang, H.; Heselpoth, R.D.; Shen, Y.; Schmelcher, M.; Eichenseher, F.; Nelson, D.C. Biochemical and biophysical characterization of PlyGRCS, a bacteriophage endolysin active against methicillin-resistant Staphylococcus aureus. Appl. Microbiol. Biotechnol. 2015, 99, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Ning, H.; Lin, H.; Wang, J.; He, X.; Lv, X.; Ju, L. Characterizations of the endolysin Lys84 and its domains from phage qdsa002 with high activities against Staphylococcus aureus and its biofilms. Enzym. Microb. Technol. 2021, 148, 109809. [Google Scholar] [CrossRef] [PubMed]
- Pennone, V.; Sanz-Gaitero, M.; O’Connor, P.; Coffey, A.; Jordan, K.; van Raaij, M.J.; McAuliffe, O. Inhibition of L. monocytogenes Biofilm Formation by the Amidase Domain of the Phage vB_LmoS_293 Endolysin. Viruses 2019, 11, 722. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, H.; Thiagarajan, V.; Walmagh, M.; Sillankorva, S.; Lavigne, R.; Neves-Petersen, M.T.; Kluskens, L.D.; Azeredo, J. A thermostable Salmonella phage endolysin, Lys68, with broad bactericidal properties against gram-negative pathogens in presence of weak acids. PLoS ONE 2014, 9, e108376. [Google Scholar] [CrossRef]
- Rahman, M.U.; Wang, W.; Sun, Q.; Shah, J.A.; Li, C.; Sun, Y.; Li, Y.; Zhang, B.; Chen, W.; Wang, S. Endolysin, a Promising Solution against Antimicrobial Resistance. Antibiotics 2021, 10, 1277. [Google Scholar] [CrossRef]
- Antonova, N.P.; Vasina, D.V.; Rubalsky, E.O.; Fursov, M.V.; Savinova, A.S.; Grigoriev, I.V.; Usachev, E.V.; Shevlyagina, N.V.; Zhukhovitsky, V.G.; Balabanyan, V.U.; et al. Modulation of Endolysin LysECD7 Bactericidal Activity by Different Peptide Tag Fusion. Biomolecules 2020, 10, 440. [Google Scholar] [CrossRef]
- Torres-Barceló, C.; Hochberg, M.E. Evolutionary Rationale for Phages as Complements of Antibiotics. Trends Microbiol. 2016, 24, 249–256. [Google Scholar] [CrossRef]
- Chatterjee, A.; Johnson, C.N.; Luong, P.; Hullahalli, K.; McBride, S.W.; Schubert, A.M.; Palmer, K.L.; Carlson, P.E., Jr.; Duerkop, B.A. Bacteriophage Resistance Alters Antibiotic-Mediated Intestinal Expansion of Enterococci. Infect Immun. 2019, 87, e00085-19. [Google Scholar] [CrossRef]
- German, G.J.; Misra, R. The TolC protein of Escherichia coli serves as a cell-surface receptor for the newly characterized TLS bacteriophage. J. Mol. Biol. 2001, 308, 579–585. [Google Scholar] [CrossRef]
- Gordillo Altamirano, F.; Forsyth, J.H.; Patwa, R.; Kostoulias, X.; Trim, M.; Subedi, D.; Archer, S.K.; Morris, F.C.; Oliveira, C.; Kielty, L.; et al. Bacteriophage-resistant Acinetobacter baumannii are resensitized to antimicrobials. Nat. Microbiol. 2021, 6, 157–161. [Google Scholar] [CrossRef]
- Bedi, M.S.; Verma, V.; Chhibber, S. Amoxicillin and specific bacteriophage can be used together for eradication of biofilm of Klebsiella pneumoniae B5055. World J. Microbiol. Biotechnol. 2009, 25, 1145. [Google Scholar] [CrossRef]
- Knecht, L.E.; Veljkovic, M.; Fieseler, L. Diversity and Function of Phage Encoded Depolymerases. Front. Microbiol. 2019, 10, 2949. [Google Scholar] [CrossRef] [PubMed]
- Canfield, G.S.; Chatterjee, A.; Espinosa, J.; Mangalea, M.R.; Sheriff, E.K.; Keidan, M.; McBride, S.W.; McCollister, B.D.; Hang, H.C.; Duerkop, B.A. Lytic bacteriophages facilitate antibiotic sensitization of Enterococcus faecium. Antimicrob. Agents Chemother. 2021. [Google Scholar] [CrossRef]
- Nikolich, M.P.; Filippov, A.A. Bacteriophage Therapy: Developments and Directions. Antibiotics 2020, 9, 135. [Google Scholar] [CrossRef]
- Sharma, U.; Vipra, A.; Channabasappa, S. Phage-derived lysins as potential agents for eradicating biofilms and persisters. Drug Discov. Today 2018, 23, 848–856. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Wang, R.; Xu, M.; Liu, Y.; Zhu, X.; Qiu, J.; Liu, Q.; He, P.; Li, Q. A Novel Polysaccharide Depolymerase Encoded by the Phage SH-KP152226 Confers Specific Activity Against Multidrug-Resistant Klebsiella pneumoniae via Biofilm Degradation. Front Microbiol. 2019, 10, 2768. [Google Scholar] [CrossRef]
- Tabassum, R.; Shafique, M.; Khawaja, K.A.; Alvi, I.A.; Rehman, Y.; Sheik, C.S.; Abbas, Z.; Rehman, S.U. Complete genome analysis of a Siphoviridae phage TSK1 showing biofilm removal potential against Klebsiella pneumoniae. Sci. Rep. 2018, 8, 17904. [Google Scholar] [CrossRef]
- Fernandes, P.; Oldach, D. Antibiotic usage and the development of resistance. Clin. Infect Dis. 2015, 60, 1139–1140. [Google Scholar] [CrossRef]
- Greene, A.C. CRISPR-Based Antibacterials: Transforming Bacterial Defense into Offense. Trends Biotechnol. 2018, 36, 127–130. [Google Scholar] [CrossRef]
- Bhaya, D.; Davison, M.; Barrangou, R. CRISPR-Cas systems in bacteria and archaea: Versatile small RNAs for adaptive defense and regulation. Annu. Rev. Genet. 2011, 45, 273–297. [Google Scholar] [CrossRef]
- Bikard, D.; Hatoum-Aslan, A.; Mucida, D.; Marraffini, L.A. CRISPR interference can prevent natural transformation and virulence acquisition during in vivo bacterial infection. Cell Host Microbe 2012, 12, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Bikard, D. Consequences of Cas9 cleavage in the chromosome of Escherichia coli. Nucleic Acids Res. 2016, 44, 4243–4251. [Google Scholar] [CrossRef] [PubMed]
- Vercoe, R.B.; Chang, J.T.; Dy, R.L.; Taylor, C.; Gristwood, T.; Clulow, J.S.; Richter, C.; Przybilski, R.; Pitman, A.R.; Fineran, P.C. Cytotoxic chromosomal targeting by CRISPR/Cas systems can reshape bacterial genomes and expel or remodel pathogenicity islands. PLoS Genet 2013, 9, e1003454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabezon, E.; Ripoll-Rozada, J.; Pena, A.; de la Cruz, F.; Arechaga, I. Towards an integrated model of bacterial conjugation. FEMS Microbiol. Rev. 2015, 39, 81–95. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Li, M.; Li, Y.; Cao, H.; Miao, L.; Xu, Z.; Higuchi, Y.; Yamasaki, S.; Nishino, K.; Woo, P.C.Y.; et al. Native CRISPR-Cas-Mediated Genome Editing Enables Dissecting and Sensitizing Clinical Multidrug-Resistant P. aeruginosa. Cell Rep. 2019, 29, 1707–1717.e1703. [Google Scholar] [CrossRef]
- Kim, J.S.; Cho, D.H.; Park, M.; Chung, W.J.; Shin, D.; Ko, K.S.; Kweon, D.H. CRISPR/Cas9-Mediated Re-Sensitization of Antibiotic-Resistant Escherichia coli Harboring Extended-Spectrum β-Lactamases. J. Microbiol. Biotechnol. 2016, 26, 394–401. [Google Scholar] [CrossRef]
- Yosef, I.; Manor, M.; Kiro, R.; Qimron, U. Temperate and lytic bacteriophages programmed to sensitize and kill antibiotic-resistant bacteria. Proc. Natl. Acad. Sci. USA 2015, 112, 7267–7272. [Google Scholar] [CrossRef]
- Citorik, R.J.; Mimee, M.; Lu, T.K. Sequence-specific antimicrobials using efficiently delivered RNA-guided nucleases. Nat. Biotechnol. 2014, 32, 1141–1145. [Google Scholar] [CrossRef]
- Gunday, C.; Anand, S.; Gencer, H.B.; Munafo, S.; Moroni, L.; Fusco, A.; Donnarumma, G.; Ricci, C.; Hatir, P.C.; Tureli, N.G.; et al. Ciprofloxacin-loaded polymeric nanoparticles incorporated electrospun fibers for drug delivery in tissue engineering applications. Drug Deliv. Transl. Res. 2020, 10, 706–720. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.K.; Kwon, K.; Ryu, J.S.; Lee, H.N.; Park, C.; Chung, H.J. Nonviral Genome Editing Based on a Polymer-Derivatized CRISPR Nanocomplex for Targeting Bacterial Pathogens and Antibiotic Resistance. Bioconjugate Chem. 2017, 28, 957–967. [Google Scholar] [CrossRef]
- Gonzalez de Aledo, M.; Gonzalez-Bardanca, M.; Blasco, L.; Pacios, O.; Bleriot, I.; Fernandez-Garcia, L.; Fernandez-Quejo, M.; Lopez, M.; Bou, G.; Tomas, M. CRISPR-Cas, a Revolution in the Treatment and Study of ESKAPE Infections: Pre-Clinical Studies. Antibiotics 2021, 10, 756. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Quiles-Puchalt, N.; Chiang, Y.N.; Bacigalupe, R.; Fillol-Salom, A.; Chee, M.S.J.; Fitzgerald, J.R.; Penades, J.R. Genome hypermobility by lateral transduction. Science 2018, 362, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Alekshun, M.N.; Levy, S.B. Molecular mechanisms of antibacterial multidrug resistance. Cell 2007, 128, 1037–1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, X.; Feng, X.; Werber, J.R.; Chu, C.; Zucker, I.; Kim, J.H.; Osuji, C.O.; Elimelech, M. Enhanced antibacterial activity through the controlled alignment of graphene oxide nanosheets. Proc. Natl. Acad. Sci. USA 2017, 114, e9793–e9801. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, A.; Ivanova, K.; Tied, A.; Heinze, T.; Tzanov, T. Layer-By-Layer Coating of Aminocellulose and Quorum Quenching Acylase on Silver Nanoparticles Synergistically Eradicate Bacteria and Their Biofilms. Adv. Funct. Mater. 2020, 30, 1–9. [Google Scholar] [CrossRef]
- Hu, D.; Li, H.; Wang, B.; Ye, Z.; Lei, W.; Jia, F.; Jin, Q.; Ren, K.F.; Ji, J. Surface-Adaptive Gold Nanoparticles with Effective Adherence and Enhanced Photothermal Ablation of Methicillin-Resistant Staphylococcus aureus Biofilm. ACS Nano 2017, 11, 9330–9339. [Google Scholar] [CrossRef]
- Chen, Z.; Wang, Z.; Ren, J.; Qu, X. Enzyme Mimicry for Combating Bacteria and Biofilms. Acc. Chem. Res. 2018, 51, 789–799. [Google Scholar] [CrossRef]
- Xu, B.; Wang, H.; Wang, W.; Gao, L.; Li, S.; Pan, X.; Wang, H.; Yang, H.; Meng, X.; Wu, Q.; et al. A Single-Atom Nanozyme for Wound Disinfection Applications. Angew. Chem. Int. Ed. Engl. 2019, 58, 4911–4916. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Compound Name | Bacteriostatic Spectrum | Action Target | Bacteriostatic Mechanism | Report |
|---|---|---|---|---|
| Teixobactin | Gram-positive bacteria | cell wall | inhibit cell wall synthesis by binding to a highly conserved motif of lipid II and lipid III | Kim Lewis 2015, [16,93] |
| Pseudouridimycin | S. aureus, etc | RNA polymerase | nucleoside triphosphate to RNA polymerase by occupying the binding site of NTP. | Sonia I Maffioli 2017, [94] |
| G907 | E. coli, etc | ATP-binding cassette transporter | inhibit E. coli MsbA physiological functions | Christopher M. Koth 2018, [95] |
| Arylomycin(G0775) | ESKAPE, etc | type I signal peptidase | inhibit the activity of type I signal peptidase | Christopher Heise, 2018, [18] |
|
Chimeric peptidomimetic | Gram-negative bacteria | cell membranes | bind to both lipopolysaccharide and the main component (BamA) of the β-barrel folding complex (BAM) | John A. Robinson 2019, [17] |
| Darobactin |
Gram-negative bacteria | cell membranes | bind to the key outer membrane protein BamA, disrupts the bacterial outer membrane and induces cell lysis | Kim Lewis 2019, [96] |
| Complestatin and Corbomycin | Gram-positive bacteria | cell wall | block the effect of cell autolysin on cell wall and preventing the collapse of cell wall | Wright, Gerard D 2020, [19] |
| Halicin | E. coli, M. tuberculosis, A. baumannii, etc | cell membranes | destroy their ability to maintain electrochemical gradients on cell membranes | James J. Collins 2020, [97] |
| SCH-79797 | broad spectrum | folate metabolism and cell membrane | simultaneously targeting folate metabolism and membrane integrity | Zemer Gitai 2020, [98] |
| Macolacin | Gram-positive Bacteria(mrc-1) | cell membranes | a homologue with different structure from colistin, and its antibacterial mechanism is similar to colistin | Sean F. Brady, 2021, [99] |
| bCSE inhibitors (NL1, NL2, NL3) | S. aureus, P. aeruginosa | H2S synthesize metabolism | inhibit the production of bacterial H2S to enhance the bactericidal efficacy of antibiotics | Nudler 2021, [100] |
| Iboxamycin (IBX) | broad spectrum | ribosome | shift methylated ribosomal nucleotides and expose drug binding sites | Andrew G. Myers, 2021, [20] |
| Menaquinone-binding antibiotic (MBA) | MRSA, etc | menaquinones | target menaquinones that play a key role in the electronic transmission of bacteria | Sean F. Brady 2021, [101] |
| Cilagicin | Gram-positive bacteria | cell walls | simultaneous binding of two molecules c55-p and c55-pp that maintain bacterial cell walls | Sean F. Brady 2022, [102] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, F.; Cheng, W. The Mechanism of Bacterial Resistance and Potential Bacteriostatic Strategies. Antibiotics 2022, 11, 1215. https://doi.org/10.3390/antibiotics11091215
Zhang F, Cheng W. The Mechanism of Bacterial Resistance and Potential Bacteriostatic Strategies. Antibiotics. 2022; 11(9):1215. https://doi.org/10.3390/antibiotics11091215
Chicago/Turabian StyleZhang, Fusheng, and Wei Cheng. 2022. "The Mechanism of Bacterial Resistance and Potential Bacteriostatic Strategies" Antibiotics 11, no. 9: 1215. https://doi.org/10.3390/antibiotics11091215
APA StyleZhang, F., & Cheng, W. (2022). The Mechanism of Bacterial Resistance and Potential Bacteriostatic Strategies. Antibiotics, 11(9), 1215. https://doi.org/10.3390/antibiotics11091215