

Antibiofilm and Anti-Quorum-Sensing Activities of Novel Pyrazole and Pyrazolo[1,5-a]pyrimidine Derivatives as Carbonic Anhydrase I and II Inhibitors: Design, Synthesis, Radiosterilization, and Molecular Docking Studies

 ,
, 
Abstract
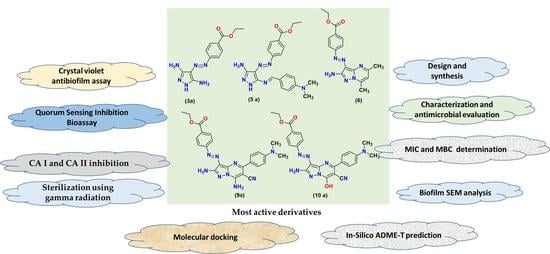
:
1. Introduction
2. Results and Discussion
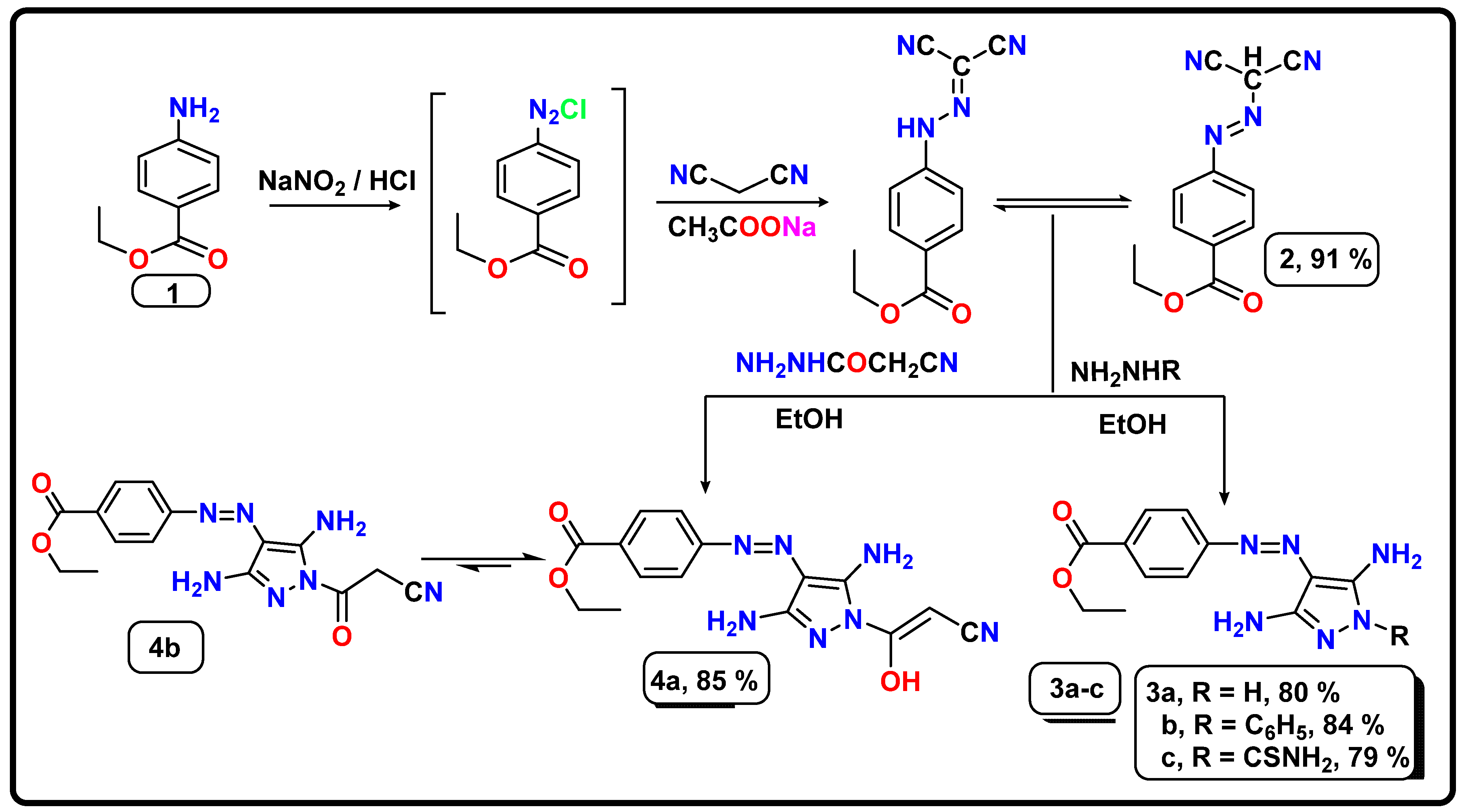
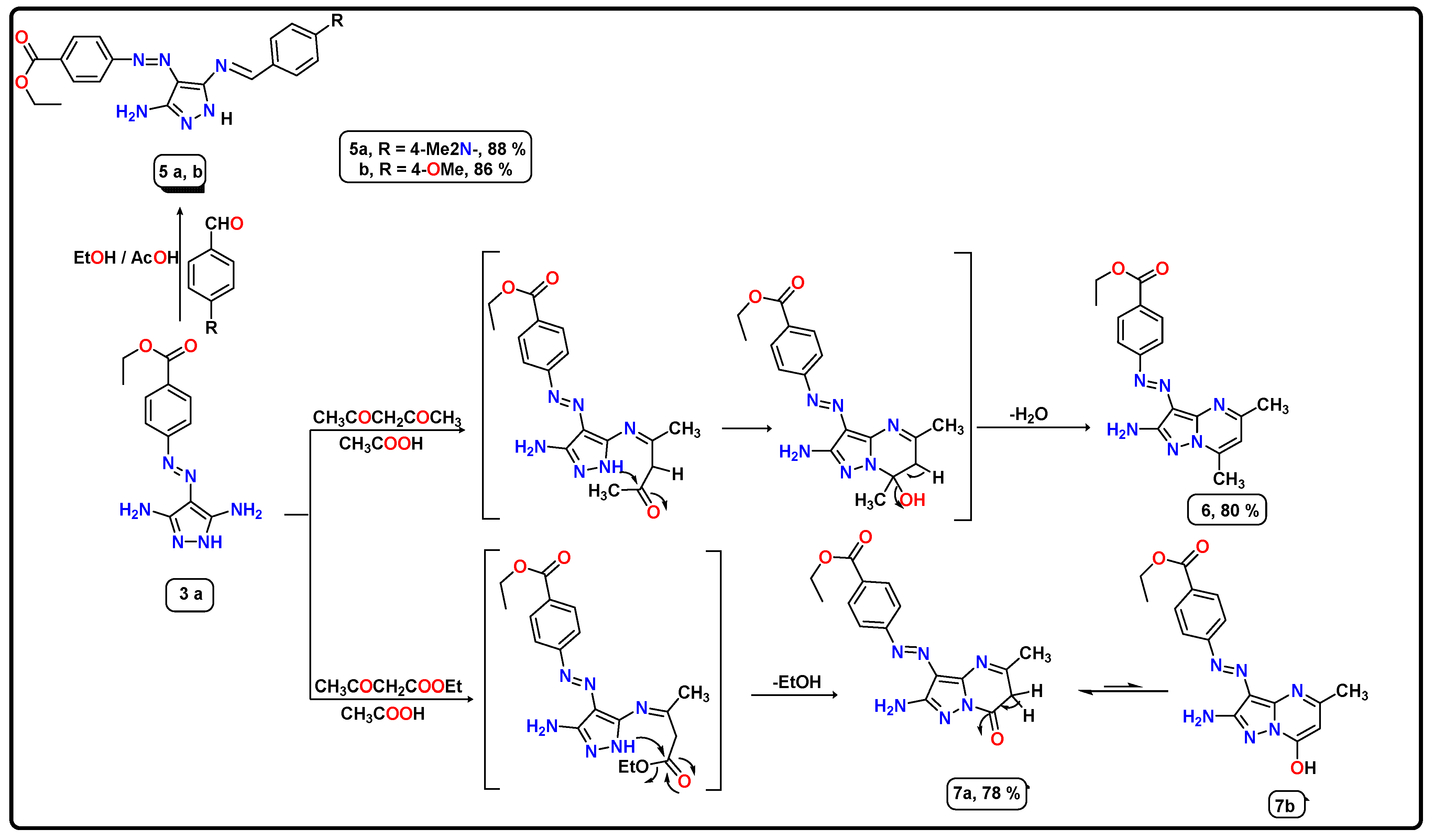
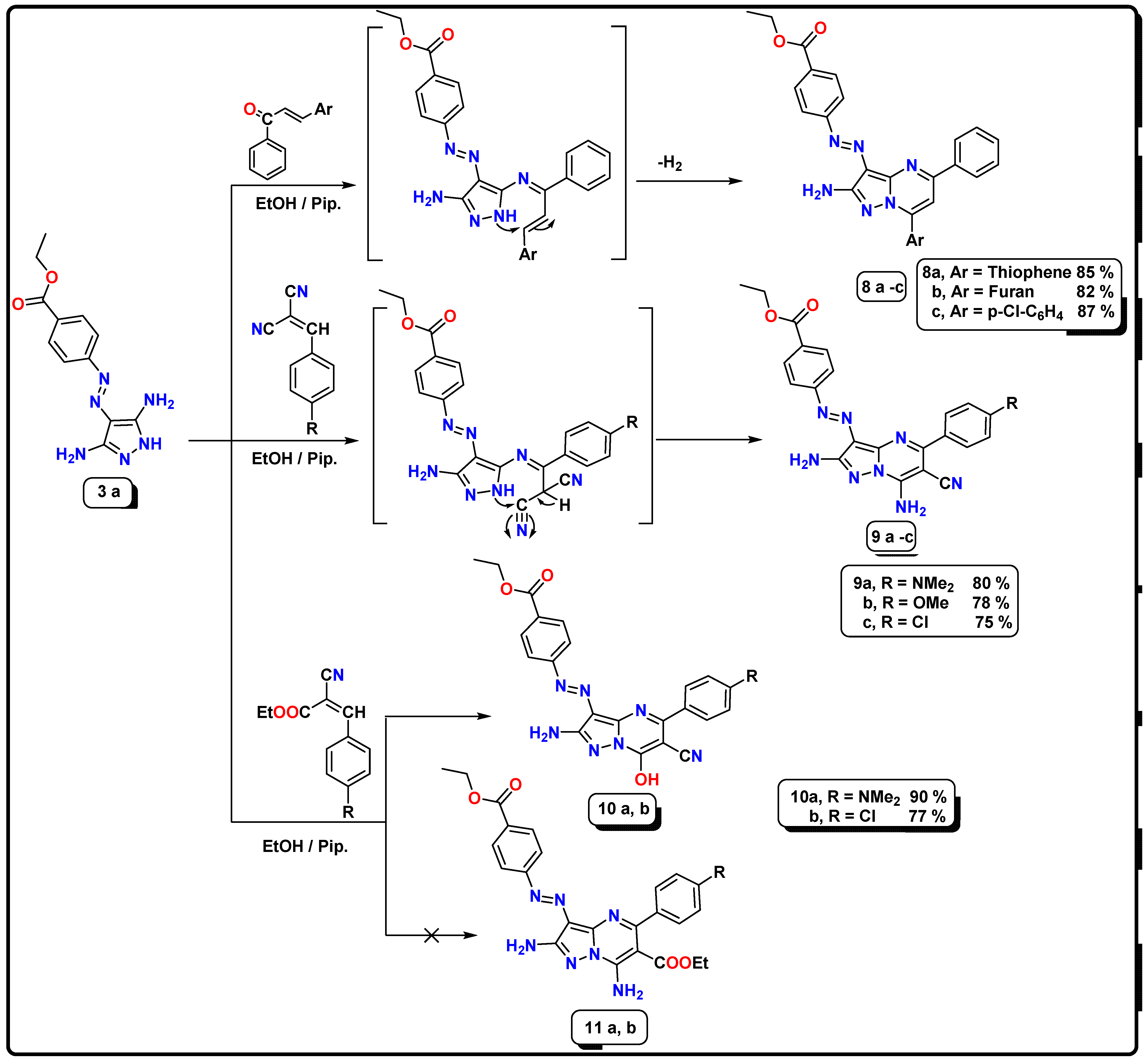
2.1. Chemistry
2.2. Biological Activity
2.2.1. Screening of Synthesized Compounds for Antibacterial Activity
2.2.2. MIC and MBC Determination
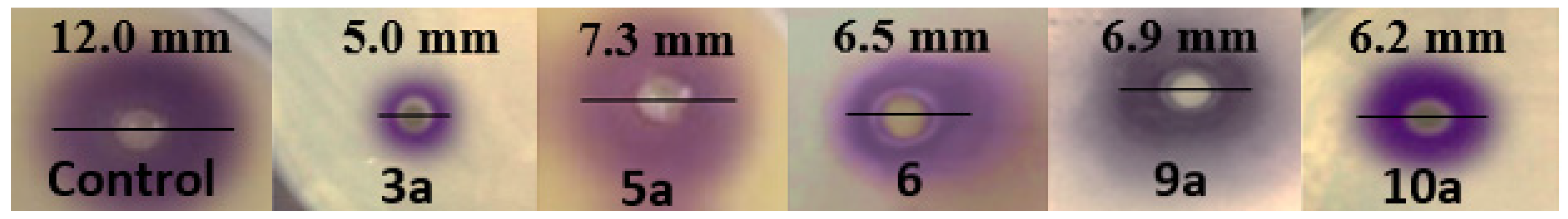
2.2.3. Antibiofilm and Anti-Infective Activities of Pyrazole and Pyrazolo[1,5-a]pyrimidine Derivatives
Crystal Violet Antibiofilm Assay
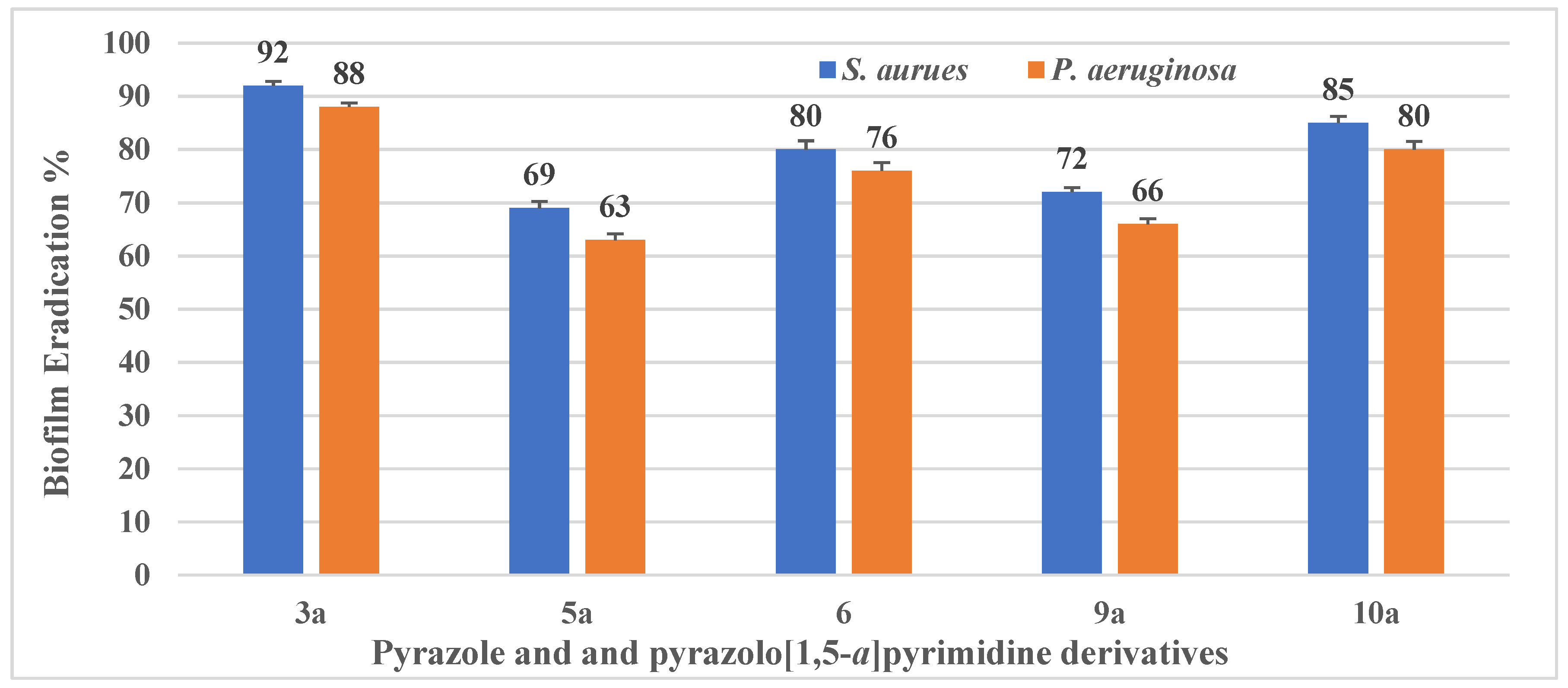
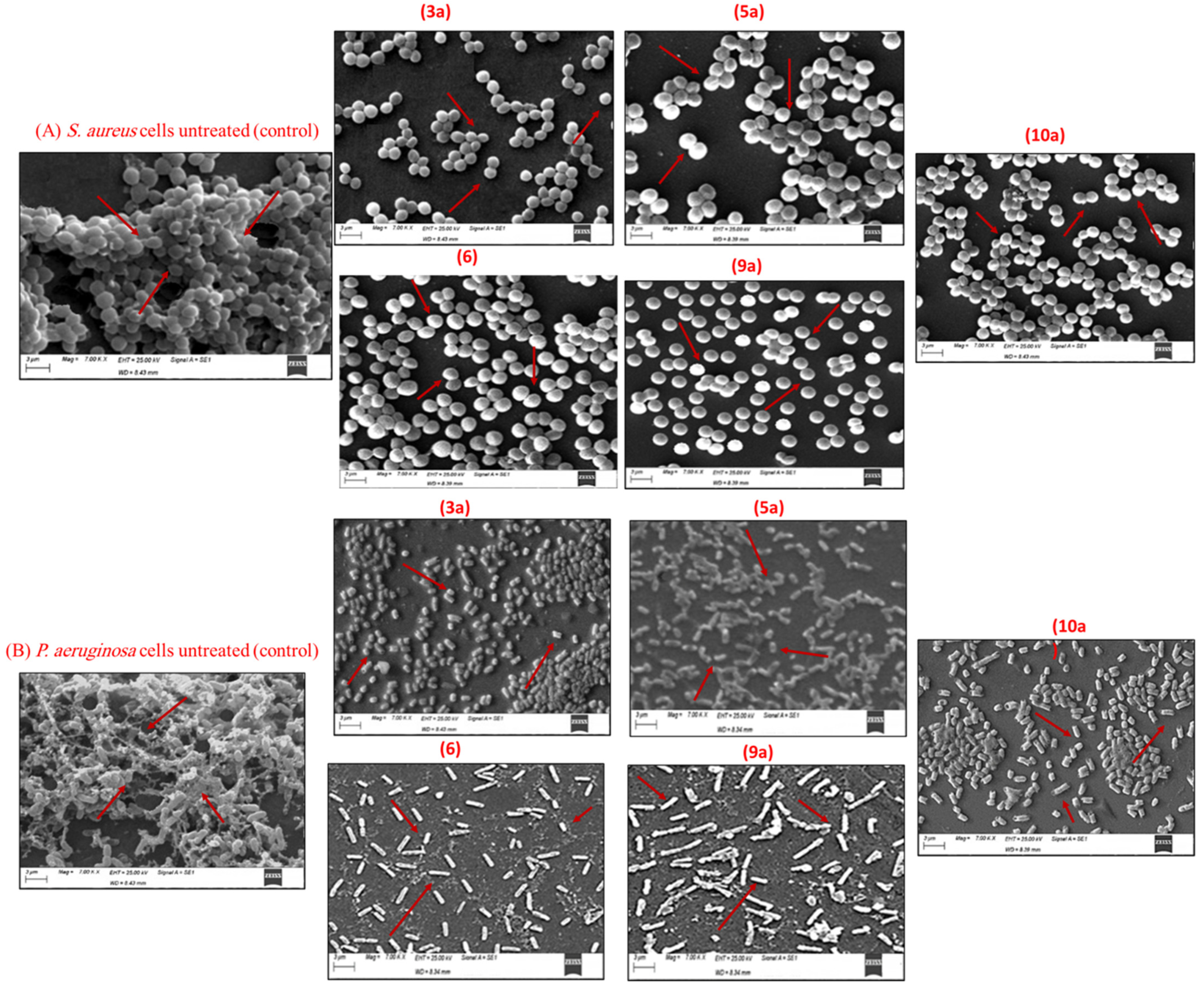
Determination of the Antibiofilm Activity of Pyrazole and Pyrazolo[1,5-a]pyrimidine Derivatives via SEM Analysis
Quorum-Sensing Inhibition Bioassay
2.2.4. CA-I and CA-II Isoenzymes Purification and Inhibition
2.2.5. Effect of Gamma Sterilization on the Antimicrobial Activity of the Most Active Compounds
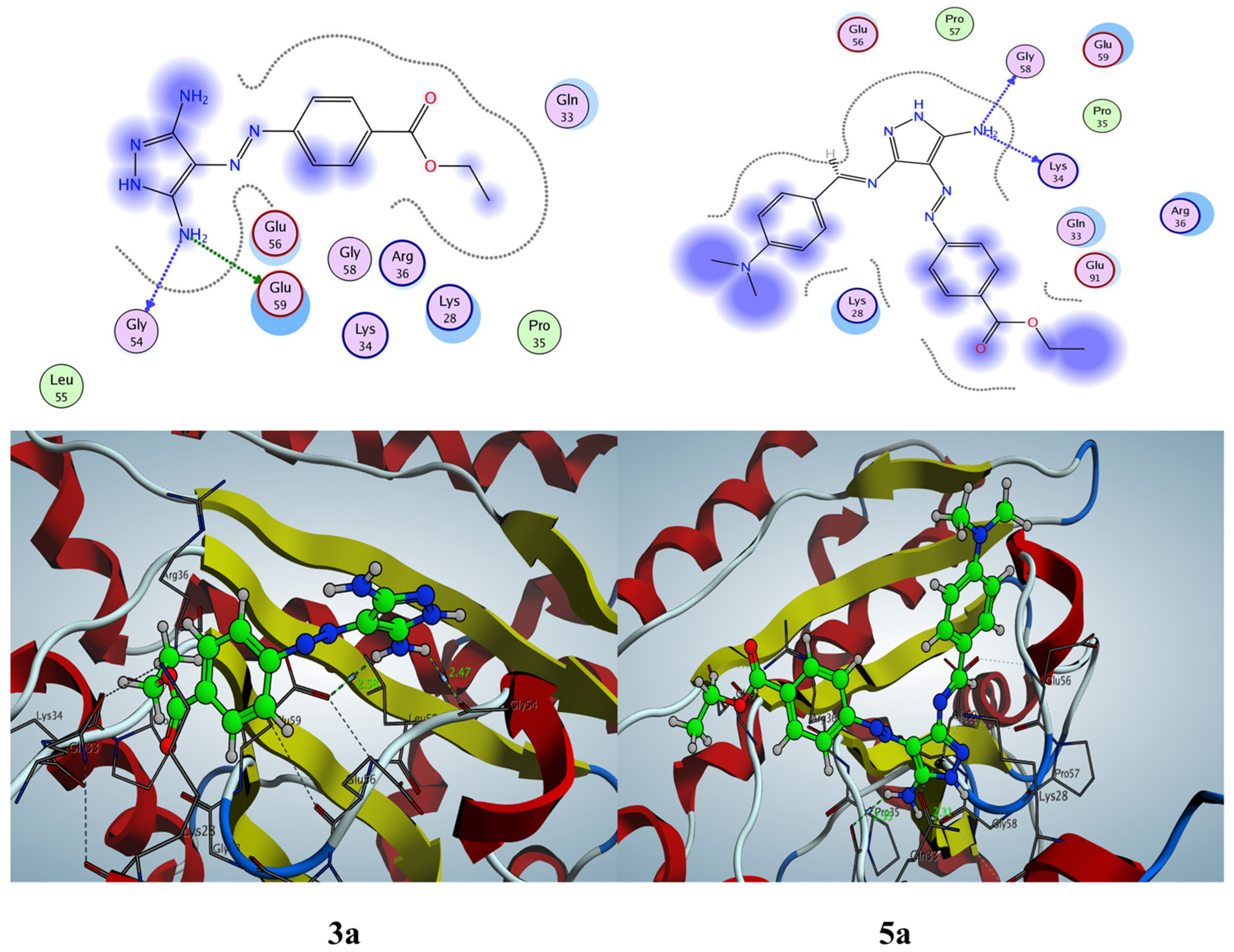
2.3. Molecular Docking Studies
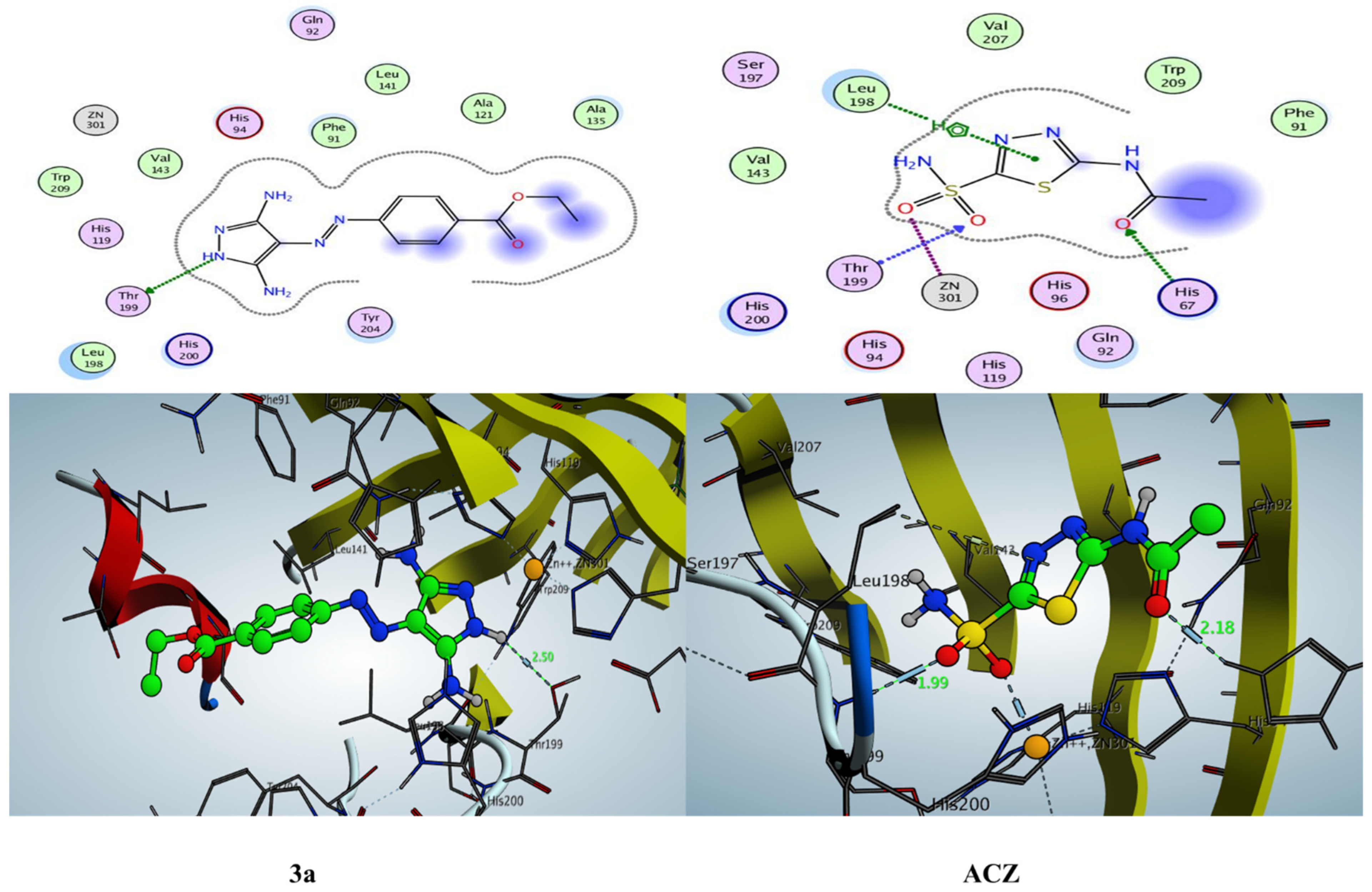
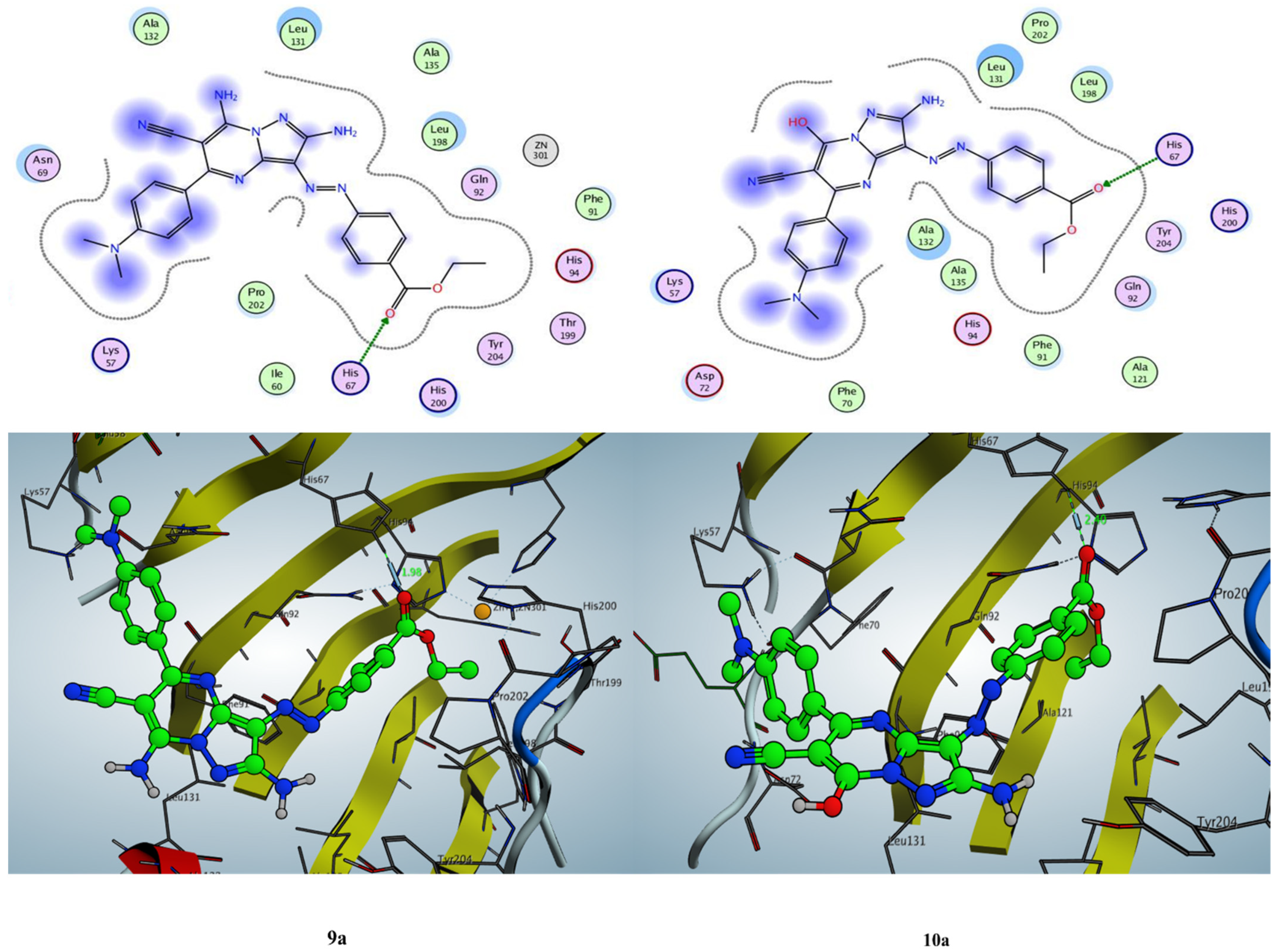
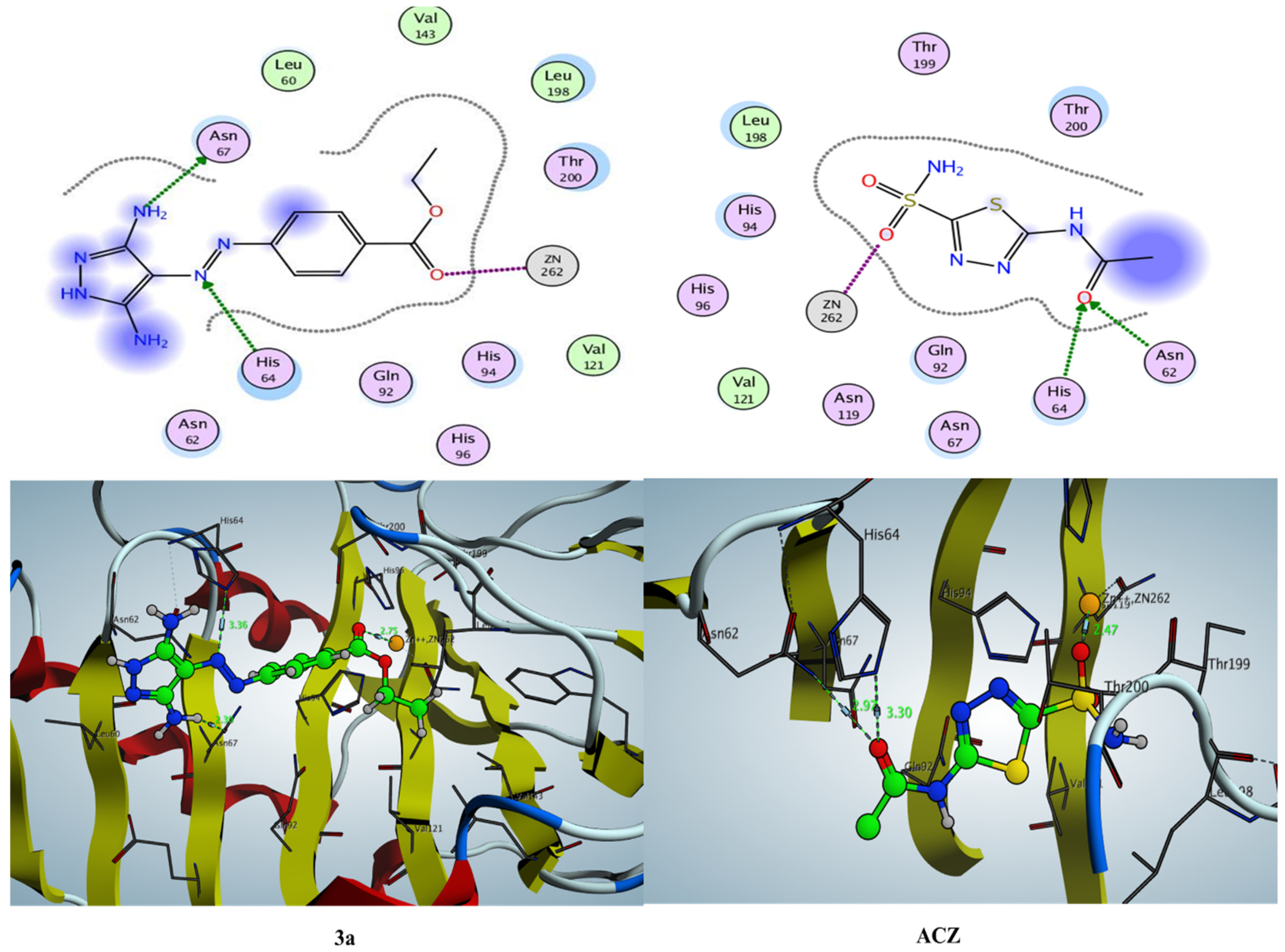
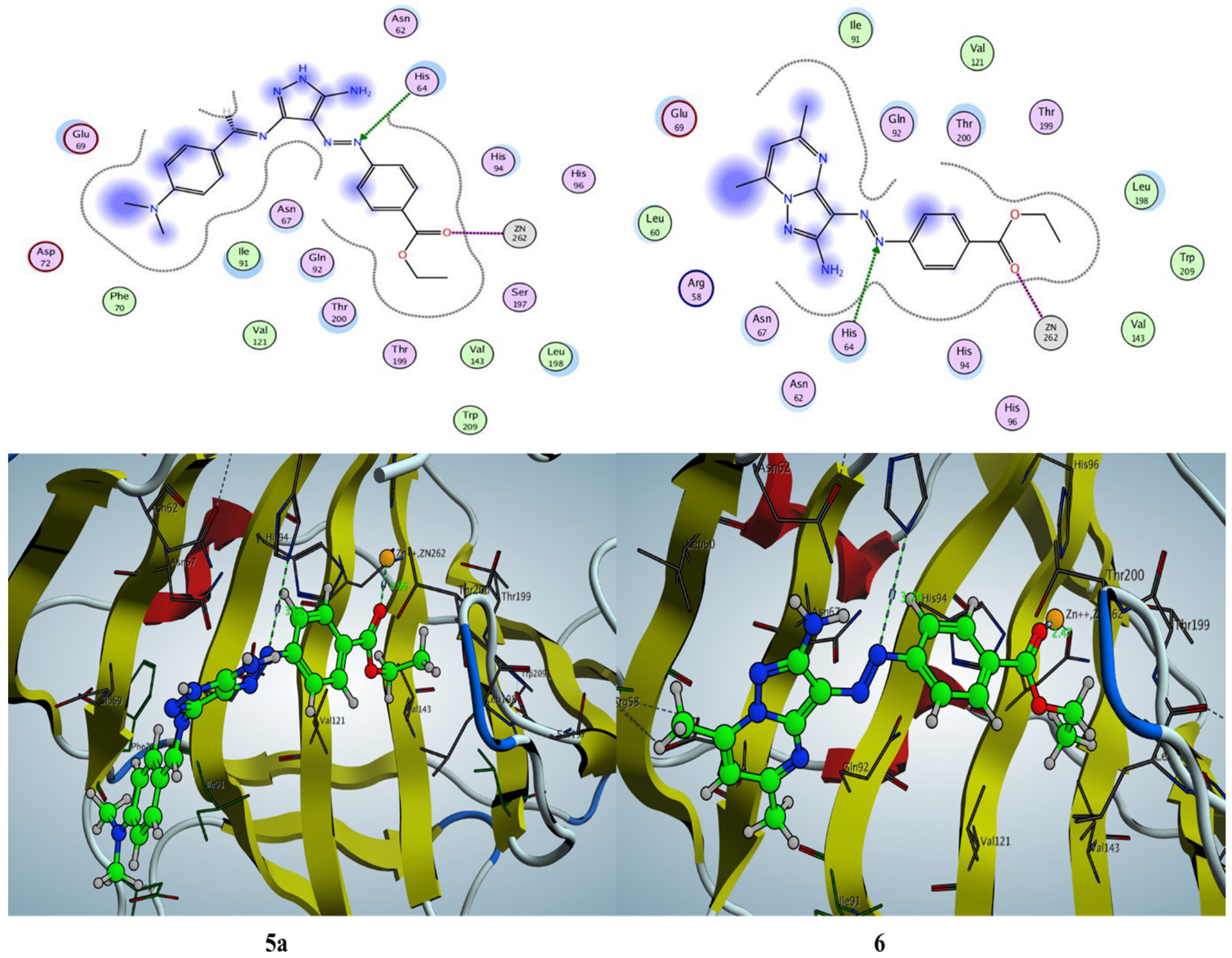
2.3.1. Docking Simulation inside the Active Site of hCA-I (PDB: 2NN7)
2.3.2. Docking Simulation inside the Active Site of hCA-II (PDB: 1H9N)
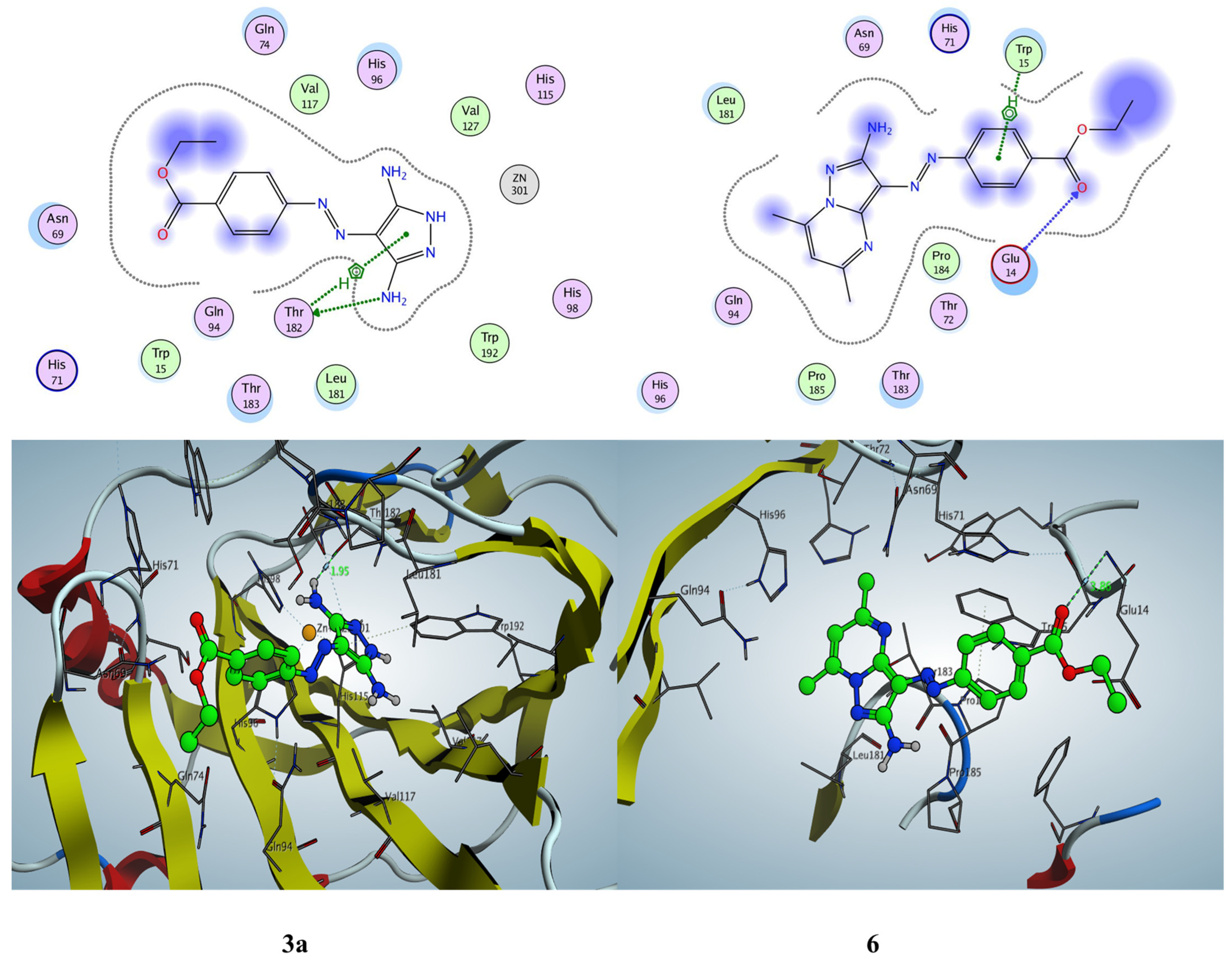
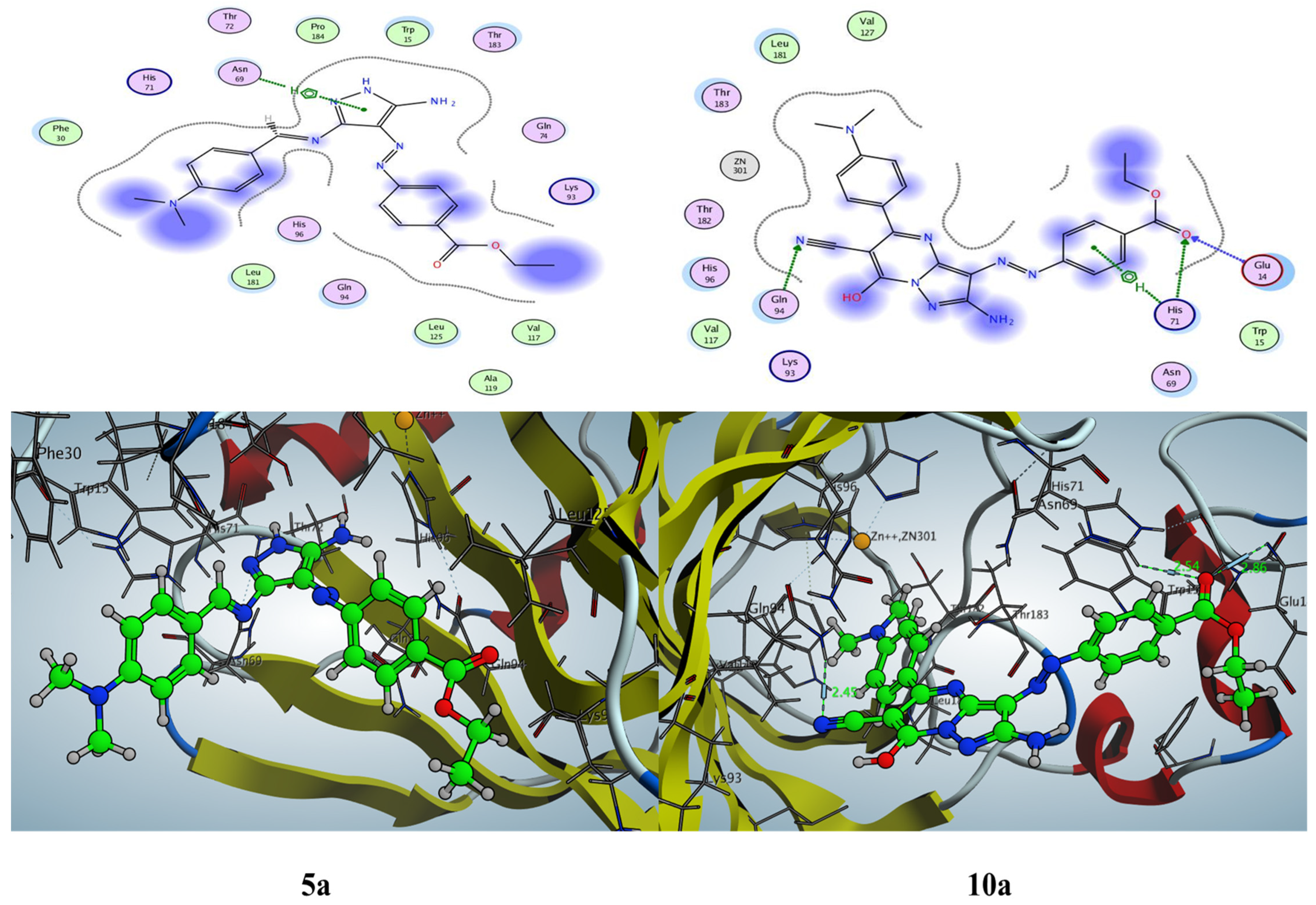
2.3.3. Docking Simulation inside the Active Site of Alpha CA (PDB: 5HPJ)
2.3.4. Docking Simulation inside the Active Site of Beta CA (PDB: 1I6P)
2.4. In Silico ADMET Prediction
3. Materials and Methods
3.1. Chemistry Materials and Equipment
- Synthesis of ethyl 4-((dicyanomethyl)diazenyl)benzoate (2)
- Synthesis of ethyl 4-((3,5-diamino-1H-pyrazol-4-yl)diazenyl)benzoates (3a–c) and (4a,b)
- Synthesis of Schiff bases derivatives (5a,b)
- Synthesis of ethyl-4-((2-amino-5,7-dimethylpyrazolo[1,5-a]pyrimidin-3-yl)diazenyl)benzoate (6)
- Synthesis of ethyl 4-((2-amino-5-methyl-7-oxo-6,7-dihydropyrazolo[1,5-a]pyrimidin-3-yl)diazenyl)benzoate (7a)
- Synthesis of ethyl 4-((2-amino-7-aryl-5-phenylpyrazolo[1,5-a]pyrimidin-3-yl)diazenyl)benzoate (8a–c)
- Synthesis of ethyl-4-((2,7-diamino-6-cyano-5-(p-substituted)pyrazolo[1,5-a]pyrimidin-3-yl)diazenyl)benzoate (9a–c)
- Synthesis of ethyl-4-((2-amino-6-cyano-7-hydroxy-5-(p-substituted)pyrazolo[1,5-a]pyrimidin-3-yl)diazenyl)benzoate (10a,b)
3.2. Biological Activity
3.2.1. Bacterial Strains, Microbiological Media, and Chemicals
3.2.2. Screening of Synthesized Compounds for Antibacterial Activity
3.2.3. Detection of Minimum Inhibitory Concentrations (MICs) and Minimum Bactericidal Concentrations (MBCs) of Pyrazoles
3.2.4. Evaluation of Antibiofilm Activities of Pyrazole and Pyrazolo[1,5-a]pyrimidine Derivatives
- Quantitative analysis of biofilm formation
- Biofilm inhibition assay
- In situ visualization of biofilm formation inhibition
- Bioassay for quorum-sensing inhibition against biofilm-forming bacteria using CV026 as an indicator strain
- (a)
- Carbonic anhydrase I and II isoenzymes purification and inhibition assay
- (b)
- Sterilization of the active pyrazolo-derivatives using gamma radiation
3.3. Molecular Docking Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Allison, A.; Kok-Fai, K.; Kalai, M. Inhibition of Quorum Sensing-Controlled Virulence Factor Production in Pseudomonas aeruginosa by South Florida Plant Extracts. Antimicrob. Agents Chemother. 2008, 52, 198–203. [Google Scholar] [CrossRef] [Green Version]
- Pulingam, T.; Parumasivam, T.; Gazzali, A.M.; Sulaiman, A.M.; Chee, J.Y.; Lakshmanan, M.; Chin, C.F.; Sudesh, K. Antimicrobial resistance: Prevalence, economic burden, mechanisms of resistance and strategies to overcome. Eur. J. Pharm. Sci. 2022, 170, 106103. [Google Scholar] [CrossRef] [PubMed]
- Baghdadi, J.D.; Coffey, K.C.; Adediran, T.; Goodman, K.E.; Pineles, L.; Magder, L.S.; O’Hara, L.M.; Pineles, B.L.; Nadimpalli, G.; Morgan, D.J.; et al. Antibiotic Use and Bacterial Infection among Inpatients in the First Wave of COVID-19: A Retrospective Cohort Study of 64,691 Patients. Antimicrob. Agents Chemother. 2021, 65, e01341-21. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, S.G.; Ahammad, S.Z. COVID-19 and antimicrobial resistance: A cross-study. Sci. Total Environ. 2022, 807, 150873. [Google Scholar] [CrossRef] [PubMed]
- Lobie, T.A.; Roba, A.A.; Booth, J.A.; Kristiansen, K.I.; Aseffa, A.; Skarstad, K.; Bjørås, M. Antimicrobial resistance: A challenge awaiting the post-COVID-19 era. Int. J. Infect. Dis. 2021, 111, 322–325. [Google Scholar] [CrossRef] [PubMed]
- Westblade, L.F.; Simon, M.S.; Satlin, M.J. Bacterial Coinfections in Coronavirus Disease 2019. Trends Microbiol. 2021, 29, 930–941. [Google Scholar] [CrossRef]
- Shafran, N.; Shafran, I.; Ben-Zvi, H.; Sofer, S.; Sheena, L.; Krause, I.; Shlomai, A.; Goldberg, E.; Sklan, E.H. Secondary bacterial infection in COVID-19 patients is a stronger predictor for death compared to influenza patients. Sci. Rep. 2021, 11, 12703. [Google Scholar] [CrossRef]
- Ibrahim, S.A.; Fayed, E.A.; Rizk, H.F.; Desouky, S.E.; Ragab, A. Hydrazonoyl bromide precursors as DHFR inhibitors for the synthesis of bis-thiazolyl pyrazole derivatives; antimicrobial activities, antibiofilm, and drug combination studies against MRSA. Bioorg. Chem. 2021, 116, 105339. [Google Scholar] [CrossRef]
- Keelara, S.; Thakur, S.; Patel, J. Biofilm Formation by Environmental Isolates of Salmonella and Their Sensitivity to Natural Antimicrobials. Foodborne Pathog. Dis. 2016, 13, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Sepandj, F.; Ceri, H.; Gibb, A.; Read, R.; Olson, M. Minimum Inhibitory Concentration versus Minimum Biofilm Eliminating Concentration in Evaluation of Antibiotic Sensitivity of Enterococci Causing Peritonitis. Perit. Dial. Int. 2007, 27, 464–465. [Google Scholar] [CrossRef]
- Tsai, C.-S.; Winans, S.C. LuxR-type quorum-sensing regulators that are detached from common scents. Mol. Microbiol. 2010, 77, 1072–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González, J.E.; Keshavan, N.D. Messing with Bacterial Quorum Sensing. Microbiol. Mol. Biol. Rev. 2006, 70, 859–875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Truchado, P.; Tomás-Barberán, F.A.; Larrosa, M.; Allende, A. Food phytochemicals act as Quorum Sensing inhibitors reducing production and/or degrading autoinducers of Yersinia enterocolitica and Erwinia carotovora. Food Control 2012, 24, 78–85. [Google Scholar] [CrossRef]
- Singh, V.K.; Mishra, A.; Jha, B. Marine bacterial extracellular polymeric substances: Characteristics and applications. In Marine Glycobiology; CRC Press: Boca Raton, FL, USA, 2016; pp. 389–398. ISBN 1315371391. [Google Scholar]
- Singh, V.K.; Kavita, K.; Prabhakaran, R.; Jha, B. Cis-9-octadecenoic acid from the rhizospheric bacterium Stenotrophomonas maltophilia BJ01 shows quorum quenching and anti-biofilm activities. Biofouling 2013, 29, 855–867. [Google Scholar] [CrossRef] [PubMed]
- Rasko, D.A.; Moreira, C.G.; Li, D.R.; Reading, N.C.; Ritchie, J.M.; Waldor, M.K.; Williams, N.; Taussig, R.; Wei, S.; Roth, M.; et al. Targeting QseC Signaling and Virulence for Antibiotic Development. Science 2008, 321, 1078–1080. [Google Scholar] [CrossRef] [Green Version]
- Paulander, W.; Varming, A.N.; Bojer, M.S.; Friberg, C.; Bæk, K.; Ingmer, H. The agr quorum sensing system in Staphylococcus aureus cells mediates death of sub-population. BMC Res. Notes 2018, 11, 503. [Google Scholar] [CrossRef]
- Khayat, M.T.; Abbas, H.A.; Ibrahim, T.S.; Khayyat, A.N.; Alharbi, M.; Darwish, K.M.; Elhady, S.S.; Khafagy, E.-S.; Safo, M.K.; Hegazy, W.A.H. Anti-Quorum Sensing Activities of Gliptins against Pseudomonas aeruginosa and Staphylococcus aureus. Biomedicines 2022, 10, 1169. [Google Scholar] [CrossRef]
- Alzahrani, A.Y.; Ammar, Y.A.; Abu-Elghait, M.; Salem, M.A.; Assiri, M.A.; Ali, T.E.; Ragab, A. Development of novel indolin-2-one derivative incorporating thiazole moiety as DHFR and quorum sensing inhibitors: Synthesis, antimicrobial, and antibiofilm activities with molecular modelling study. Bioorg. Chem. 2022, 119, 105571. [Google Scholar] [CrossRef]
- Supuran, C.T.; Capasso, C. Antibacterial carbonic anhydrase inhibitors: An update on the recent literature. Expert Opin. Ther. Pat. 2020, 30, 963–982. [Google Scholar] [CrossRef]
- Supuran, C.T.; Capasso, C. An Overview of the Bacterial Carbonic Anhydrases. Metabolites 2017, 7, 56. [Google Scholar] [CrossRef]
- Wassel, M.M.S.; Ragab, A.; Elhag Ali, G.A.M.; Mehany, A.B.M.; Ammar, Y.A. Novel adamantane-pyrazole and hydrazone hybridized: Design, synthesis, cytotoxic evaluation, SAR study and molecular docking simulation as carbonic anhydrase inhibitors. J. Mol. Struct. 2021, 1223, 128966. [Google Scholar] [CrossRef]
- Alterio, V.; di Fiore, A.; D’Ambrosio, K.; Supuran, C.T.; De Simone, G. Multiple Binding Modes of Inhibitors to Carbonic Anhydrases: How to Design Specific Drugs Targeting 15 Different Isoforms? Chem. Rev. 2012, 112, 4421–4468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Supuran, C. Bacterial Carbonic Anhydrases as Drug Targets: Toward Novel Antibiotics? Front. Pharmacol. 2011, 2, 34–42. [Google Scholar] [CrossRef] [Green Version]
- Morishita, S.; Nishimori, I.; Minakuchi, T.; Onishi, S.; Takeuchi, H.; Sugiura, T.; Vullo, D.; Scozzafava, A.; Supuran, C.T. Cloning, polymorphism, and inhibition of beta-carbonic anhydrase of Helicobacter pylori. J. Gastroenterol. 2008, 43, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Kivelä, A.-J.; Kivelä, J.; Saarnio, J.; Parkkila, S. Carbonic anhydrases in normal gastrointestinal tract and gastrointestinal tumours. World J. Gastroenterol. 2005, 11, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Ekinci, D.; Beydemir, S.; Alim, Z. Some drugs inhibit in vitro hydratase and esterase activities of human carbonic anhydrase-I and II. Pharmacol. Rep. 2007, 59, 580–587. [Google Scholar] [PubMed]
- Del Prete, S.; Vullo, D.; De Luca, V.; Carginale, V.; Osman, S.M.; AlOthman, Z.; Supuran, C.T.; Capasso, C. Comparison of the sulfonamide inhibition profiles of the α-, β- and γ-carbonic anhydrases from the pathogenic bacterium Vibrio cholerae. Bioorg. Med. Chem. Lett. 2016, 26, 1941–1946. [Google Scholar] [CrossRef]
- Abdel Gawad, N.M.; Amin, N.H.; Elsaadi, M.T.; Mohamed, F.M.M.; Angeli, A.; De Luca, V.; Capasso, C.; Supuran, C.T. Synthesis of 4-(thiazol-2-ylamino)-benzenesulfonamides with carbonic anhydrase I, II and IX inhibitory activity and cytotoxic effects against breast cancer cell lines. Bioorg. Med. Chem. 2016, 24, 3043–3051. [Google Scholar] [CrossRef]
- Ozensoy, O.; Arslan, O.; Oznur Sinan, S. A New Method for Purification of Carbonic Anhydrase Isozymes by Affinity Chromatography. Biochemistry 2004, 69, 216–219. [Google Scholar] [CrossRef]
- Kaishap, P.P.; Baruah, S.; Shekarrao, K.; Gogoi, S.; Boruah, R.C. A facile method for the synthesis of steroidal and nonsteroidal 5-methyl pyrazolo[1,5-a]pyrimidines. Tetrahedron Lett. 2014, 55, 3117–3121. [Google Scholar] [CrossRef]
- Engers, D.W.; Frist, A.Y.; Lindsley, C.W.; Hong, C.C.; Hopkins, C.R. Synthesis and structure–activity relationships of a novel and selective bone morphogenetic protein receptor (BMP) inhibitor derived from the pyrazolo[1.5-a]pyrimidine scaffold of Dorsomorphin: The discovery of ML347 as an ALK2 versus ALK3 selective MLPCN. Bioorg. Med. Chem. Lett. 2013, 23, 3248–3252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayman, R.; Radwan, A.M.; Elmetwally, A.M.; Ammar, Y.A.; Ragab, A. Discovery of novel pyrazole and pyrazolo[1,5-a]pyrimidine derivatives as cyclooxygenase inhibitors (COX-1 and COX-2) using molecular modeling simulation. Arch. Pharm. 2022, e2200395. [Google Scholar] [CrossRef] [PubMed]
- Marinescu, M. Synthesis of Antimicrobial Benzimidazole–Pyrazole Compounds and Their Biological Activities. Antibiotics 2021, 10, 1002. [Google Scholar] [CrossRef] [PubMed]
- Ali Mohamed, H.; Ammar, Y.A.; Elhagali, G.A.M.; Eyada, H.A.; Aboul-Magd, D.S.; Ragab, A. In Vitro Antimicrobial Evaluation, Single-Point Resistance Study, and Radiosterilization of Novel Pyrazole Incorporating Thiazol-4-one/Thiophene Derivatives as Dual DNA Gyrase and DHFR Inhibitors against MDR Pathogens. ACS Omega 2022, 7, 4970–4990. [Google Scholar] [CrossRef] [PubMed]
- Da Costa, L.; Scheers, E.; Coluccia, A.; Casulli, A.; Roche, M.; di Giorgio, C.; Neyts, J.; Terme, T.; Cirilli, R.; La Regina, G.; et al. Structure-Based Drug Design of Potent Pyrazole Derivatives against Rhinovirus Replication. J. Med. Chem. 2018, 61, 8402–8416. [Google Scholar] [CrossRef] [Green Version]
- Corona, A.; Onnis, V.; Deplano, A.; Bianco, G.; Demurtas, M.; Distinto, S.; Cheng, Y.-C.; Alcaro, S.; Esposito, F.; Tramontano, E. Design, synthesis and antiviral evaluation of novel heteroarylcarbothioamide derivatives as dual inhibitors of HIV-1 reverse transcriptase-associated RNase H and RDDP functions. Pathog. Dis. 2017, 75, ftx078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Zharani, M.; Al-Eissa, M.S.; Rudayni, H.A.; Ali, D.; Alkahtani, S.; Surendrakumar, R.; Idhayadhulla, A. Pyrazolo[3,4-b]pyridin-3(2H)-one derivatives: Synthesis and their investigation of mosquito larvicidal activity. J. King Saud Univ.-Sci. 2022, 34, 101767. [Google Scholar] [CrossRef]
- Naim, M.J.; Alam, O.; Alam, M.J.; Hassan, M.Q.; Siddiqui, N.; Naidu, V.G.M.; Alam, M.I. Design, synthesis and molecular docking of thiazolidinedione based benzene sulphonamide derivatives containing pyrazole core as potential anti-diabetic agents. Bioorg. Chem. 2018, 76, 98–112. [Google Scholar] [CrossRef]
- Faidallah, H.M.; Al-Mohammadi, M.M.; Alamry, K.A.; Khan, K.A. Synthesis and biological evaluation of fluoropyrazolesulfonylurea and thiourea derivatives as possible antidiabetic agents. J. Enzyme Inhib. Med. Chem. 2016, 31, 157–163. [Google Scholar] [CrossRef] [Green Version]
- Meta, E.; Brullo, C.; Tonelli, M.; Franzblau, S.G.; Wang, Y.; Ma, R.; Baojie, W.; Orena, B.S.; Pasca, M.R.; Bruno, O. Pyrazole and imidazo [1,2-b] pyrazole derivatives as new potential anti-tuberculosis agents. Med. Chem. 2019, 15, 17–27. [Google Scholar] [CrossRef]
- Xu, Z.; Gao, C.; Ren, Q.-C.; Song, X.-F.; Feng, L.-S.; Lv, Z.-S. Recent advances of pyrazole-containing derivatives as anti-tubercular agents. Eur. J. Med. Chem. 2017, 139, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, F.; Naureen, S.; Choudhry, S.; Huma, R.; Ashraf, M.; Al-Rashida, M.; Jahan, B.; Hyder Khan, M.; Iqbal, F.; Ali Munawar, M.; et al. Evaluation of α-glucosidase inhibiting potentials with docking calculations of synthesized arylidene-pyrazolones. Bioorg. Chem. 2018, 77, 507–514. [Google Scholar] [CrossRef] [PubMed]
- De Grauw, J.C.; van Loon, J.P.A.M.; van de Lest, C.H.A.; Brunott, A.; van Weeren, P.R. In vivo effects of phenylbutazone on inflammation and cartilage-derived biomarkers in equine joints with acute synovitis. Vet. J. 2014, 201, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Ortega, J.E.; Gonzalez-Lira, V.; Horrillo, I.; Herrera-Marschitz, M.; Callado, L.F.; Meana, J.J. Additive effect of rimonabant and citalopram on extracellular serotonin levels monitored with in vivo microdialysis in rat brain. Eur. J. Pharmacol. 2013, 709, 13–19. [Google Scholar] [CrossRef]
- Fioravanti, R.; Bolasco, A.; Manna, F.; Rossi, F.; Orallo, F.; Ortuso, F.; Alcaro, S.; Cirilli, R. Synthesis and biological evaluation of N-substituted-3,5-diphenyl-2-pyrazoline derivatives as cyclooxygenase (COX-2) inhibitors. Eur. J. Med. Chem. 2010, 45, 6135–6138. [Google Scholar] [CrossRef]
- Penning, T.D.; Talley, J.J.; Bertenshaw, S.R.; Carter, J.S.; Collins, P.W.; Docter, S.; Graneto, M.J.; Lee, L.F.; Malecha, J.W.; Miyashiro, J.M.; et al. Synthesis and Biological Evaluation of the 1,5-Diarylpyrazole Class of Cyclooxygenase-2 Inhibitors: Identification of 4-[5-(4-Methylphenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl]benzenesulfonamide (SC-58635, Celecoxib). J. Med. Chem. 1997, 40, 1347–1365. [Google Scholar] [CrossRef] [PubMed]
- Devi, N.; Shankar, R.; Singh, V. 4-Formyl-Pyrazole-3-Carboxylate: A Useful Aldo-X Bifunctional Precursor for the Syntheses of Pyrazole-fused/Substituted Frameworks. J. Heterocycl. Chem. 2018, 55, 373–390. [Google Scholar] [CrossRef]
- Chalyk, B.A.; Khutorianskyi, A.; Lysenko, A.; Fil, Y.; Kuchkovska, Y.O.; Gavrilenko, K.S.; Bakanovych, I.; Moroz, Y.S.; Gorlova, A.O.; Grygorenko, O.O. Regioselective Synthesis of Functionalized 3- or 5-Fluoroalkyl Isoxazoles and Pyrazoles from Fluoroalkyl Ynones and Binucleophiles. J. Org. Chem. 2019, 84, 15212–15225. [Google Scholar] [CrossRef]
- Mao, F.; Ni, W.; Xu, X.; Wang, H.; Wang, J.; Ji, M.; Li, J. Chemical structure-related drug-like criteria of global approved drugs. Molecules 2016, 21, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kramer, C.S. Chapter 5 Pyrazoles. In Privileged Scaffolds in Medicinal Chemistry; Drug Discovery; The Royal Society of Chemistry: London, UK, 2015; pp. 115–131. ISBN 978-1-78262-030-3. [Google Scholar]
- Khokhlova, O.N.; Borozdina, N.A.; Sadovnikova, E.S.; Pakhomova, I.A.; Rudenko, P.A.; Korolkova, Y.V.; Kozlov, S.A.; Dyachenko, I.A. Comparative Study of the Aftereffect of CO2 Inhalation or Tiletamine–Zolazepam–Xylazine Anesthesia on Laboratory Outbred Rats and Mice. Biomedicines 2022, 10, 512. [Google Scholar]
- Abdelgawad, M.A.; Elkanzi, N.A.A.; Musa, A.; Ghoneim, M.M.; Ahmad, W.; Elmowafy, M.; Abdelhaleem Ali, A.M.; Abdelazeem, A.H.; Bukhari, S.N.A.; El-Sherbiny, M.; et al. Optimization of pyrazolo[1,5-a]pyrimidine based compounds with pyridine scaffold: Synthesis, biological evaluation and molecular modeling study. Arab. J. Chem. 2022, 15, 104015. [Google Scholar] [CrossRef]
- Fraley, M.E.; Rubino, R.S.; Hoffman, W.F.; Hambaugh, S.R.; Arrington, K.L.; Hungate, R.W.; Bilodeau, M.T.; Tebben, A.J.; Rutledge, R.Z.; Kendall, R.L.; et al. Optimization of a pyrazolo[1,5-a]pyrimidine class of KDR kinase inhibitors: Improvements in physical properties enhance cellular activity and pharmacokinetics. Bioorg. Med. Chem. Lett. 2002, 12, 3537–3541. [Google Scholar] [CrossRef] [PubMed]
- Ivachtchenko, A.V.; Dmitriev, D.E.; Golovina, E.S.; Kadieva, M.G.; Koryakova, A.G.; Kysil, V.M.; Mitkin, O.D.; Okun, I.M.; Tkachenko, S.E.; Vorobiev, A.A. (3-Phenylsulfonylcycloalkano[e and d]pyrazolo[1,5-a]pyrimidin-2-yl)amines: Potent and Selective Antagonists of the Serotonin 5-HT6 Receptor. J. Med. Chem. 2010, 53, 5186–5196. [Google Scholar] [CrossRef] [PubMed]
- Hassan, A.S.; Morsy, N.M.; Awad, H.M.; Ragab, A. Synthesis, molecular docking, and in silico ADME prediction of some fused pyrazolo[1,5-a]pyrimidine and pyrazole derivatives as potential antimicrobial agents. J. Iran. Chem. Soc. 2022, 19, 521–545. [Google Scholar] [CrossRef]
- Selleri, S.; Bruni, F.; Costagli, C.; Costanzo, A.; Guerrini, G.; Ciciani, G.; Gratteri, P.; Besnard, F.; Costa, B.; Montali, M.; et al. A Novel Selective GABAA α1 Receptor Agonist Displaying Sedative and Anxiolytic-like Properties in Rodents. J. Med. Chem. 2005, 48, 6756–6760. [Google Scholar] [CrossRef]
- Hwang, J.Y.; Windisch, M.P.; Jo, S.; Kim, K.; Kong, S.; Kim, H.C.; Kim, S.; Kim, H.; Lee, M.E.; Kim, Y.; et al. Discovery and characterization of a novel 7-aminopyrazolo[1,5-a]pyrimidine analog as a potent hepatitis C virus inhibitor. Bioorg. Med. Chem. Lett. 2012, 22, 7297–7301. [Google Scholar] [CrossRef]
- Selleri, S.; Bruni, F.; Costagli, C.; Costanzo, A.; Guerrini, G.; Ciciani, G.; Costa, B.; Martini, C. 2-Arylpyrazolo[1,5-a]pyrimidin-3-yl acetamides. New potent and selective peripheral benzodiazepine receptor ligands. Bioorg. Med. Chem. 2001, 9, 2661–2671. [Google Scholar] [CrossRef]
- Hassan, A.S.; Morsy, N.M.; Aboulthana, W.M.; Ragab, A. In vitro enzymatic evaluation of some pyrazolo[1,5-a]pyrimidine derivatives: Design, synthesis, antioxidant, anti-diabetic, anti-Alzheimer, and anti-arthritic activities with molecular modeling simulation. Drug Dev. Res. 2022; Early View. [Google Scholar] [CrossRef]
- Asati, V.; Anant, A.; Patel, P.; Kaur, K.; Gupta, G.D. Pyrazolopyrimidines as anticancer agents: A review on structural and target-based approaches. Eur. J. Med. Chem. 2021, 225, 113781. [Google Scholar] [CrossRef]
- Cherukupalli, S.; Hampannavar, G.A.; Chinnam, S.; Chandrasekaran, B.; Sayyad, N.; Kayamba, F.; Reddy Aleti, R.; Karpoormath, R. An appraisal on synthetic and pharmaceutical perspectives of pyrazolo[4,3-d]pyrimidine scaffold. Bioorg. Med. Chem. 2018, 26, 309–339. [Google Scholar] [CrossRef]
- Cherukupalli, S.; Karpoormath, R.; Chandrasekaran, B.; Hampannavar, G.A.; Thapliyal, N.; Palakollu, V.N. An insight on synthetic and medicinal aspects of pyrazolo[1,5-a]pyrimidine scaffold. Eur. J. Med. Chem. 2017, 126, 298–352. [Google Scholar] [CrossRef]
- Van Cauwenbergh, T.; Theys, E.; Stroeykens, D.; Croonenborghs, B.; Gillet, A.; DeMent, A.; Van Schepdael, A.; Haghedooren, E. The effect of Gamma and Ethylene Oxide Sterilization on a Selection of Active Pharmaceutical Ingredients for Ophthalmics. J. Pharm. Sci. 2022, 111, 2011–2017. [Google Scholar] [CrossRef] [PubMed]
- Ammar, Y.A.; Micky, J.A.; Aboul-Magd, D.S.; Abd El-Hafez, S.M.A.; Hessein, S.A.; Ali, A.M.; Ragab, A. Development and radiosterilization of new hydrazono-quinoline hybrids as DNA gyrase and topoisomerase IV inhibitors: Antimicrobial and hemolytic activities against uropathogenic isolates with molecular docking study. Chem. Biol. Drug Des. 2022; Early View. [Google Scholar] [CrossRef]
- Fayed, E.A.; Mohsen, M.; El-Gilil, S.M.A.; Aboul-Magd, D.S.; Ragab, A. Novel cyclohepta[b]thiophene derivative incorporating pyrimidine, pyridine, and chromene moiety as potential antimicrobial agents targeting DNA gyrase. J. Mol. Struct. 2022, 1262, 133028. [Google Scholar] [CrossRef]
- Ragab, A.; Abusaif, M.S.; Gohar, N.A.; Aboul-Magd, D.S.; Fayed, E.A.; Ammar, Y.A. Development of new spiro[1,3]dithiine-4,11′-indeno[1,2-b]quinoxaline derivatives as S. aureus Sortase A inhibitors and radiosterilization with molecular modeling simulation. Bioorg. Chem. 2022, 131, 106307. [Google Scholar] [CrossRef] [PubMed]
- Abdelgalil, M.M.; Ammar, Y.A.; Elhag Ali, G.A.M.; Ali, A.K.; Ragab, A. A novel of quinoxaline derivatives tagged with pyrrolidinyl scaffold as a new class of antimicrobial agents: Design, synthesis, antimicrobial activity, and molecular docking simulation. J. Mol. Struct. 2023, 1274, 134443. [Google Scholar] [CrossRef]
- Elsisi, D.M.; Ragab, A.; Elhenawy, A.A.; Farag, A.A.; Ali, A.M.; Ammar, Y.A. Experimental and theoretical investigation for 6-Morpholinosulfonylquinoxalin-2(1H)-one and its haydrazone derivate: Synthesis, characterization, tautomerization and antimicrobial evaluation. J. Mol. Struct. 2022, 1247, 131314. [Google Scholar] [CrossRef]
- Ibrahim, S.A.; Ragab, A.; El-Ghamry, H.A. Coordination compounds of pyrazolone-based ligand: Design, characterization, biological evaluation, antitumor efficiency, and DNA binding evaluation supported by in silico studies. Appl. Organomet. Chem. 2022, 36, e6508. [Google Scholar] [CrossRef]
- Ragab, A.; Elsisi, D.M.; Abu Ali, O.A.; Abusaif, M.S.; Askar, A.A.; Farag, A.A.; Ammar, Y.A. Design, synthesis of new novel quinoxalin-2(1H)-one derivatives incorporating hydrazone, hydrazine, and pyrazole moieties as antimicrobial potential with in-silico ADME and molecular docking simulation. Arab. J. Chem. 2022, 15, 103497. [Google Scholar] [CrossRef]
- Supuran, C.T. Carbonic anhydrase inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 3467–3474. [Google Scholar] [CrossRef]
- Arabaci, B.; Gulcin, I.; Alwasel, S. Capsaicin: A Potent Inhibitor of Carbonic Anhydrase Isoenzymes. Molecules 2014, 19, 10103–10114. [Google Scholar] [CrossRef]
- Akıncıoğlu, A.; Topal, M.; Gülçin, İ.; Göksu, S. Novel Sulphamides and Sulphonamides Incorporating the Tetralin Scaffold as Carbonic Anhydrase and Acetylcholine Esterase Inhibitors. Arch. Pharm. 2014, 347, 68–76. [Google Scholar] [CrossRef]
- Çolak, Ş. Ionizing Radiation Used in Drug Sterilization, Characterization of Radical Intermediates by Electron Spin Resonance (ESR) Analyses. In Evolution of Ionizing Radiation Research; IntechOpen: Rijeka, Croatia, 2015; p. 281. [Google Scholar] [CrossRef] [Green Version]
- Dziedzic-Goclawska, A.; Kaminski, A.; Uhrynowska-Tyszkiewicz, I.; Michalik, J.; Stachowicz, W. Trends in Radiation Sterilization of Health Care Products; International Atomic Energy Agency: Vienna, Austria, 2008; pp. 231–256. [Google Scholar]
- Ragab, A.; Abusaif, M.S.; Aboul-Magd, D.S.; Wassel, M.M.S.; Elhagali, G.A.M.; Ammar, Y.A. A new exploration toward adamantane derivatives as potential anti-MDR agents: Design, synthesis, antimicrobial, and radiosterilization activity as potential topoisomerase IV and DNA gyrase inhibitors. Drug Dev. Res. 2022, 83, 1305–1330. [Google Scholar] [CrossRef] [PubMed]
- Ragab, A.; Ammar, Y.A.; Ezzat, A.; Mahmoud, A.M.; Basseem, M.; Mohamed, I.; El-tabl, A.S.; Farag, R.S. Synthesis, characterization, thermal properties, antimicrobial evaluation, ADMET study, and molecular docking simulation of new mono Cu (II) and Zn (II) complexes with 2-oxoindole derivatives. Comput. Biol. Med. 2022, 145, 105473. [Google Scholar] [CrossRef]
- Hughes, J.P.; Rees, S.; Kalindjian, S.B.; Philpott, K.L. Principles of early drug discovery. Br. J. Pharmacol. 2011, 162, 1239–1249. [Google Scholar] [CrossRef] [Green Version]
- Saadon, K.E.; Taha, N.M.H.; Mahmoud, N.A.; Elhagali, G.A.M.; Ragab, A. Synthesis, characterization, and in vitro antibacterial activity of some new pyridinone and pyrazole derivatives with some in silico ADME and molecular modeling study. J. Iran. Chem. Soc. 2022, 19, 3899–3917. [Google Scholar] [CrossRef]
- Esser, C. Biology and function of the aryl hydrocarbon receptor: Report of an international and interdisciplinary conference. Arch. Toxicol. 2012, 86, 1323–1329. [Google Scholar] [CrossRef]
- Ramanathan, M.; Wan, J.; Liu, Y.-H.; Peng, S.-M.; Liu, S.-T. Synthesis of 2-arylamino-3-cyanoquinolines via a cascade reaction through a nitrilium intermediate. Org. Biomol. Chem. 2020, 18, 975–982. [Google Scholar] [CrossRef]
- Alzahrani, A.Y.; Ammar, Y.A.; Salem, M.A.; Abu-Elghait, M.; Ragab, A. Design, synthesis, molecular modeling, and antimicrobial potential of novel 3-[(1H-pyrazol-3-yl)imino]indolin-2-one derivatives as DNA gyrase inhibitors. Arch. Pharm. 2022, 355, 2100266. [Google Scholar] [CrossRef]
- Wayne, P.A. Clinical and Laboratory Standards Institute; Performance Standards for Antimicrobial Susceptibility Testing; Scientific Information Database: Tehran, Iran, 2011. [Google Scholar]
- Wayne, P.A. Clinical and Laboratory Standards Institute; Performance Standards for Antimicrobial Susceptibility Testing; Scientific Information Database: Tehran, Iran, 2020. [Google Scholar]
- Saqr, A.A.; Aldawsari, M.F.; Khafagy, E.-S.; Shaldam, M.A.; Hegazy, W.A.H.; Abbas, H.A. A Novel Use of Allopurinol as A Quorum-Sensing Inhibitor in Pseudomonas aeruginosa. Antibiotics 2021, 10, 1385. [Google Scholar] [CrossRef]
- Gatsing, D.; Tchakoute, V.; Ngamga, D.; Kuiate, J.R.; Tamokou, J.D.D.; NJI, N.B.; Tchouanguep, F.M.; Fodouop, S.P. In Vitro Antibacterial Activity Of Crinum Purpurascens Herb Leaf Extract Against The Salmonella Species Causing Typhoid Fever And Its Toxicological Evaluation. Iran. J. Med. Sci. 2009, 34, 126–136. [Google Scholar]
- Kouidhi, B.; Zmantar, T.; Hentati, H.; Bakhrouf, A. Cell surface hydrophobicity, biofilm formation, adhesives properties and molecular detection of adhesins genes in Staphylococcus aureus associated to dental caries. Microb. Pathog. 2010, 49, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Adukwu, E.C.; Allen, S.C.H.; Phillips, C.A. The anti-biofilm activity of lemongrass (Cymbopogon flexuosus) and grapefruit (Citrus paradisi) essential oils against five strains of Staphylococcus aureus. J. Appl. Microbiol. 2012, 113, 1217–1227. [Google Scholar] [CrossRef] [PubMed]
- Tutar, U.; Koçyiğit, Ü.M.; Gezegen, H. Evaluation of antimicrobial, antibiofilm and carbonic anhydrase inhibition profiles of 1,3-bis-chalcone derivatives. J. Biochem. Mol. Toxicol. 2019, 33, e22281. [Google Scholar] [CrossRef]
- Chaieb, K.; Kouidhi, B.; Jrah, H.; Mahdouani, K.; Bakhrouf, A. Antibacterial activity of Thymoquinone, an active principle of Nigella sativa and its potency to prevent bacterial biofilm formation. BMC Complement. Altern. Med. 2011, 11, 29. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-H.; Cho, M.H.; Lee, J. 3-Indolylacetonitrile Decreases Escherichia coli O157:H7 Biofilm Formation and Pseudomonas aeruginosa Virulence. Environ. Microbiol. 2011, 13, 62–73. [Google Scholar] [CrossRef]
- Husain, F.M.; Ahmad, I.; Asif, M.; Tahseen, Q. Influence of clove oil on certain quorum-sensing-regulated functions and biofilm of Pseudomonas aeruginosa and Aeromonas hydrophila. J. Biosci. 2013, 38, 835–844. [Google Scholar] [CrossRef]
- McLean, R.J.C.; Pierson, L.S.; Fuqua, C. A simple screening protocol for the identification of quorum signal antagonists. J. Microbiol. Methods 2004, 58, 351–360. [Google Scholar] [CrossRef]
- Koh, K.H.; Tham, F.-Y. Screening of traditional Chinese medicinal plants for quorum-sensing inhibitors activity. J. Microbiol. Immunol. Infect. 2011, 44, 144–148. [Google Scholar] [CrossRef] [Green Version]
- Aktaş, A.; Taslimi, P.; Gülçin, İ.; Gök, Y. Novel NHC Precursors: Synthesis, Characterization, and Carbonic Anhydrase and Acetylcholinesterase Inhibitory Properties. Arch. Pharm. 2017, 350, e201700045. [Google Scholar] [CrossRef]
- Verpoorte, J.A.; Mehta, S.; Edsall, J.T. Esterase activities of human carbonic anhydrases B and C. J. Biol. Chem. 1967, 242, 4221–4229. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Şentürk, M.; Gülçin, İ.; Ciftci, M.; Küfrevioğlu, Ö.İ. Dantrolene Inhibits Human Erythrocyte Glutathione Reductase. Biol. Pharm. Bull. 2008, 31, 2036–2039. [Google Scholar] [CrossRef] [Green Version]
- Sarikaya, S.B.O.; Sisecioglu, M.; Cankaya, M.; Gulcin, İ.; Ozdemir, H. Inhibition profile of a series of phenolic acids on bovine lactoperoxidase enzyme. J. Enzyme Inhib. Med. Chem. 2015, 30, 479–483. [Google Scholar] [CrossRef]
- Özer, A.; Turker, S.; Çolak, S.; Korkmaz, M.; Kiliç, E.; Özalp, M. The effects of gamma irradiation on diclofenac sodium, liposome and niosome ingredients for rheumatoid arthritis. Interv. Med. Appl. Sci. 2013, 5, 122–130. [Google Scholar] [CrossRef] [Green Version]
- Husseiny, S.H.M.; Helimish, F.A. Contamination of Eye Drops with Bacillus Species and Evaluation of Their Virulence Factors. World Appl. Sci. J. 2012, 19, 847–855. [Google Scholar] [CrossRef]
- Khattab, E.S.A.E.H.; Ragab, A.; Abol-Ftouh, M.A.; Elhenawy, A.A. Therapeutic strategies for Covid-19 based on molecular docking and dynamic studies to the ACE-2 receptors, Furin, and viral spike proteins. J. Biomol. Struct. Dyn. 2022, 40, 13291–13309. [Google Scholar] [CrossRef]



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound No. | Diameter of Inhibition Zones in mm | |||||
|---|---|---|---|---|---|---|
| Gram-Positive Bacteria | Gram-Negative Bacteria | |||||
| E. faecalis DH-5478 | S. aureus DH-432 | S. pyogenes DH-3467 | A. baumannii DH-243 | E. coli DH-5987 | P. aeruginosa DH-5698 | |
| 3a | 30.6 | 31.1 | 31.5 | 29.3 | 32 | 30.8 |
| 3b | 24.4 | 25.2 | 24.7 | 23.1 | 26.9 | 25.1 |
| 3c | 21.3 | 23.8 | 22.6 | 22.5 | 24.7 | 23.2 |
| 4 | 22.1 | 24.7 | 23.4 | 21.3 | 22.4 | 20.5 |
| 5a | 28.2 | 28.6 | 28.9 | 28.0 | 29.9 | 28.3 |
| 5b | 25.4 | 26.2 | 24.1 | 23.4 | 25.7 | 24.7 |
| 6 | 29.5 | 29.5 | 30.2 | 28.8 | 31.0 | 28.8 |
| 7 | 23.0 | 25.3 | 22.8 | 24.5 | 25.8 | 24.0 |
| 8a | 20.6 | 22.0 | 21.4 | 19.7 | 23.4 | 21.6 |
| 8b | 17.3 | 19.5 | 18.7 | 16.5 | 20.1 | 18.4 |
| 8c | 15.1 | 16.1 | 15.4 | 15.2 | 17.8 | 16.3 |
| 9a | 28.7 | 29.1 | 29.3 | 28.4 | 30.4 | 28.5 |
| 9b | 24.3 | 25.0 | 25.7 | 23.0 | 26.5 | 25.4 |
| 9c | 23.1 | 24.2 | 23.5 | 23.9 | 25.2 | 24.1 |
| 10a | 29.8 | 30.6 | 30.7 | 29.1 | 31.5 | 30.1 |
| 10b | 25.1 | 26.8 | 26.3 | 24.2 | 27.1 | 26.3 |
| E | 11 | 12 | 10.5 | NA | NA | NA |
| AK | NA | NA | NA | 13 | 11 | 14 |
| DMSO | -ve | -ve | -ve | -ve | -ve | -ve |
| Compound No. | MIC (Mean ± SEM) in µg/mL (MBC/MIC) | |||||
|---|---|---|---|---|---|---|
| Gram-Positive Bacteria | Gram-Negative Bacteria | |||||
| E. faecalis DH-5478 | S. aureus DH-432 | S. pyogenes DH-3467 | A. baumannii DH-243 | E. coli DH-5987 | P. aeruginosa DH-5698 | |
| 3a | 0.125 ± 0.1(0.5) | 0.125 ± 0.3(0.5) | 0.125 ± 0.3(0.5) | 0.25 ± 0.2(0.5) | 0.0625 ± 0.4(0.5) | 0.125 ± 0.2(0.5) |
| 3b | 0.75 ± 0.2(0.5) | 0.5 ± 0.1(1) | 0.75 ± 0.4(1) | 1 ± 0.3(0.5) | 0.5 ± 0.2(1) | 1 ± 0.2(0.5) |
| 3c | 1 ± 0.3 (1) | 0.75 ± 0.2 (1) | 0.75 ± 0.2(0.5) | 2 ± 0.2(1) | 0.75 ± 0.5(0.5) | 1 ± 0.3(1) |
| 4 | 2 ± 0.3(0.5) | 1 ± 0.4(0.5) | 1.5 ± 0.3(0.5) | 2 ± 0.4(0.5) | 0.75 ± 0.3(1) | 1.5 ± 0.2(1) |
| 5a | 0.5 ± 0.2(0.5) | 0.25 ± 0.2(0.5) | 0.5 ± 0.1(0.5) | 0.5 ± 0.3(0.5) | 0.25 ± 0.2(0.5) | 0.5 ± 0.1(0.5) |
| 5b | 1.5 ± 0.3(1) | 1 ± 0.3(1) | 1 ± 0.2(1) | 2 ± 0.1(0.5) | 1 ± 0.2(0.5) | 2 ± 0.3(1) |
| 6 | 0.375 ± 0.2(0.5) | 0.187 ± 0.1(0.5) | 0.25 ± 0.3(0.5) | 0.5 ± 0.1(0.5) | 0.25 ± 0.1(0.5) | 0.375 ± 0.4(0.5) |
| 7 | 2 ± 0.4(1) | 1 ± 0.3(1) | 1.5 ± 0.2(1) | 2 ± 0.3(1) | 1.5 ± 0.2(0.5) | 2 ± 0.3(0.5) |
| 8a | 4 ± 0.3(1) | 2 ± 0.2(1) | 4 ± 0.4(1) | 4 ± 0.2(2) | 2 ± 0.2(1) | 2 ± 0.3(1) |
| 8b | 4 ± 0.1(2) | 4 ± 0.3(2) | 8 ± 0.2(1) | 8 ± 0.2(2) | 2 ± 0.3(1) | 4 ± 0.1(2) |
| 8c | 8 ± 0.2(2) | 4 ± 0.1(1) | 4 ± 0.2(2) | 8 ± 0.4(2) | 4 ± 0.1(2) | 8 ± 0.3(2) |
| 9a | 0.5 ± 0.1(0.5) | 0.25 ± 0.2(0.5) | 0.25 ± 0.2(0.5) | 0.375 ± 0.2(0.5) | 0.125 ± 0.1(0.5) | 0.375 ± 0.3(0.5) |
| 9b | 2 ± 0.2(1) | 1.5 ± 0.2(1) | 2 ± 0.3(0.5) | 2 ± 0.1(1) | 1 ± 0.3(0.5) | 1.5 ± 0.2(0.5) |
| 9c | 4 ± 0.2(1) | 2 ± 0.3(1) | 2 ± 0.1(1) | 4 ± 0.4(1) | 1 ± 0.3(1) | 2 ± 0.2(1) |
| 10a | 0.187 ± 0.1(0.5) | 0.125 ± 0.2(0.5) | 0.187 ± 0.2(0.5) | 0.375 ± 0.4(0.5) | 0.125 ± 0.2(.5) | 0.187 ± 0.1(0.5) |
| 10b | 2 ± 0.3(1) | 1.5 ± 0.3(0.5) | 1.5 ± 0.2(0.5) | 2 ± 0.2(1) | 0.75 ± 0.1(0.5) | 1.5 ± 0.1(0.5) |
| E | 32 ± 0.1(4) | 8 ± 0.2(2) | 64 ± 0.3(2) | NA | NA | NA |
| AK | NA | NA | NA | 128 ± 0.2(4) | 32 ± 0.2(4) | 128 ± 0.1(4) |
| DMSO | -ve | -ve | -ve | -ve | -ve | -ve |
| Purification Step | Activity (EU/mL) | Total Volume (mL) | Protein (mg/mL) | Total Protein (mg) | Total Activity (EU) | Specific Activity (EU/mg) | Yield (%) | Purification Factor |
|---|---|---|---|---|---|---|---|---|
| Hemolysate | 140 | 90 | 15.9 | 1370 | 12,122 | 9.23 | 100 | 1 |
| hCA-I | 372 | 15 | 0.5 | 4.5 | 3691 | 950 | 55 | 100 |
| hCA-II | 800 | 10 | 0.2 | 0.85 | 4857 | 8300 | 62 | 900 |
| Compound No. | hCA-I | hCA-II | ||
|---|---|---|---|---|
| IC50 (nM) | Ki (nM) | IC50 (nM) | Ki (nM) | |
| 3a | 92.34 | 88.78 | 73.2 | 91.85 |
| 5a | 137.99 | 176.21 | 161.22 | 144.27 |
| 6 | 168.84 | 153.46 | 130.29 | 145.58 |
| 9a | 123.41 | 101.55 | 142.28 | 135.78 |
| 10a | 153.45 | 143.59 | 121.98 | 139.37 |
| ACZ a | 994.78 | 1052.22 | 900.33 | 981.64 |
| Dose (kGy) | Total Count CFU/g | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 3a | 5a | 6 | 9a | 10a | ||||||
| Fungi | Bacteria | Fungi | Bacteria | Fungi | Bacteria | Fungi | Bacteria | Fungi | Bacteria | |
| Control | 1.5 × 101 | 2 × 101 | 2 × 102 | 0 | 2 × 101 | 3 × 101 | 1.5 × 102 | 0 | 1 × 102 | 1 × 101 |
| 1 | 1.5 × 101 | 0 | 1 × 102 | 0 | 1.5 × 101 | 0 | 1 × 102 | 0 | 1 × 101 | 1 × 101 |
| 3 | 1 × 101 | 0 | 1 × 101 | 0 | 0 | 0 | 0 | 0 | 1 × 101 | 0 |
| 5 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 7 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 10 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 15 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 20 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Test Items | Most-Active Pyrazole and Pyrazolo[1,5-a]pyrimidine Derivatives Compared with Acetazolamide | ||||||
|---|---|---|---|---|---|---|---|
| 3a | 5a | 6 | 9a | 10a | ACZ | ||
| Molecular Properties | |||||||
| SwissADME | Xlog P3 | 1.52 | 3.62 | 2.77 | 3.68 | 4.01 | -0.26 |
| TPSA (°A2) | 131.74 | 121.32 | 107.23 | 160.28 | 154.49 | 151.66 | |
| M. Wt. | 274.28 | 405.45 | 338.36 | 469.50 | 470.48 | 222.25 | |
| nHBA (NO) | 5 | 6 | 6 | 7 | 8 | 6 | |
| nHBD (OHNH) | 3 | 2 | 1 | 2 | 2 | 2 | |
| NRB | 5 | 8 | 5 | 7 | 7 | 3 | |
| Log S (ESOL) | −2.58 | −4.53 | −3.80 | −5.05 | −5.27 | −1.14 | |
| Solubility Class | Soluble | M Soluble | Soluble | M Soluble | M Soluble | Soluble | |
| Drug-Likeness and Medicinal Chemistry Prediction | |||||||
| PAINS | 1 (N=N) | 1 (N=N) | 1 (N=N) | 1 (N=N) | 1 (N=N) | 0 | |
| Lead-Likeness | Yes | No (2) | Yes | No (2) | No (2) | No (1) | |
| Synthetic Accessibility | 2.68 | 2.89 | 3.22 | 3.84 | 3.91 | 3.00 | |
| Bioavailability Score | 0.55 | 0.55 | 0.55 | 0.56 | 0.55 | 0.55 | |
| Lipinski Rule (Violation) | Yes (0) | Yes (0) | Yes (0) | Yes (0) | Yes (0) | Yes (0) | |
| Veber Rule (Violation) | Yes (0) | Yes (0) | Yes (0) | No (1) | No (1) | No (1) | |
| Oral Toxicity Prediction | Most-Active Pyrazole and Pyrazolo[1,5-a]pyrimidine Derivatives and Acetazolamide as Positive Control | ||||||
|---|---|---|---|---|---|---|---|
| 3a | 5a | 6 | 9a | 10a | ACZ | ||
| Oral Toxicity Prediction | |||||||
| ProTox-II prediction | LD50 mg/kg | 5000 | 500 | 928 | 300 | 300 | 4300 |
| Toxicity Class | V | IV | IV | III | III | V | |
| Hepatotoxicity | Inactive 0.50 | Inactive 0.59 | Inactive 0.62 | Inactive 0.62 | Inactive 0.60 | Inactive 0.56 | |
| Carcinogenicity | Active 0.60 | Active 0.54 | Active 0.65 | Active 0.55 | Inactive 0.56 | Active 0.51 | |
| Immunotoxicity | Inactive 0.99 | Inactive 0.97 | Inactive 0.99 | Inactive 0.99 | Inactive 0.99 | Inactive 0.99 | |
| Cytotoxicity | Inactive 0.74 | Inactive 0.60 | Inactive 0.61 | Inactive 0.53 | Inactive 0.70 | Inactive 0.54 | |
| Aryl Hydrocarbon Receptor (AhR) | Inactive 0.50 | Inactive 0.54 | Active 0.64 | Inactive 0.94 | Inactive 0.62 | Inactive 0.99 | |
| Phosphoprotein (Tumor Suppressor) p53 | Inactive 0.84 | Inactive 0.85 | Inactive 0.80 | Inactive 0.77 | Inactive 0.86 | Inactive 0.99 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ragab, A.; Fouad, S.A.; Ammar, Y.A.; Aboul-Magd, D.S.; Abusaif, M.S. Antibiofilm and Anti-Quorum-Sensing Activities of Novel Pyrazole and Pyrazolo[1,5-a]pyrimidine Derivatives as Carbonic Anhydrase I and II Inhibitors: Design, Synthesis, Radiosterilization, and Molecular Docking Studies. Antibiotics 2023, 12, 128. https://doi.org/10.3390/antibiotics12010128
Ragab A, Fouad SA, Ammar YA, Aboul-Magd DS, Abusaif MS. Antibiofilm and Anti-Quorum-Sensing Activities of Novel Pyrazole and Pyrazolo[1,5-a]pyrimidine Derivatives as Carbonic Anhydrase I and II Inhibitors: Design, Synthesis, Radiosterilization, and Molecular Docking Studies. Antibiotics. 2023; 12(1):128. https://doi.org/10.3390/antibiotics12010128
Chicago/Turabian StyleRagab, Ahmed, Sawsan A. Fouad, Yousry A. Ammar, Dina S. Aboul-Magd, and Moustafa S. Abusaif. 2023. "Antibiofilm and Anti-Quorum-Sensing Activities of Novel Pyrazole and Pyrazolo[1,5-a]pyrimidine Derivatives as Carbonic Anhydrase I and II Inhibitors: Design, Synthesis, Radiosterilization, and Molecular Docking Studies" Antibiotics 12, no. 1: 128. https://doi.org/10.3390/antibiotics12010128
APA StyleRagab, A., Fouad, S. A., Ammar, Y. A., Aboul-Magd, D. S., & Abusaif, M. S. (2023). Antibiofilm and Anti-Quorum-Sensing Activities of Novel Pyrazole and Pyrazolo[1,5-a]pyrimidine Derivatives as Carbonic Anhydrase I and II Inhibitors: Design, Synthesis, Radiosterilization, and Molecular Docking Studies. Antibiotics, 12(1), 128. https://doi.org/10.3390/antibiotics12010128