
Gene–Gene Interactions Reduce Aminoglycoside Susceptibility of Pseudomonas aeruginosa through Efflux Pump-Dependent and -Independent Mechanisms



Abstract
:1. Introduction
2. Results
2.1. Identifying Frequent FusA1 and AmgS Sequence Variants in Clinical and Environmental Isolates of P. aeruginosa
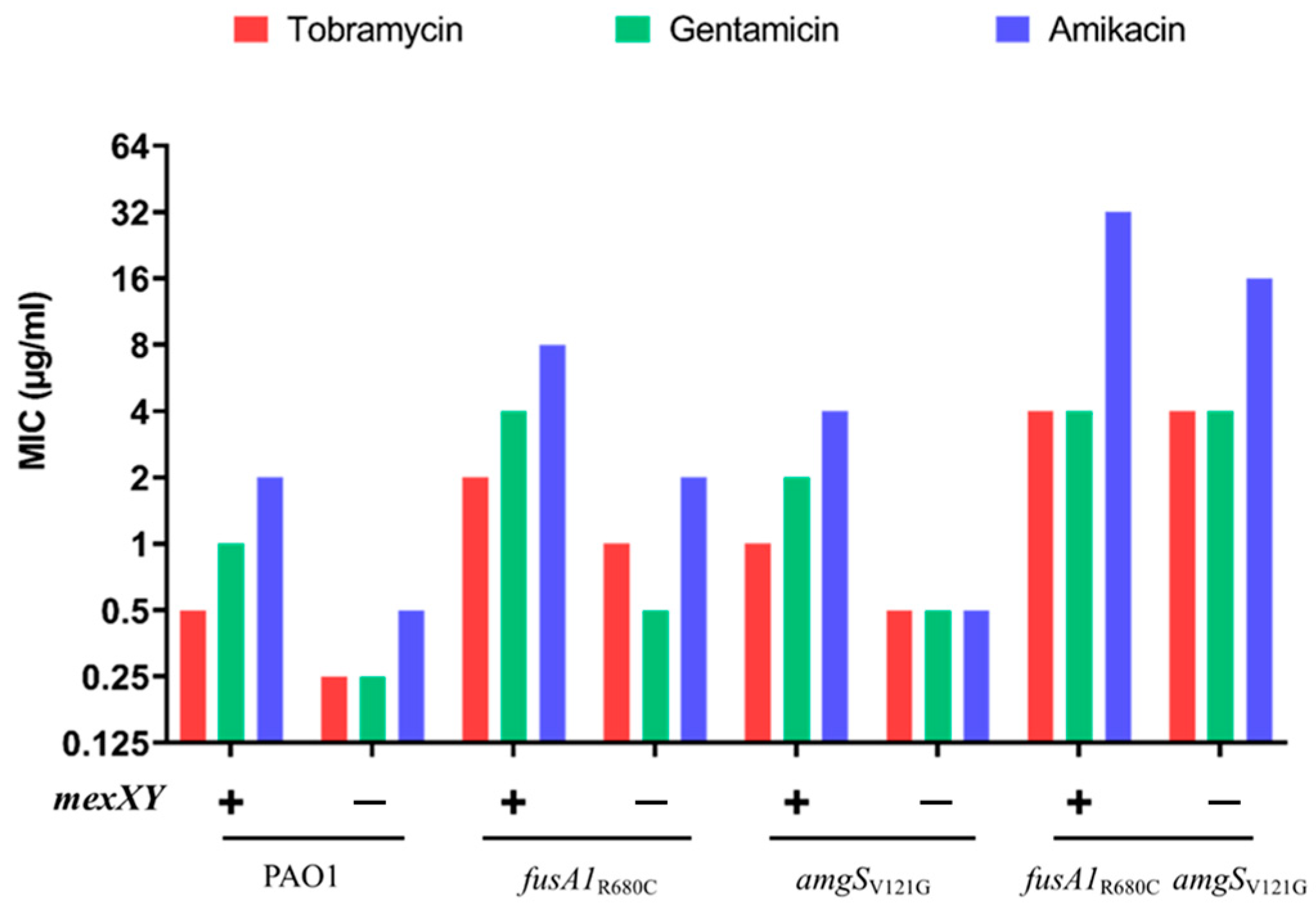
2.2. Effects of fusA1, amgS and mexY Mutations on Aminoglycoside Resistance
2.3. Effects of Combinations of Mutations
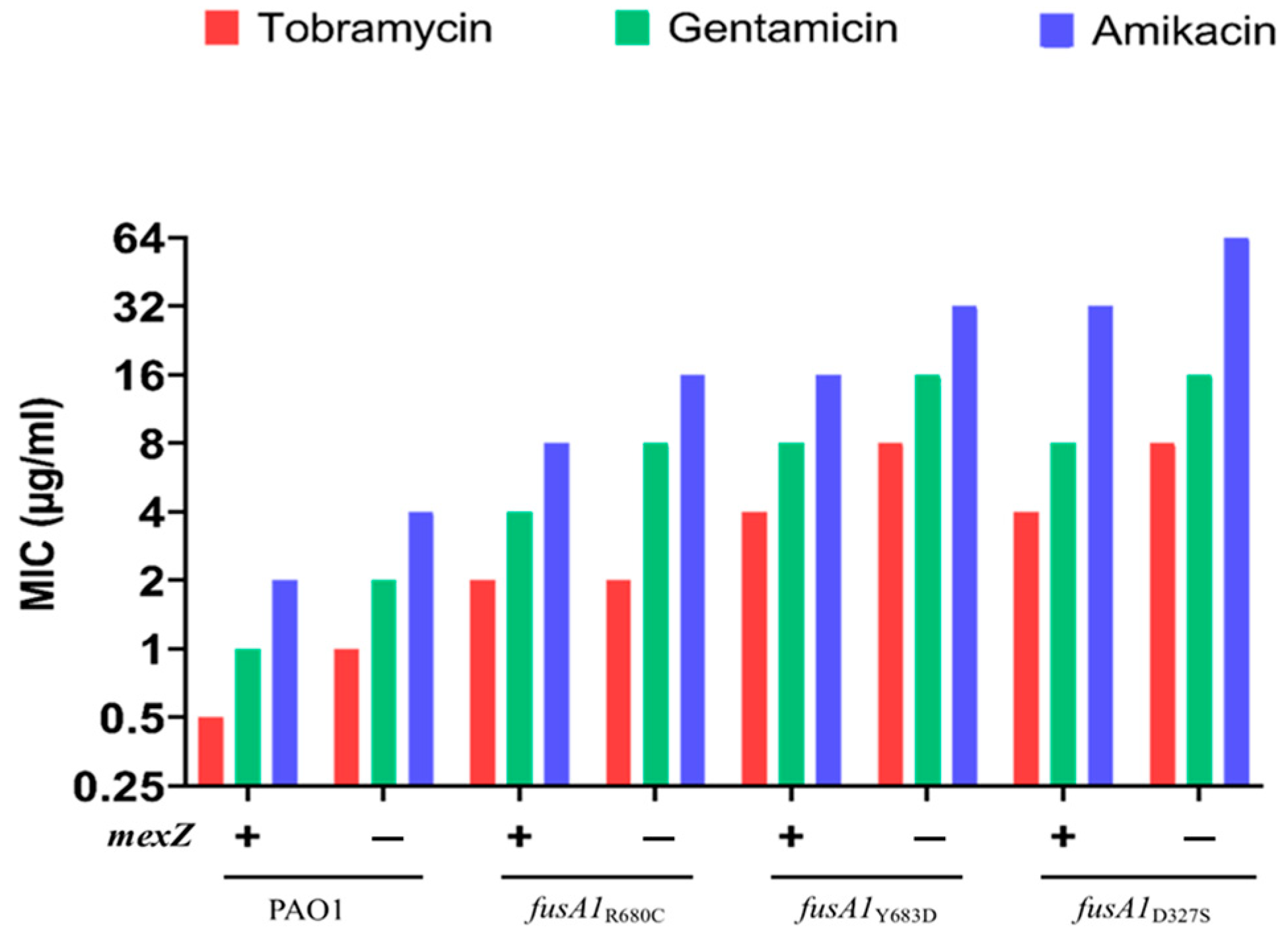
2.4. The Effects of Mutations on mexXY Gene Expression
2.5. The Effects of fusA1 and amgS Mutations in the Absence of mexXY Genes
2.6. The Effects of Mutations on Growth Rates
3. Discussion
4. Materials and Methods
4.1. Growth Conditions and Bacterial Strains Used in the Study
4.2. DNA Methods
4.3. Allelic Replacement and Genetic Mutants
4.4. Gene Expressional Analysis
4.5. Whole Genome Sequences and Variant Calling
4.6. Measuring Minimum Inhibitory Concentration (MIC)
4.7. Bacterial Growth Kinetics
4.8. Statistical Analysis and Homology Model
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bassetti, M.; Vena, A.; Croxatto, A.; Righi, E.; Guery, B. How to manage Pseudomonas aeruginosa infections. Drugs Context 2018, 7, 212527. [Google Scholar] [CrossRef] [PubMed]
- Driscoll, J.A.; Brody, S.L.; Kollef, M.H. The epidemiology, pathogenesis and treatment of Pseudomonas aeruginosa infections. Drugs 2007, 67, 351–368. [Google Scholar] [CrossRef] [PubMed]
- Ratjen, F.; Brockhaus, F.; Angyalosi, G. Aminoglycoside therapy against Pseudomonas aeruginosa in cystic fibrosis: A review. J. Cyst. Fibros. 2009, 8, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Shteinberg, M.; Elborn, J.S. Use of inhaled tobramycin in cystic fibrosis. Adv. Ther. 2015, 32, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ehsan, Z.; Wetzel, J.D.; Clancy, J.P. Nebulized liposomal amikacin for the treatment of Pseudomonas aeruginosa infection in cystic fibrosis patients. Expert Opin. Investig. Drugs 2014, 23, 743–749. [Google Scholar] [CrossRef]
- Taccetti, G.; Campana, S.; Neri, A.; Boni, V.; Festini, F. Antibiotic therapy against Pseudomonas aeruginosa in cystic fibrosis. J. Chemother. 2008, 20, 166–169. [Google Scholar] [CrossRef]
- Garcia-Clemente, M.; de la Rosa, D.; Maiz, L.; Giron, R.; Blanco, M.; Olveira, C.; Canton, R.; Martinez-Garcia, M.A. Impact of Pseudomonas aeruginosa Infection on Patients with Chronic Inflammatory Airway Diseases. J. Clin. Med. 2020, 9, 3800. [Google Scholar] [CrossRef]
- Appaneal, H.J.; Caffrey, A.R.; Jiang, L.; Dosa, D.; Mermel, L.A.; LaPlante, K.L. Antibiotic resistance rates for Pseudomonas aeruginosa clinical respiratory and bloodstream isolates among the Veterans Affairs Healthcare System from 2009 to 2013. Diagn. Microbiol. Infect. Dis. 2018, 90, 311–315. [Google Scholar] [CrossRef] [Green Version]
- Mustafa, M.H.; Chalhoub, H.; Denis, O.; Deplano, A.; Vergison, A.; Rodriguez-Villalobos, H.; Tunney, M.M.; Elborn, J.S.; Kahl, B.C.; Traore, H.; et al. Antimicrobial Susceptibility of Pseudomonas aeruginosa Isolated from Cystic Fibrosis Patients in Northern Europe. Antimicrob. Agents Chemother. 2016, 60, 6735–6741. [Google Scholar] [CrossRef] [Green Version]
- Thacharodi, A.; Lamont, I.L. Aminoglycoside-Modifying Enzymes Are Sufficient to Make Pseudomonas aeruginosa Clinically Resistant to Key Antibiotics. Antibiotics 2022, 11, 884. [Google Scholar] [CrossRef]
- Thacharodi, A.; Lamont, I.L. Aminoglycoside resistance in Pseudomonas aeruginosa: The contribution of the MexXY-OprM efflux pump varies between isolates. J. Med. Microbiol. 2022, 71, 001551. [Google Scholar] [CrossRef]
- Morita, Y.; Tomida, J.; Kawamura, Y. MexXY multidrug efflux system of Pseudomonas aeruginosa. Front. Microbiol. 2012, 3, 408. [Google Scholar] [CrossRef] [Green Version]
- Aires, J.R.; Kohler, T.; Nikaido, H.; Plésiat, P. Involvement of an active efflux system in the natural resistance of Pseudomonas aeruginosa to aminoglycosides. Antimicrob. Agents Chemother. 1999, 43, 2624–2628. [Google Scholar] [CrossRef] [Green Version]
- Matsuo, Y.; Eda, S.; Gotoh, N.; Yoshihara, E.; Nakae, T. MexZ-mediated regulation of mexXY multidrug efflux pump expression in Pseudomonas aeruginosa by binding on the mexZ-mexX intergenic DNA. FEMS Microbiol. Lett. 2004, 238, 23–28. [Google Scholar]
- Masuda, N.; Sakagawa, E.; Ohya, S.; Gotoh, N.; Tsujimoto, H.; Nishino, T. Contribution of the MexX-MexY-OprM efflux system to intrinsic resistance in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2000, 44, 2242–2246. [Google Scholar] [CrossRef] [Green Version]
- Jeannot, K.; Sobel, M.L.; El Garch, F.; Poole, K.; Plésiat, P. Induction of the MexXY efflux pump in Pseudomonas aeruginosa is dependent on drug-ribosome interaction. J. Bacteriol. 2005, 187, 5341–5346. [Google Scholar] [CrossRef] [Green Version]
- Vogne, C.; Aires, J.R.; Bailly, C.; Hocquet, D.; Plésiat, P. Role of the multidrug efflux system MexXY in the emergence of moderate resistance to aminoglycosides among Pseudomonas aeruginosa isolates from patients with cystic fibrosis. Antimicrob. Agents Chemother. 2004, 48, 1676–1680. [Google Scholar] [CrossRef] [Green Version]
- Sobel, M.L.; McKay, G.A.; Poole, K. Contribution of the MexXY multidrug transporter to aminoglycoside resistance in Pseudomonas aeruginosa clinical isolates. Antimicrob. Agents Chemother. 2003, 47, 3202–3207. [Google Scholar] [CrossRef] [Green Version]
- Llanes, C.; Hocquet, D.; Vogne, C.; Benali-Baitich, D.; Neuwirth, C.; Plésiat, P. Clinical strains of Pseudomonas aeruginosa overproducing MexAB-OprM and MexXY efflux pumps simultaneously. Antimicrob. Agents Chemother. 2004, 48, 1797–1802. [Google Scholar] [CrossRef] [Green Version]
- Lau, C.H.-F.; Hughes, D.; Poole, K. MexY-promoted aminoglycoside resistance in Pseudomonas aeruginosa: Involvement of a putative proximal binding pocket in aminoglycoside recognition. MBio 2014, 5, e01068-14. [Google Scholar] [CrossRef] [Green Version]
- Maunders, E.A.; Triniman, R.C.; Western, J.; Rahman, T.; Welch, M. Global reprogramming of virulence and antibiotic resistance in Pseudomonas aeruginosa by a single nucleotide polymorphism in elongation factor, fusA1. J. Biol. Chem. 2020, 295, 16411–16426. [Google Scholar] [CrossRef] [PubMed]
- Lau, C.H.-F.; Fraud, S.; Jones, M.; Peterson, S.N.; Poole, K. Mutational activation of the AmgRS two-component system in aminoglycoside-resistant Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2013, 57, 2243–2251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, C.H.F.; Krahn, T.; Gilmour, C.; Mullen, E.; Poole, K. AmgRS-mediated envelope stress-inducible expression of the mexXY multidrug efflux operon of Pseudomonas aeruginosa. Microbiologyopen 2015, 4, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Bolard, A.; Plésiat, P.; Jeannot, K. Mutations in gene fusA1 as a novel mechanism of aminoglycoside resistance in clinical strains of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2017, 62, e01835-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savelsbergh, A.; Rodnina, M.V.; Wintermeyer, W. Distinct functions of elongation factor G in ribosome recycling and translocation. RNA 2009, 15, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Hinz, A.; Bauerle, E.; Angermeyer, A.; Juhaszova, K.; Kaneko, Y.; Singh, P.K.; Manoil, C. Targeting a bacterial stress response to enhance antibiotic action. Proc. Natl. Acad. Sci. USA 2009, 106, 14570–14575. [Google Scholar] [CrossRef] [Green Version]
- Hinz, A.; Lee, S.; Jacoby, K.; Manoil, C. Membrane proteases and aminoglycoside antibiotic resistance. J. Bacteriol. 2011, 193, 4790–4797. [Google Scholar] [CrossRef] [Green Version]
- Krahn, T.; Gilmour, C.; Tilak, J.; Fraud, S.; Kerr, N.; Lau, C.H.-F.; Poole, K. Determinants of intrinsic aminoglycoside resistance in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2012, 56, 5591–5602. [Google Scholar] [CrossRef] [Green Version]
- Chuanchuen, R.; Wannaprasat, W.; Ajariyakhajorn, K.; Schweizer, H.P. Role of the MexXY multidrug efflux pump in moderate aminoglycoside resistance in Pseudomonas aeruginosa isolates from Pseudomonas mastitis. Microbiol. Immunol. 2008, 52, 392–398. [Google Scholar] [CrossRef]
- Poonsuk, K.; Chuanchuen, R. Contribution of the MexXY multidrug efflux pump and other chromosomal mechanisms on aminoglycoside resistance in Pseudomonas aeruginosa isolates from canine and feline infections. J. Vet. Med. Sci. 2012, 74, 1575–1582. [Google Scholar] [CrossRef] [Green Version]
- Martin, L.W.; Robson, C.L.; Watts, A.M.; Gray, A.R.; Wainwright, C.E.; Bell, S.C.; Ramsay, K.A.; Kidd, T.J.; Reid, D.W.; Brockway, B. Expression of Pseudomonas aeruginosa antibiotic resistance genes varies greatly during infections in cystic fibrosis patients. Antimicrob. Agents Chemother. 2018, 62, e01789-18. [Google Scholar] [CrossRef]
- Ramsay, K.A.; Wardell, S.J.T.; Patrick, W.M.; Brockway, B.; Reid, D.W.; Winstanley, C.; Bell, S.C.; Lamont, I.L. Genomic and phenotypic comparison of environmental and patient-derived isolates of Pseudomonas aeruginosa suggest that antimicrobial resistance is rare within the environment. J. Med. Microbiol. 2019, 68, 1591–1595. [Google Scholar] [CrossRef]
- Muller, C.; Plésiat, P.; Jeannot, K. A two-component regulatory system interconnects resistance to polymyxins, aminoglycosides, fluoroquinolones, and β-lactams in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2011, 55, 1211–1221. [Google Scholar] [CrossRef] [Green Version]
- Salsi, E.; Farah, E.; Netter, Z.; Dann, J.; Ermolenko, D.N. Movement of elongation factor G between compact and extended conformations. J. Mol. Biol. 2015, 427, 454–467. [Google Scholar] [CrossRef] [Green Version]
- CLSI. Performance Standards for Antimicrobial Susceptibility Testing, 28th ed.; Clinical and Laboratory Standards Insitute: Wayne, PA, USA, 2018. [Google Scholar]
- Wardell, S.J.; Rehman, A.; Martin, L.W.; Winstanley, C.; Patrick, W.M.; Lamont, I.L. A large-scale whole-genome comparison shows that experimental evolution in response to antibiotics predicts changes in naturally evolved clinical Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2019, 63, e01619-19. [Google Scholar] [CrossRef]
- Andersson, D.I.; Hughes, D. Antibiotic resistance and its cost: Is it possible to reverse resistance? Nat. Rev. Microbiol. 2010, 8, 260–271. [Google Scholar] [CrossRef]
- Pang, Z.; Raudonis, R.; Glick, B.R.; Lin, T.-J.; Cheng, Z. Antibiotic resistance in Pseudomonas aeruginosa: Mechanisms and alternative therapeutic strategies. Biotechnol. Adv. 2019, 37, 177–192. [Google Scholar] [CrossRef]
- Poole, K. Pseudomonas aeruginosa: Resistance to the max. Front. Microbiol. 2011, 2, 65. [Google Scholar] [CrossRef] [Green Version]
- Krause, K.M.; Serio, A.W.; Kane, T.R.; Connolly, L.E. Aminoglycosides: An overview. Cold Spring Harb. Perspect. Med. 2016, 6, a027029. [Google Scholar] [CrossRef] [Green Version]
- Andersson, D.I.; Patin, S.M.; Nilsson, A.I.; Kugelberg, E. The biological cost of antibiotic resistance. In Enzyme-Mediated Resistance to Antibiotics: Mechanisms, Dissemination, and Prospects for Inhibition; John Wiley & Sons: Hoboken, NJ, USA, 2007; pp. 339–348. [Google Scholar]
- Melnyk, A.H.; Wong, A.; Kassen, R. The fitness costs of antibiotic resistance mutations. Evol. Appl. 2015, 8, 273–283. [Google Scholar] [CrossRef]
- Macvanin, M.; Björkman, J.; Eriksson, S.; Rhen, M.; Andersson, D.I.; Hughes, D. Fusidic acid-resistant mutants of Salmonella enterica serovar Typhimurium with low fitness in vivo are defective in RpoS induction. Antimicrob. Agents Chemother. 2003, 47, 3743–3749. [Google Scholar] [CrossRef] [PubMed]
- Hmelo, L.R.; Borlee, B.R.; Almblad, H.; Love, M.E.; Randall, T.E.; Tseng, B.S.; Lin, C.; Irie, Y.; Storek, K.M.; Yang, J.J. Precision-engineering the Pseudomonas aeruginosa genome with two-step allelic exchange. Nat. Protoc. 2015, 10, 1820–1841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thoma, S.; Schobert, M. An improved Escherichia coli donor strain for diparental mating. FEMS Microbiol. Lett. 2009, 294, 127–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konings, A.F.; Martin, L.W.; Sharples, K.J.; Roddam, L.F.; Latham, R.; Reid, D.W.; Lamont, I.L. Pseudomonas aeruginosa uses multiple pathways to acquire iron during chronic infection in cystic fibrosis lungs. Infect. Immun. 2013, 81, 2697–2704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freschi, L.; Vincent, A.T.; Jeukens, J.; Emond-Rheault, J.-G.; Kukavica-Ibrulj, I.; Dupont, M.-J.; Charette, S.J.; Boyle, B.; Levesque, R.C. The Pseudomonas aeruginosa pan-genome provides new insights on its population structure, horizontal gene transfer, and pathogenicity. Genome Biol. Evol. 2019, 11, 109–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hilliam, Y.; Moore, M.P.; Lamont, I.L.; Bilton, D.; Haworth, C.S.; Foweraker, J.; Walshaw, M.J.; Williams, D.; Fothergill, J.L.; De Soyza, A. Pseudomonas aeruginosa adaptation and diversification in the non-cystic fibrosis bronchiectasis lung. Eur. Respir. J. 2017, 49, 1602108. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.; Chan, A.P. PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 2015, 31, 2745–2747. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.; Sims, G.E.; Murphy, S.; Miller, J.R.; Chan, A.P. Predicting the functional effect of amino acid substitutions and indels. PLoS ONE 2012, 7, e46688. [Google Scholar] [CrossRef] [Green Version]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [Green Version]
- Holloway, B. Genetic recombination in Pseudomonas aeruginosa. J. Gen. Microbiol. 1955, 13, 572–581. [Google Scholar] [CrossRef] [Green Version]
- Yanisch-Perron, C.; Vieira, J.; Messing, J. Improved M13 phage cloning vectors and host strains: Nucleotide sequences of the M13mpl8 and pUC19 vectors. Gene 1985, 33, 103–119. [Google Scholar] [CrossRef]
- Hoang, T.T.; Karkhoff-Schweizer, R.R.; Kutchma, A.J.; Schweizer, H.P. A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences: Application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 1998, 212, 77–86. [Google Scholar] [CrossRef]
- Rehman, A.; Jeukens, J.; Levesque, R.C.; Lamont, I.L. Gene-Gene Interactions Dictate Ciprofloxacin Resistance in Pseudomonas aeruginosa and Facilitate Prediction of Resistance Phenotype from Genome Sequence Data. Antimicrob Agents Chemother 2021, 65, e0269620. [Google Scholar] [CrossRef]
- Freschi, L.; Jeukens, J.; Kukavica-Ibrulj, I.; Boyle, B.; Dupont, M.J.; Laroche, J.; Larose, S.; Maaroufi, H.; Fothergill, J.L.; Moore, M.; et al. Clinical utilization of genomics data produced by the international Pseudomonas aeruginosa consortium. Front. Microbiol. 2015, 6, 1036. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Gene | C a | E a | Function | Frequent Variants b | PROVEAN Score c | Frequent Variants | PROVEAN Score | |
|---|---|---|---|---|---|---|---|---|
| Clinical | Environmental | |||||||
| 1 | fusA1 | 28 | 1 | EF-G: translation elongation factor G | R680C (14) A481V (14) M648V (13) Y552C (10) | −7.502 −3.364 −3.35 −7.533 | E245I (2) C266L (2) | −3.869 −8.554 |
| 2 | amgS | 10 | 5 | Part of two-component regulator system | V121G (26) R389S (3) R182C (3) D292G (3) P124Q (3) | −6.471 −5.431 −4.833 −3.386 −6.891 | R389H (2) | −4.513 |
| Strains | Aminoglycoside MICs a (µg/mL) | ||
|---|---|---|---|
| Tob | Gen | Amik | |
| Wild Type (WT) | |||
| PAO1 | 0.5 | 1 | 2 |
| Single mutants | |||
| ∆mexZ | 1 | 2 | 4 |
| mexYG287S | 0.5 | 1 | 2 |
| fusA1R680C | 2 | 4 | 8 |
| amgSV121G | 1 | 2 | 4 |
| Double mutants | |||
| ∆mexZ mexYG287S | 1 | 2 | 4 |
| ∆mexZ fusA1R680C | 2 | 8 | 16 |
| ∆mexZ amgSV121G | 1 | 2 | 4 |
| amgSV121GmexYG287S | 1 | 2 | 8 |
| mexYG287SfusA1R680C | 2 | 4 | 16 |
| fusA1R680CamgSV121G | 4 | 4 | 32 |
| Triple mutants | |||
| ∆mexZ fusA1R680C amgSV121G | 4 | 8 | 32 |
| ∆mexZ fusA1R680C mexYG287S | 4 | 8 | 32 |
| ∆mexZ amgSV121G mexYG287S | 1 | 2 | 8 |
| fusA1R680CamgSV121GmexYG287S | 4 | 8 | 64 |
| Quadruple mutant | |||
| fusA1R680CamgSV121GmexYG287S ∆mexZ | 4 | 16 | 64 |
| Strain | mexX Expression | Fold Change a | Fold Changes in MIC a | ||
|---|---|---|---|---|---|
| Tob | Gen | Ami | |||
| Reference strain | |||||
| PAO1 | 0.0007 | n/a | n/a | n/a | n/a |
| Single mutants | |||||
| fusA1R680C | 0.00142 | 2 | 4 | 4 | 4 |
| amgSV121G | 0.00173 | 2.5 | 2 | 2 | 2 |
| ∆mexZ | 0.00379 | 5 | 2 | 2 | 2 |
| Double mutants | |||||
| ∆mexZ fusA1R680C | 0.02497 | 35.5 | 4 | 8 | 8 |
| ∆mexZ amgSV121G | 0.01743 | 25 | 2 | 2 | 2 |
| fusA1R680C amgSV121G | 0.01894 | 27 | 8 | 4 | 16 |
| Triple mutant | |||||
| ∆mexZ fusA1R680C amgSV121G | 0.03383 | 48 | 8 | 8 | 16 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thacharodi, A.; Lamont, I.L. Gene–Gene Interactions Reduce Aminoglycoside Susceptibility of Pseudomonas aeruginosa through Efflux Pump-Dependent and -Independent Mechanisms. Antibiotics 2023, 12, 152. https://doi.org/10.3390/antibiotics12010152
Thacharodi A, Lamont IL. Gene–Gene Interactions Reduce Aminoglycoside Susceptibility of Pseudomonas aeruginosa through Efflux Pump-Dependent and -Independent Mechanisms. Antibiotics. 2023; 12(1):152. https://doi.org/10.3390/antibiotics12010152
Chicago/Turabian StyleThacharodi, Aswin, and Iain L. Lamont. 2023. "Gene–Gene Interactions Reduce Aminoglycoside Susceptibility of Pseudomonas aeruginosa through Efflux Pump-Dependent and -Independent Mechanisms" Antibiotics 12, no. 1: 152. https://doi.org/10.3390/antibiotics12010152
APA StyleThacharodi, A., & Lamont, I. L. (2023). Gene–Gene Interactions Reduce Aminoglycoside Susceptibility of Pseudomonas aeruginosa through Efflux Pump-Dependent and -Independent Mechanisms. Antibiotics, 12(1), 152. https://doi.org/10.3390/antibiotics12010152