
Dynamics-Based Regulatory Switches of Type II Antitoxins: Insights into New Antimicrobial Discovery

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
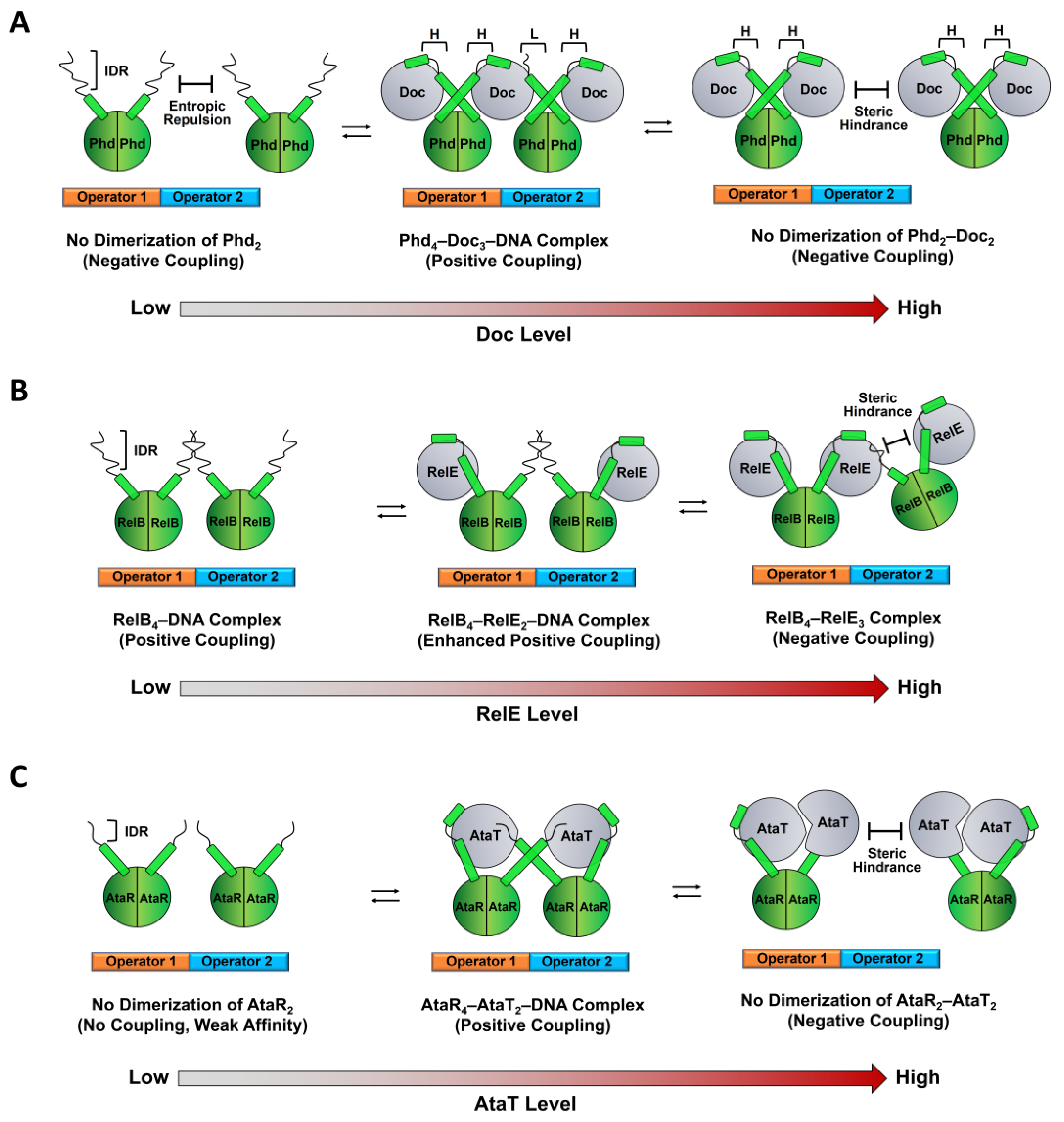
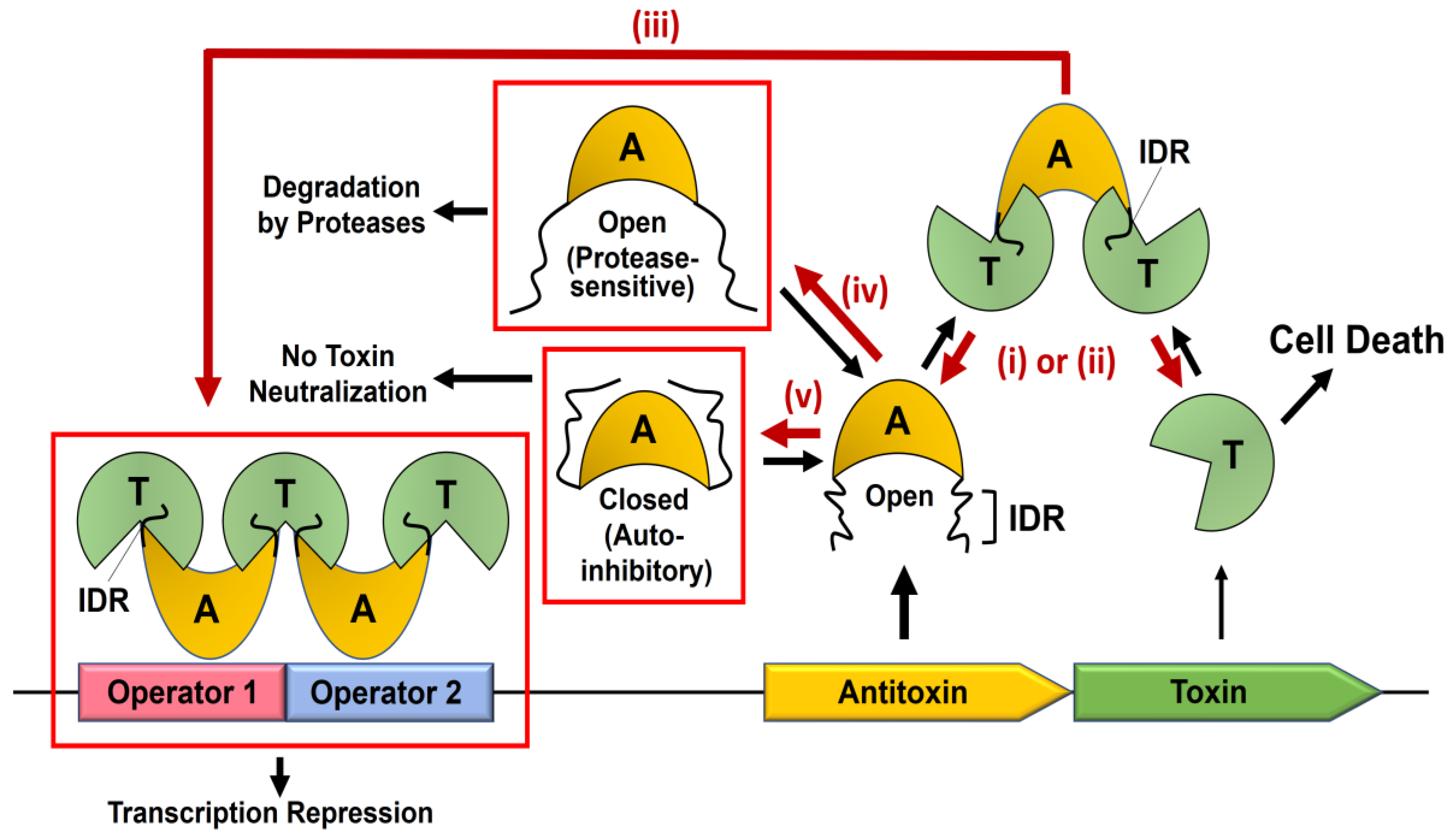
2. Dynamics-Based Regulatory Switches of Type II Antitoxins
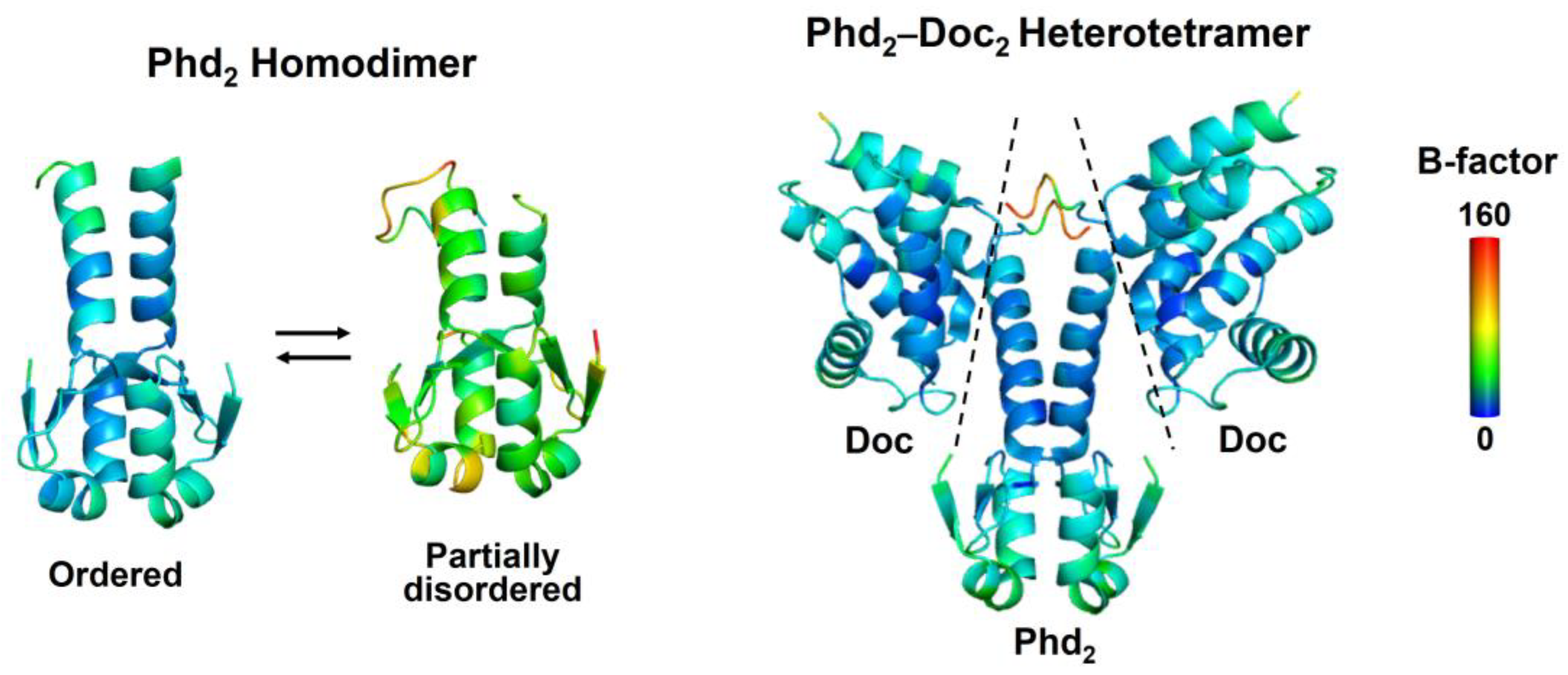
2.1. Coupling between Binding and Folding of Antitoxin IDRs
2.2. Regulation of TA Transcription by Antitoxin IDR
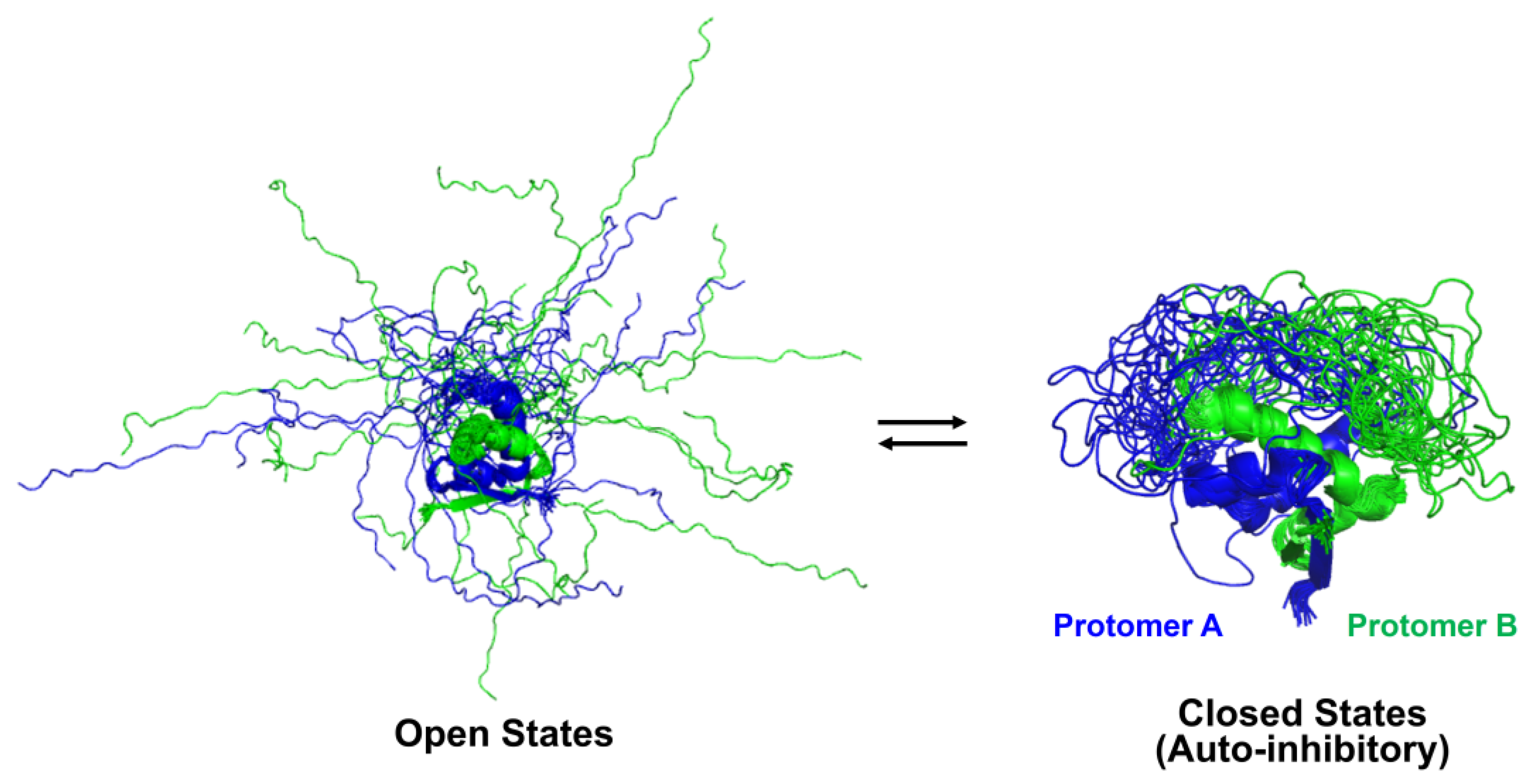
2.3. Auto-Inhibitory States of Antitoxin
2.4. Proteolysis of Antitoxin IDRs by Proteases
3. Utilization of Antitoxin IDR Dynamics for New Antibiotic Discovery
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fraikin, N.; Goormaghtigh, F.; Van Melderen, L. Type II Toxin-Antitoxin Systems: Evolution and Revolutions. J. Bacteriol. 2020, 202, e00763-19. [Google Scholar] [CrossRef] [Green Version]
- Jurėnas, D.; Fraikin, N.; Goormaghtigh, F.; Van Melderen, L. Biology and evolution of bacterial toxin-antitoxin systems. Nat. Rev. Genet. 2022, 20, 335–350. [Google Scholar] [CrossRef]
- Qiu, J.; Zhai, Y.; Wei, M.; Zheng, C.; Jiao, X. Toxin-antitoxin systems: Classification, biological roles, and applications. Microbiol. Res. 2022, 264, 127159. [Google Scholar] [CrossRef]
- De Bruyn, P.; Girardin, Y.; Loris, R. Prokaryote toxin-antitoxin modules: Complex regulation of an unclear function. Protein Sci. 2021, 30, 1103–1113. [Google Scholar] [CrossRef] [PubMed]
- Paul, P.; Sahu, B.R.; Suar, M. Plausible role of bacterial toxin-antitoxin system in persister cell formation and elimination. Mol. Oral Microbiol. 2019, 34, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Równicki, M.; Lasek, R.; Trylska, J.; Bartosik, D. Targeting Type II Toxin–Antitoxin Systems as Antibacterial Strategies. Toxins 2020, 12, 568. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.J.; Hergenrother, P.J. Artificial activation of toxin-antitoxin systems as an antibacterial strategy. Trends Microbiol. 2012, 20, 291–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slayden, R.A.; Dawson, C.C.; Cummings, J.E. Toxin-antitoxin systems and regulatory mechanisms in Mycobacterium tuberculosis. Pathog. Dis. 2018, 76, fty039. [Google Scholar] [CrossRef] [PubMed]
- Sala, A.; Bordes, P.; Genevaux, P. Multiple Toxin-Antitoxin Systems in Mycobacterium tuberculosis. Toxins 2014, 6, 1002–1020. [Google Scholar] [CrossRef] [Green Version]
- Kamruzzaman, M.; Wu, A.; Iredell, J. Biological Functions of Type II Toxin-Antitoxin Systems in Bacteria. Microorganisms 2021, 9, 1276. [Google Scholar] [CrossRef]
- Loris, R.; Garcia-Pino, A. Disorder- and Dynamics-Based Regulatory Mechanisms in Toxin–Antitoxin Modules. Chem. Rev. 2014, 114, 6933–6947. [Google Scholar] [CrossRef] [PubMed]
- Iakoucheva, L.M.; Brown, C.J.; Lawson, J.D.; Obradovic, Z.; Dunker, A.K. Intrinsic Disorder in Cell-signaling and Cancer-associated Proteins. J. Mol. Biol. 2002, 323, 573–584. [Google Scholar] [CrossRef] [Green Version]
- Dyson, H.J.; Wright, P.E. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 2005, 6, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Smock, R.G.; Gierasch, L.M. Sending Signals Dynamically. Science 2009, 324, 198–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hilser, V.J.; Thompson, E.B. Intrinsic disorder as a mechanism to optimize allosteric coupling in proteins. Proc. Natl. Acad. Sci. USA 2007, 104, 8311–8315. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Li, M.; Liu, Z. A comprehensive ensemble model for comparing the allosteric effect of ordered and disordered proteins. PLoS Comput. Biol. 2018, 14, e1006393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babu, M.M. The contribution of intrinsically disordered regions to protein function, cellular complexity, and human disease. Biochem. Soc. Trans. 2016, 44, 1185–1200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruan, H.; Sun, Q.; Zhang, W.; Liu, Y.; Lai, L. Targeting intrinsically disordered proteins at the edge of chaos. Drug Discov. Today 2018, 24, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Cao, H.; Liu, Z. Binding cavities and druggability of intrinsically disordered proteins. Protein Sci. 2015, 24, 688–705. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Liu, X.; Chen, J. Targeting Intrinsically Disordered Proteins through Dynamic Interactions. Biomolecules 2020, 10, 743. [Google Scholar] [CrossRef]
- Burger, V.M.; Vandervelde, A.; Hendrix, J.; Konijnenberg, A.; Sobott, F.; Loris, R.; Stultz, C.M. Hidden States within Disordered Regions of the CcdA Antitoxin Protein. J. Am. Chem. Soc. 2017, 139, 2693–2701. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Pino, A.; Balasubramanian, S.; Wyns, L.; Gazit, E.; De Greve, H.; Magnuson, R.D.; Charlier, D.; van Nuland, N.A.; Loris, R. Allostery and Intrinsic Disorder Mediate Transcription Regulation by Conditional Cooperativity. Cell 2010, 142, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Pino, A.; De Gieter, S.S.; Talavera, A.A.; De Greve, H.; Efremov, R.; Loris, R. An intrinsically disordered entropic switch determines allostery in Phd–Doc regulation. Nat. Chem. Biol. 2016, 12, 490–496. [Google Scholar] [CrossRef]
- De Gieter, S.; Konijnenberg, A.; Talavera, A.; Butterer, A.; Haesaerts, S.; De Greve, H.; Sobott, F.; Loris, R.; Garcia-Pino, A. The Intrinsically Disordered Domain of the Antitoxin Phd Chaperones the Toxin Doc against Irreversible Inactivation and Misfolding. J. Biol. Chem. 2014, 289, 34013–34023. [Google Scholar] [CrossRef] [Green Version]
- Page, R.; Peti, W. Toxin-antitoxin systems in bacterial growth arrest and persistence. Nat. Chem. Biol. 2016, 12, 208–214. [Google Scholar] [CrossRef]
- Shammas, S.L. Mechanistic roles of protein disorder within transcription. Curr. Opin. Struct. Biol. 2017, 42, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Drobnak, I.; De Jonge, N.; Haesaerts, S.; Vesnaver, G.; Loris, R.; Lah, J. Energetic Basis of Uncoupling Folding from Binding for an Intrinsically Disordered Protein. J. Am. Chem. Soc. 2013, 135, 1288–1294. [Google Scholar] [CrossRef]
- Boehr, D.D.; Nussinov, R.; Wright, P.E. The role of dynamic conformational ensembles in biomolecular recognition. Nat. Chem. Biol. 2009, 5, 789–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.-Y.; Lee, B.-J. Structure, Biology, and Therapeutic Application of Toxin–Antitoxin Systems in Pathogenic Bacteria. Toxins 2016, 8, 305. [Google Scholar] [CrossRef] [Green Version]
- Park, S.J.; Son, W.S.; Lee, B.-J. Structural overview of toxin-antitoxin systems in infectious bacteria: A target for developing antimicrobial agents. Biochim. et Biophys. Acta (BBA) Proteins Proteom. 2013, 1834, 1155–1167. [Google Scholar] [CrossRef]
- Bax, A.; Clore, G.M. Protein NMR: Boundless opportunities. J. Magn. Reson. 2019, 306, 187–191. [Google Scholar] [CrossRef]
- Garcia-Pino, A.; Christensen-Dalsgaard, M.; Wyns, L.; Yarmolinsky, M.; Magnuson, R.D.; Gerdes, K.; Loris, R. Doc of Prophage P1 Is Inhibited by Its Antitoxin Partner Phd through Fold Complementation. J. Biol. Chem. 2008, 283, 30821–30827. [Google Scholar] [CrossRef] [Green Version]
- Smith, J.A.; Magnuson, R.D. Modular Organization of the Phd Repressor/Antitoxin Protein. J. Bacteriol. 2004, 186, 2692–2698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Jonge, N.; Garcia-Pino, A.; Buts, L.; Haesaerts, S.; Charlier, D.; Zangger, K.; Wyns, L.; De Greve, H.; Loris, R. Rejuvenation of CcdB-Poisoned Gyrase by an Intrinsically Disordered Protein Domain. Mol. Cell 2009, 35, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Vandervelde, A.; Drobnak, I.; Hadži, S.; Sterckx, Y.; Welte, T.; De Greve, H.; Charlier, D.; Efremov, R.; Loris, R.; Lah, J. Molecular mechanism governing ratio-dependent transcription regulation in the ccdAB operon. Nucleic Acids Res. 2017, 45, 2937–2950. [Google Scholar] [CrossRef]
- Bøggild, A.; Sofos, N.; Andersen, K.R.; Feddersen, A.; Easter, A.D.; Passmore, L.A.; Brodersen, D.E. The Crystal Structure of the Intact E. coli RelBE Toxin-Antitoxin Complex Provides the Structural Basis for Conditional Cooperativity. Structure 2012, 20, 1641–1648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Overgaard, M.; Borch, J.; Gerdes, K. RelB and RelE of Escherichia coli Form a Tight Complex That Represses Transcription via the Ribbon–Helix–Helix Motif in RelB. J. Mol. Biol. 2009, 394, 183–196. [Google Scholar] [CrossRef]
- Xue, L.; Yue, J.; Ke, J.; Khan, M.H.; Wen, W.; Sun, B.; Zhu, Z.; Niu, L. Distinct oligomeric structures of the YoeB–YefM complex provide insights into the conditional cooperativity of type II toxin-antitoxin system. Nucleic Acids Res. 2020, 48, 10527–10541. [Google Scholar] [CrossRef]
- Jurėnas, D.; Van Melderen, L.; Garcia-Pino, A. Mechanism of regulation and neutralization of the AtaR–AtaT toxin-antitoxin system. Nat. Chem. Biol. 2019, 15, 285–294. [Google Scholar] [CrossRef] [Green Version]
- Yashiro, Y.; Yamashita, S.; Tomita, K. Crystal Structure of the Enterohemorrhagic Escherichia coli AtaT-AtaR Toxin-Antitoxin Complex. Structure 2019, 27, 476–484.e3. [Google Scholar] [CrossRef] [Green Version]
- Qian, H.; Yu, H.; Li, P.; Zhu, E.; Yao, Q.; Tai, C.; Deng, Z.; Gerdes, K.; He, X.; Gan, J.; et al. Toxin-antitoxin operon kacAT of Klebsiella pneumoniae is regulated by conditional cooperativity via a W-shaped KacA–KacT complex. Nucleic Acids Res. 2019, 47, 7690–7702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winter, A.J.; Williams, C.; Isupov, M.; Crocker, H.; Gromova, M.; Marsh, P.; Wilkinson, O.J.; Dillingham, M.; Harmer, N.J.; Titball, R.W.; et al. The molecular basis of protein toxin HicA–dependent binding of the protein antitoxin HicB to DNA. J. Biol. Chem. 2018, 293, 19429–19440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, B.L.; Lord, D.M.; Grigoriu, S.; Peti, W.; Page, R. The Escherichia coli Toxin MqsR Destabilizes the Transcriptional Repression Complex Formed between the Antitoxin MqsA and the mqsRA Operon Promoter. J. Biol. Chem. 2013, 288, 1286–1294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talavera, A.; Tamman, H.; Ainelo, A.; Konijnenberg, A.; Hadži, S.; Sobott, F.; Garcia-Pino, A.; Hõrak, R.; Loris, R. A dual role in regulation and toxicity for the disordered N-terminus of the toxin GraT. Nat. Commun. 2019, 10, 972. [Google Scholar] [CrossRef] [Green Version]
- Schureck, M.A.; Meisner, J.; Hoffer, E.D.; Wang, D.; Onuoha, N.; Cho, S.E.; Lollar, P.; Dunham, C.M. Structural basis of transcriptional regulation by the HigA antitoxin. Mol. Microbiol. 2019, 111, 1449–1462. [Google Scholar] [CrossRef]
- Deter, H.S.; Jensen, R.V.; Mather, W.H.; Butzin, N.C. Mechanisms for Differential Protein Production in Toxin–Antitoxin Systems. Toxins 2017, 9, 211. [Google Scholar] [CrossRef] [Green Version]
- Fraikin, N.; Rousseau, C.J.; Goeders, N.; Van Melderen, L. Reassessing the Role of the Type II MqsRA Toxin-Antitoxin System in Stress Response and Biofilm Formation: mqsA Is Transcriptionally Uncoupled from mqsR. Mbio 2019, 10, e02678-19. [Google Scholar] [CrossRef] [Green Version]
- Jadhav, P.V.; Sinha, V.K.; Chugh, S.; Kotyada, C.; Bachhav, D.; Singh, R.; Rothweiler, U.; Singh, M. 2.09 Å Resolution structure of E. coli HigBA toxin-antitoxin complex reveals an ordered DNA-binding domain and intrinsic dynamics in antitoxin. Biochem. J. 2020, 477, 4001–4019. [Google Scholar] [CrossRef]
- Ruangprasert, A.; Maehigashi, T.; Miles, S.J.; Giridharan, N.; Liu, J.X.; Dunham, C.M. Mechanisms of Toxin Inhibition and Transcriptional Repression by Escherichia coli DinJ-YafQ. J. Biol. Chem. 2014, 289, 20559–20569. [Google Scholar] [CrossRef] [Green Version]
- Huang, Q.; Lai, L.; Liu, Z. Quantitative Analysis of Dynamic Allostery. J. Chem. Inf. Model. 2022, 62, 2538–2549. [Google Scholar] [CrossRef] [PubMed]
- Madl, T.; Van Melderen, L.; Mine, N.; Respondek, M.; Oberer, M.; Keller, W.; Khatai, L.; Zangger, K. Structural Basis for Nucleic Acid and Toxin Recognition of the Bacterial Antitoxin CcdA. J. Mol. Biol. 2006, 364, 170–185. [Google Scholar] [CrossRef]
- Fonin, A.V.; Darling, A.L.; Kuznetsova, I.M.; Turoverov, K.K.; Uversky, V.N. Intrinsically disordered proteins in crowded milieu: When chaos prevails within the cellular gumbo. Cell. Mol. Life Sci. 2018, 75, 3907–3929. [Google Scholar] [CrossRef]
- Zhou, H.-X.; Rivas, G.; Minton, A.P. Macromolecular Crowding and Confinement: Biochemical, Biophysical, and Potential Physiological Consequences. Annu. Rev. Biophys. 2008, 37, 375–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuznetsova, I.M.; Zaslavsky, B.Y.; Breydo, L.; Turoverov, K.K.; Uversky, V.N. Beyond the Excluded Volume Effects: Mechanistic Complexity of the Crowded Milieu. Molecules 2015, 20, 1377–1409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonucci, A.; Palomino-Schätzlein, M.; de Molina, P.M.; Arbe, A.; Pierattelli, R.; Rizzuti, B.; Iovanna, J.L.; Neira, J.L. Crowding Effects on the Structure and Dynamics of the Intrinsically Disordered Nuclear Chromatin Protein NUPR1. Front. Mol. Biosci. 2021, 8, 684622. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.-L.; Nesbitt, D.J. Effects of Molecular Crowders on Single-Molecule Nucleic Acid Folding: Temperature-Dependent Studies Reveal True Crowding vs Enthalpic Interactions. J. Phys. Chem. B 2021, 125, 13147–13157. [Google Scholar] [CrossRef] [PubMed]
- Popielec, A.; Ostrowska, N.; Wojciechowska, M.; Feig, M.; Trylska, J. Crowded environment affects the activity and inhibition of the NS3/4A protease. Biochimie 2020, 176, 169–180. [Google Scholar] [CrossRef]
- Maisonneuve, E.; Gerdes, K. Molecular Mechanisms Underlying Bacterial Persisters. Cell 2014, 157, 539–548. [Google Scholar] [CrossRef] [Green Version]
- Salmon, M.A.; Van Melderen, L.; Bernard, P.; Couturier, M. The antidote and autoregulatory functions of the F plasmid CcdA protein: A genetic and biochemical survey. Mol. Genet. Genom. 1994, 244, 530–538. [Google Scholar] [CrossRef] [PubMed]
- Diago-Navarro, E.; Hernández-Arriaga, A.M.; Kubik, S.; Konieczny, I.; Díaz-Orejas, R. Cleavage of the antitoxin of the parD toxin-antitoxin system is determined by the ClpAP protease and is modulated by the relative ratio of the toxin and the antitoxin. Plasmid 2013, 70, 78–85. [Google Scholar] [CrossRef]
- Lehnherr, H.; Yarmolinsky, M.B. Addiction protein Phd of plasmid prophage P1 is a substrate of the ClpXP serine protease of Escherichia coli. Proc. Natl. Acad. Sci. USA 1995, 92, 3274–3277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LeRoux, M.; Culviner, P.H.; Liu, Y.J.; Littlehale, M.L.; Laub, M.T. Stress Can Induce Transcription of Toxin-Antitoxin Systems without Activating Toxin. Mol. Cell 2020, 79, 280–292.e8. [Google Scholar] [CrossRef] [PubMed]
- Mutschler, H.; Reinstein, J.; Meinhart, A. Assembly Dynamics and Stability of the Pneumococcal Epsilon Zeta Antitoxin Toxin (PezAT) System from Streptococcus pneumoniae. J. Biol. Chem. 2010, 285, 21797–21806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bordes, P.; Genevaux, P. Control of Toxin-Antitoxin Systems by Proteases in Mycobacterium Tuberculosis. Front. Mol. Biosci. 2021, 8, 458. [Google Scholar] [CrossRef] [PubMed]
- Olivares, A.O.; Baker, T.A.; Sauer, R.T. Mechanistic insights into bacterial AAA+ proteases and protein-remodelling machines. Nat. Rev. Genet. 2015, 14, 33–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neher, S.B.; Villén, J.; Oakes, E.C.; Bakalarski, C.E.; Sauer, R.T.; Gygi, S.P.; Baker, T.A. Proteomic Profiling of ClpXP Substrates after DNA Damage Reveals Extensive Instability within SOS Regulon. Mol. Cell 2006, 22, 193–204. [Google Scholar] [CrossRef]
- Katz, C.; Rasouly, A.; Gur, E.; Shenhar, Y.; Biran, D.; Ron, E.Z. Temperature-dependent proteolysis as a control element in Escherichia coli metabolism. Res. Microbiol. 2009, 160, 684–686. [Google Scholar] [CrossRef]
- Muthuramalingam, M.; White, J.C.; Bourne, C.R. Toxin-Antitoxin Modules Are Pliable Switches Activated by Multiple Protease Pathways. Toxins 2016, 8, 214. [Google Scholar] [CrossRef]
- Ronneau, S.; Helaine, S. Clarifying the Link between Toxin–Antitoxin Modules and Bacterial Persistence. J. Mol. Biol. 2019, 431, 3462–3471. [Google Scholar] [CrossRef]
- Engelberg-Kulka, H.; Hazan, R.; Amitai, S. mazEF: A chromosomal toxin-antitoxin module that triggers programmed cell death in bacteria. J. Cell Sci. 2005, 118, 4327–4332. [Google Scholar] [CrossRef] [Green Version]
- Christensen, S.K.; Pedersen, K.; Hansen, F.G.; Gerdes, K. Toxin-antitoxin Loci as Stress-response-elements: ChpAK/MazF and ChpBK Cleave Translated RNAs and are Counteracted by tmRNA. J. Mol. Biol. 2003, 332, 809–819. [Google Scholar] [CrossRef] [PubMed]
- Ramisetty, B.C.M.; Raj, S.; Ghosh, D. Escherichia coli MazEF toxin-antitoxin system does not mediate programmed cell death. J. Basic Microbiol. 2016, 56, 1398–1402. [Google Scholar] [CrossRef]
- Lehnherr, H.; Maguin, E.; Jafri, S.; Yarmolinsky, M.B. Plasmid Addiction Genes of Bacteriophage P1: Doc, which Causes Cell Death on Curing of Prophage, and phd, which Prevents Host Death when Prophage is Retained. J. Mol. Biol. 1993, 233, 414–428. [Google Scholar] [CrossRef] [Green Version]
- Heller, G.T.; Sormanni, P.; Vendruscolo, M. Targeting disordered proteins with small molecules using entropy. Trends Biochem. Sci. 2015, 40, 491–496. [Google Scholar] [CrossRef] [Green Version]
- Ambadipudi, S.; Zweckstetter, M. Targeting intrinsically disordered proteins in rational drug discovery. Expert Opin. Drug Discov. 2015, 11, 65–77. [Google Scholar] [CrossRef]
- Santofimia-Castaño, P.; Rizzuti, B.; Xia, Y.; Abian, O.; Peng, L.; Velázquez-Campoy, A.; Neira, J.L.; Iovanna, J. Targeting intrinsically disordered proteins involved in cancer. Cell. Mol. Life Sci. 2019, 77, 1695–1707. [Google Scholar] [CrossRef] [Green Version]
- Tsafou, K.; Tiwari, P.; Forman-Kay, J.; Metallo, S.; Toretsky, J. Targeting Intrinsically Disordered Transcription Factors: Changing the Paradigm. J. Mol. Biol. 2018, 430, 2321–2341. [Google Scholar] [CrossRef]
- Heller, G.T.; Bonomi, M.; Vendruscolo, M. Structural Ensemble Modulation upon Small-Molecule Binding to Disordered Proteins. J. Mol. Biol. 2018, 430, 2288–2292. [Google Scholar] [CrossRef] [PubMed]
- Buckton, L.K.; Rahimi, M.N.; McAlpine, S.R. Cyclic Peptides as Drugs for Intracellular Targets: The Next Frontier in Peptide Therapeutic Development. Chem. A Eur. J. 2020, 27, 1487–1513. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Guo, J.; Cheng, J.; Zhang, Z.; Kang, F.; Wu, X.; Chu, Q. High-Throughput Screening of Stapled Helical Peptides in Drug Discovery. J. Med. Chem. 2022, 66, 95–106. [Google Scholar] [CrossRef]
- Wichapong, K.; Silvestre-Roig, C.; Braster, Q.; Schumski, A.; Soehnlein, O.; Nicolaes, G.A. Structure-based peptide design targeting intrinsically disordered proteins: Novel histone H4 and H2A peptidic inhibitors. Comput. Struct. Biotechnol. J. 2021, 19, 934–948. [Google Scholar] [CrossRef]
- Leplae, R.; Geeraerts, D.; Hallez, R.; Guglielmini, J.; Drèze, P.; Van Melderen, L. Diversity of bacterial type II toxin-antitoxin systems: A comprehensive search and functional analysis of novel families. Nucleic Acids Res. 2011, 39, 5513–5525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, S.; Mishra, N.K.; Bhatnagar, S.; Bhatnagar, R. PemK Toxin of Bacillus anthracis Is a Ribonuclease. J. Biol. Chem. 2010, 285, 7254–7270. [Google Scholar] [CrossRef] [Green Version]
- Chopra, N.; Agarwal, S.; Verma, S.; Bhatnagar, S.; Bhatnagar, R. Modeling of the structure and interactions of the B. anthracis antitoxin, MoxX: Deletion mutant studies highlight its modular structure and repressor function. J. Comput. Mol. Des. 2011, 25, 275–291. [Google Scholar] [CrossRef]
- Lee, I.-G.; Lee, S.J.; Chae, S.; Lee, K.-Y.; Kim, J.-H.; Lee, B.-J. Structural and functional studies of the Mycobacterium tuberculosis VapBC30 toxin-antitoxin system: Implications for the design of novel antimicrobial peptides. Nucleic Acids Res. 2015, 43, 7624–7637. [Google Scholar] [CrossRef] [Green Version]
- Van Melderen, L.; Bernard, P.; Couturier, M. Lon-dependent proteolysis of CcdA is the key control for activation of CcdB in plasmid-free segregant bacteria. Mol. Microbiol. 1994, 11, 1151–1157. [Google Scholar] [CrossRef] [PubMed]
- Hallez, R.; Geeraerts, D.; Sterckx, Y.; Mine, N.; Loris, R.; Van Melderen, L. New toxins homologous to ParE belonging to three-component toxin-antitoxin systems in Escherichia coli O157:H7. Mol. Microbiol. 2010, 76, 719–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zielenkiewicz, U.; Cegłowski, P. The Toxin-Antitoxin System of the Streptococcal Plasmid pSM19035. J. Bacteriol. 2005, 187, 6094–6105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, P.; Issac, B.; Dodson, E.J.; Turkenburg, J.P.; Mande, S.C. Crystal Structure of Mycobacterium tuberculosis YefM Antitoxin Reveals that it is Not an Intrinsically Unstructured Protein. J. Mol. Biol. 2008, 383, 482–493. [Google Scholar] [CrossRef]
- Turnbull, K.J.; Gerdes, K. HicA toxin of Escherichia coli derepresses hicAB transcription to selectively produce HicB antitoxin. Mol. Microbiol. 2017, 104, 781–792. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, K.-Y.; Lee, B.-J. Dynamics-Based Regulatory Switches of Type II Antitoxins: Insights into New Antimicrobial Discovery. Antibiotics 2023, 12, 637. https://doi.org/10.3390/antibiotics12040637
Lee K-Y, Lee B-J. Dynamics-Based Regulatory Switches of Type II Antitoxins: Insights into New Antimicrobial Discovery. Antibiotics. 2023; 12(4):637. https://doi.org/10.3390/antibiotics12040637
Chicago/Turabian StyleLee, Ki-Young, and Bong-Jin Lee. 2023. "Dynamics-Based Regulatory Switches of Type II Antitoxins: Insights into New Antimicrobial Discovery" Antibiotics 12, no. 4: 637. https://doi.org/10.3390/antibiotics12040637
APA StyleLee, K. -Y., & Lee, B. -J. (2023). Dynamics-Based Regulatory Switches of Type II Antitoxins: Insights into New Antimicrobial Discovery. Antibiotics, 12(4), 637. https://doi.org/10.3390/antibiotics12040637