
DXP Synthase Function in a Bacterial Metabolic Adaptation and Implications for Antibacterial Strategies


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
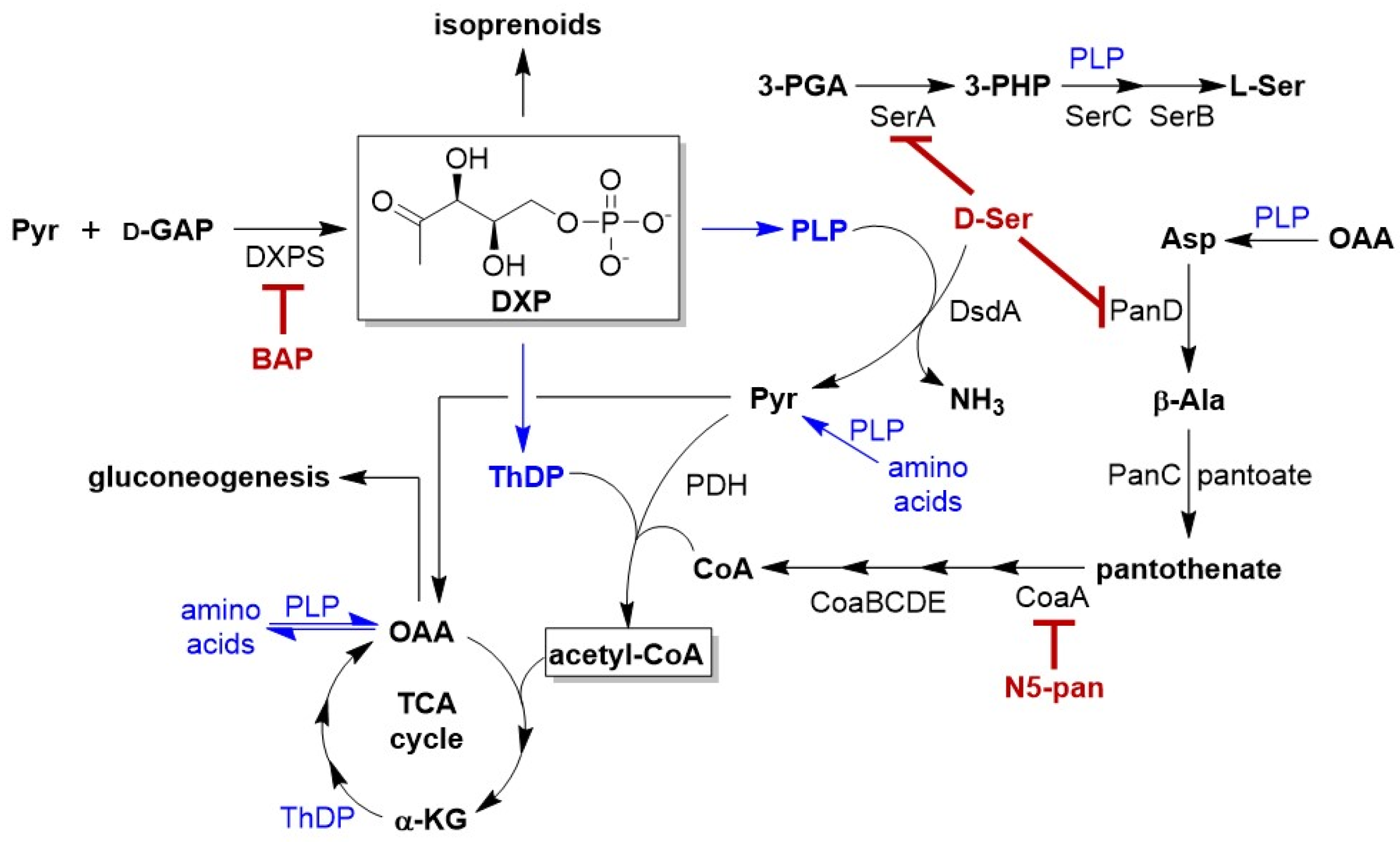
2. Results
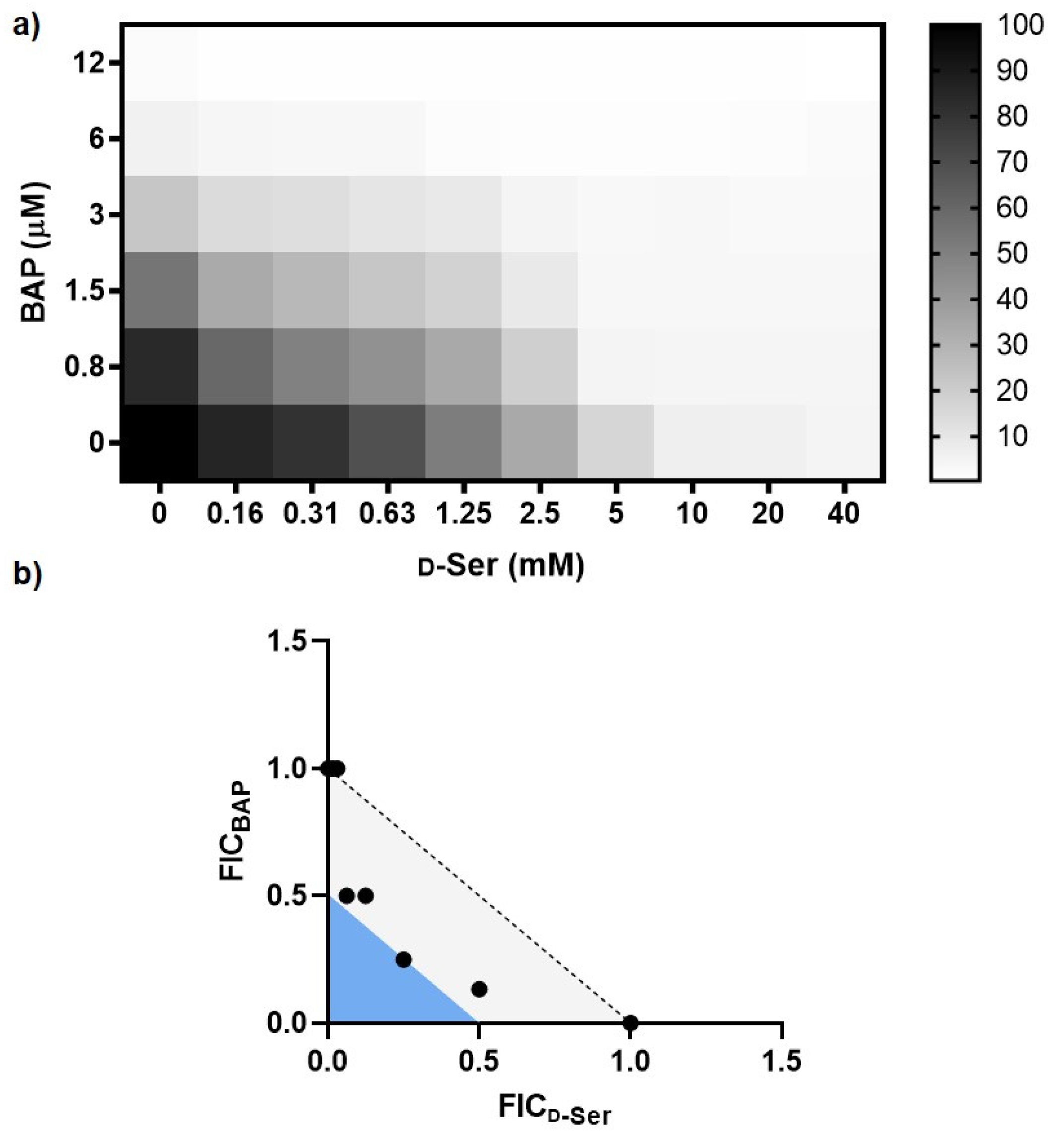
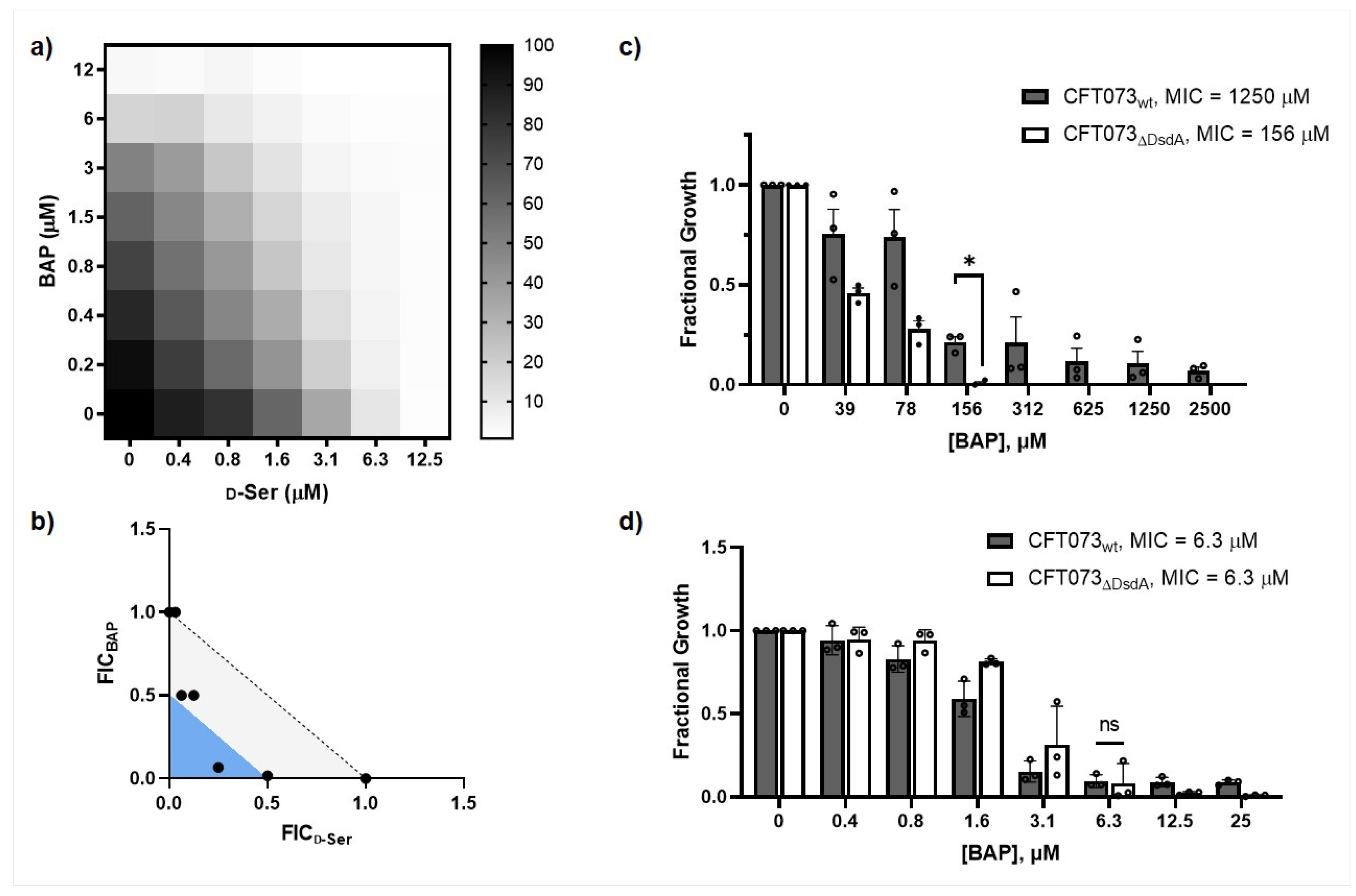
2.1. Impairing DXPS Increases UPEC Sensitivity to d-Ser
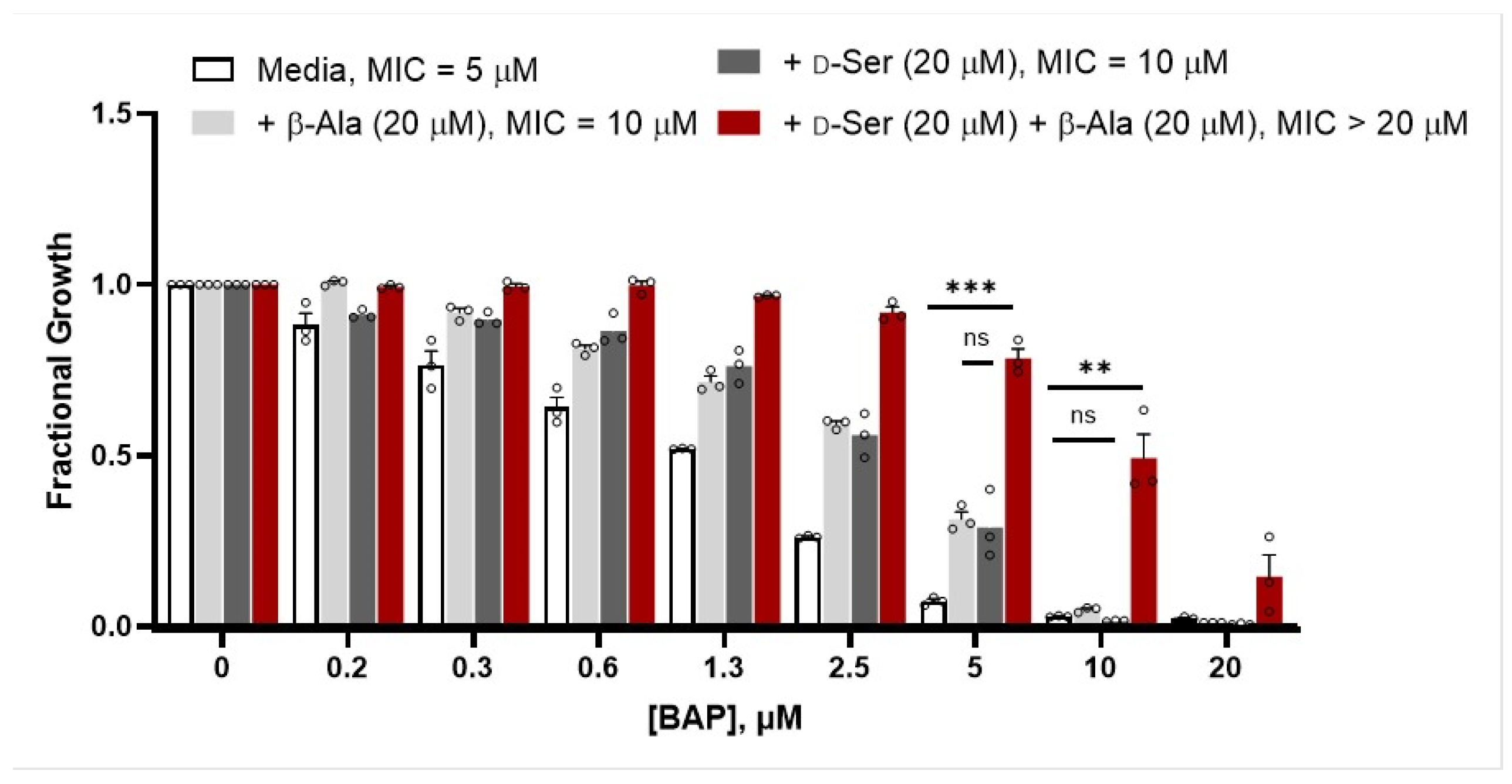
2.2. β-Alanine Suppresses BAP Activity in the Presence of d-Ser
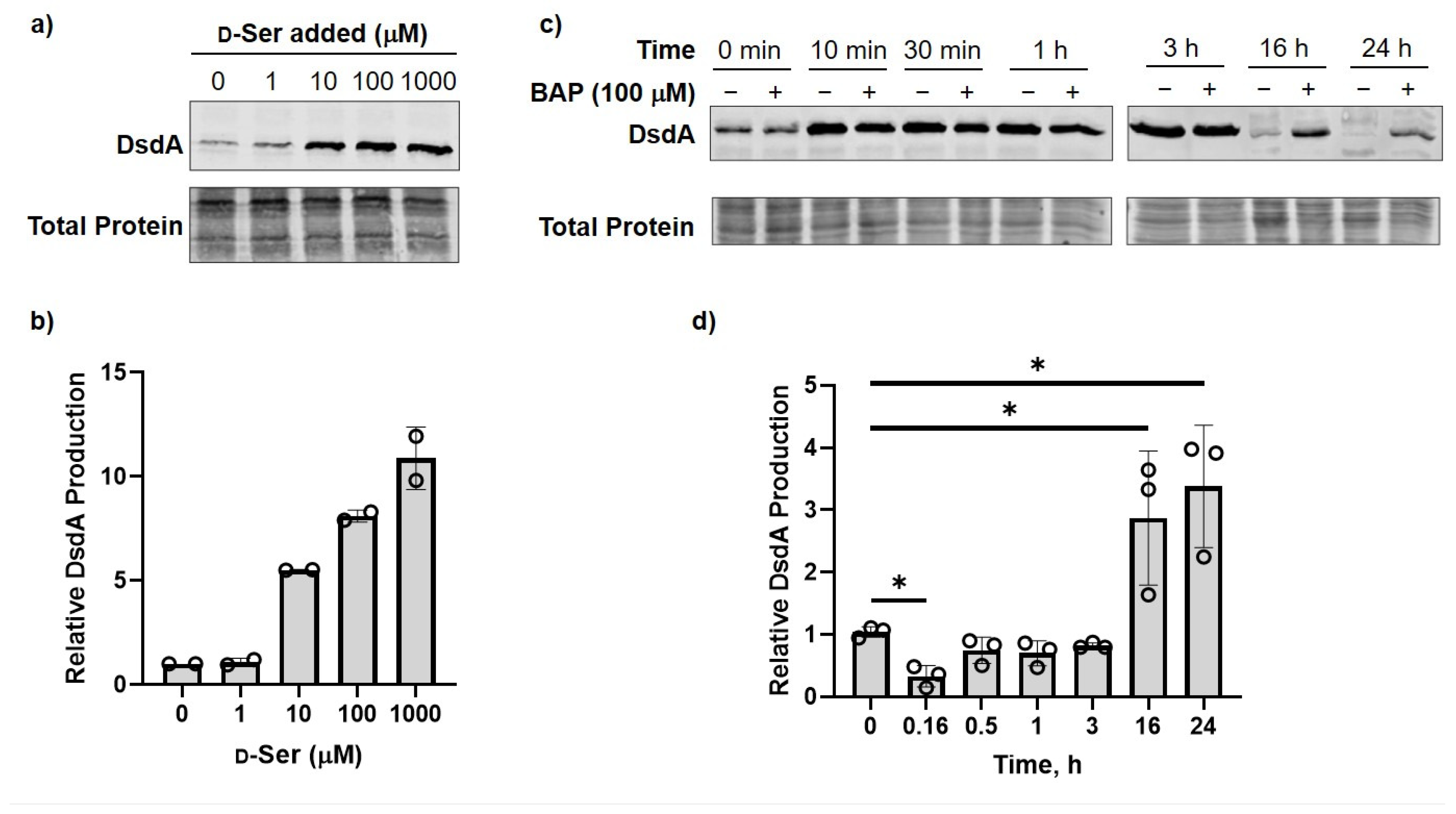
2.3. DXPS Inhibition Leads to Increased DsdA Production in the Presence of d-Ser
2.4. CFT073ΔDsdA Is Hypersensitized to d-Serine under DXPS Inhibition
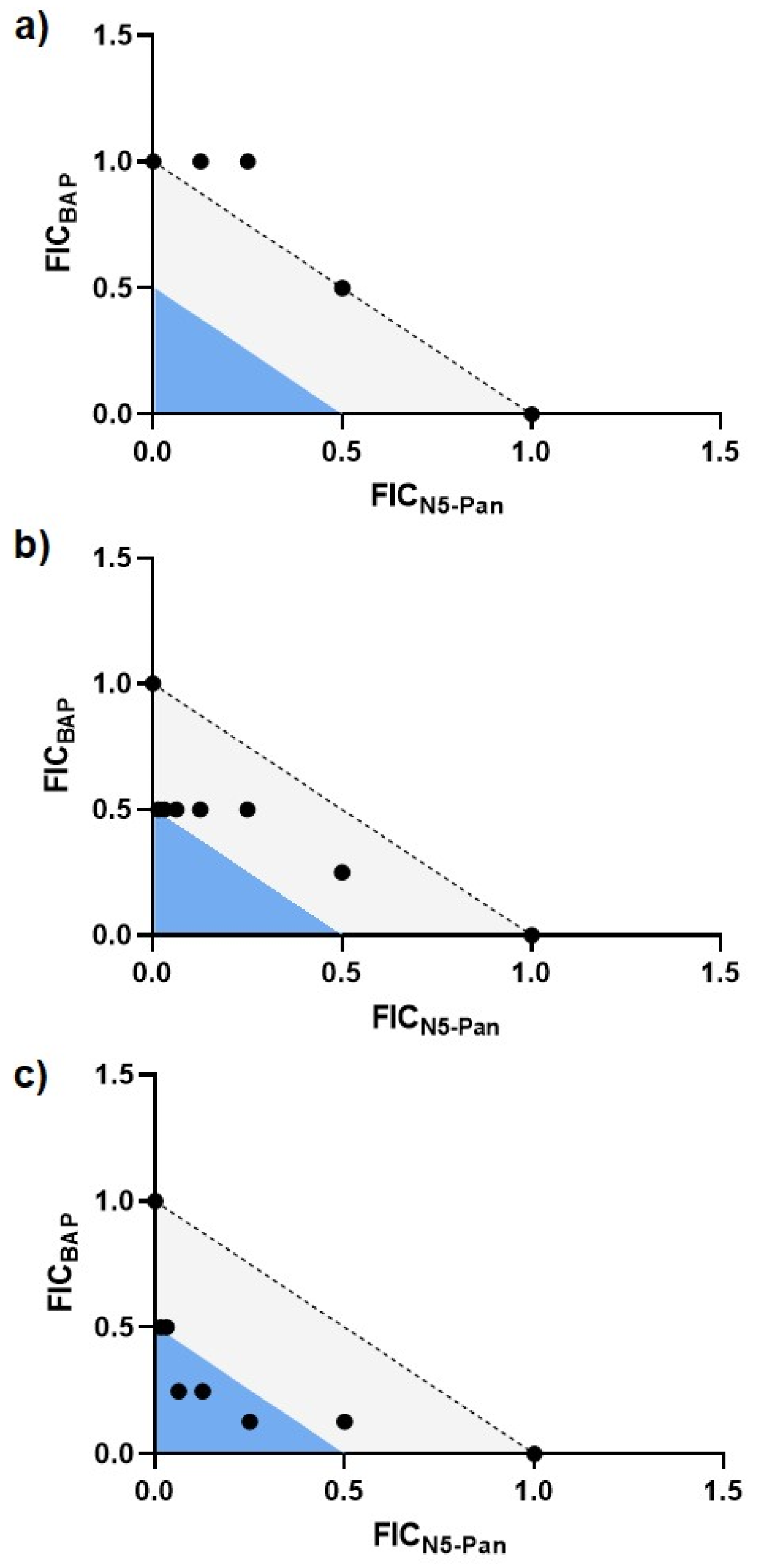
2.5. BAP-Treated CFT073 Grown in Urine Are Sensitized to Inhibition of CoA Synthesis
3. Discussion
4. Methods
4.1. General Methods
4.2. Antimicrobial Susceptibility Studies
4.3. Checkerboard Analysis
4.4. Metabolite Suppression Analysis
4.5. DsdA Expression Analysis
4.6. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Passalacqua, K.D.; Charbonneau, M.-E.; O’Riordan, M.X.D. Bacterial Metabolism Shapes the Host-Pathogen Interface. Microbiol. Spectr. 2016, 4, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohmer, L.; Hocquet, D.; Miller, S.I. Are Pathogenic Bacteria Just Looking for Food? Metabolism and Microbial Pathogenesis. Trends Microbiol. 2011, 19, 341–348. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, T.M.; Eisenreich, W.; Heesemann, J.; Goebel, W. Metabolic Adaptation of Human Pathogenic and Related Nonpathogenic Bacteria to Extra- and Intracellular Habitats. FEMS Microbiol. Rev. 2012, 36, 435–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alteri, C.J.; Mobley, H.L.T. Metabolism and Fitness of Urinary Tract Pathogens. Microbiol. Spectr. 2015, 3, 3. [Google Scholar] [CrossRef] [Green Version]
- Alteri, C.J.; Mobley, H.L.T. Escherichia coli Physiology and Metabolism Dictates Adaptation to Diverse Host Microenvironments. Curr. Opin. Microbiol. 2012, 15, 3–9. [Google Scholar] [CrossRef] [Green Version]
- Ang, M.L.T.; Murima, P.; Pethe, K. Next-Generation Antimicrobials: From Chemical Biology to First-in-Class Drugs. Arch. Pharm. Res. 2015, 38, 1702–1717. [Google Scholar] [CrossRef] [Green Version]
- Bishai, W.R. Drug Development: Locking down Metabolism. Nat. Chem. Biol. 2017, 13, 925–926. [Google Scholar] [CrossRef] [PubMed]
- Wellington, S.; Nag, P.P.; Michalska, K.; Johnston, S.E.; Jedrzejczak, R.P.; Kaushik, V.K.; Clatworthy, A.E.; Siddiqi, N.; McCarren, P.; Bajrami, B.; et al. A Small-Molecule Allosteric Inhibitor of Mycobacterium Tuberculosis Tryptophan Synthase. Nat. Chem. Biol. 2017, 13, 943–950. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.A.; Gallagher, L.A.; Thongdee, M.; Staudinger, B.J.; Lippman, S.; Singh, P.K.; Manoil, C. General and Condition-Specific Essential Functions of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 2015, 112, 5189–5194. [Google Scholar] [CrossRef] [Green Version]
- D’Elia, M.A.; Pereira, M.P.; Brown, E.D. Are Essential Genes Really Essential? Trends Microbiol. 2009, 17, 433–438. [Google Scholar] [CrossRef]
- Turner, K.H.; Wessel, A.K.; Palmer, G.C.; Murray, J.L.; Whiteley, M. Essential Genome of Pseudomonas aeruginosa in Cystic Fibrosis Sputum. Proc. Natl. Acad. Sci. USA 2015, 112, 4110–4115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carfrae, L.A.; Brown, E.D. Nutrient Stress Is a Target for New Antibiotics. Trends Microbiol. 2023. Epub ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Concepción, M.; Boronat, A. Elucidation of the Methylerythritol Phosphate Pathway for Isoprenoid Biosynthesis in Bacteria and Plastids. A Metabolic Milestone Achieved through Genomics. Plant Physiol. 2002, 130, 1079–1089. [Google Scholar] [CrossRef] [Green Version]
- Hill, R.E.; Sayer, B.G.; Spenser, I.D. Spenser Biosynthesis of Vitamin B6: Incorporation of d-1-Deoxyxylulose. J. Am. Chem. Soc. 1989, 111, 1916–1917. [Google Scholar] [CrossRef]
- Rohmer, M.; Knani, M.; Simonin, P.; Sutter, B.; Sahm, H. Isoprenoid Biosynthesis in Bacteria: A Novel Pathway for the Early Steps Leading to Isopentenyl Diphosphate. Biochem. J. 1993, 295 Pt 2, 517–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- David, S.; Estramareix, B.; Fischer, J.C.; Therisod, M. 1-Deoxy-D-Threo-2-Pentulose: The Precursor of the Five-Carbon Chain of the Thiazole of Thiamine. J. Am. Chem. Soc. 1981, 103, 7341–7342. [Google Scholar] [CrossRef]
- Sanders, S.; Bartee, D.; Harrison, M.J.; Phillips, P.D.; Koppisch, A.T.; Freel Meyers, C.L. Growth Medium-Dependent Antimicrobial Activity of Early Stage MEP Pathway Inhibitors. PLoS ONE 2018, 13, e0197638. [Google Scholar] [CrossRef] [Green Version]
- Sanders, S.; Vierling, R.J.; Bartee, D.; DeColli, A.A.; Harrison, M.J.; Aklinski, J.L.; Koppisch, A.T.; Freel Meyers, C.L. Challenges and Hallmarks of Establishing Alkylacetylphosphonates as Probes of Bacterial 1-Deoxy-d-Xylulose 5-Phosphate Synthase. ACS Infect. Dis. 2017, 3, 467–478. [Google Scholar] [CrossRef]
- Bartee, D.; Freel Meyers, C.L. Toward Understanding the Chemistry and Biology of 1-Deoxy-d-Xylulose 5-Phosphate (DXP) Synthase: A Unique Antimicrobial Target at the Heart of Bacterial Metabolism. Acc. Chem. Res. 2018, 51, 2546–2555. [Google Scholar] [CrossRef]
- Bartee, D.; Morris, F.; Al-Khouja, A.; Freel Meyers, C.L. Hydroxybenzaldoximes Are D-GAP-Competitive Inhibitors of E. Coli 1-Deoxy-D-Xylulose-5-Phosphate Synthase. Chembiochem 2015, 16, 1771–1781. [Google Scholar] [CrossRef] [Green Version]
- Morris, F.; Vierling, R.; Boucher, L.; Bosch, J.; Freel Meyers, C.L. DXP Synthase-Catalyzed C-N Bond Formation: Nitroso Substrate Specificity Studies Guide Selective Inhibitor Design. Chembiochem 2013, 14, 1309–1315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flores-Mireles, A.L.; Walker, J.N.; Caparon, M.; Hultgren, S.J. Urinary Tract Infections: Epidemiology, Mechanisms of Infection and Treatment Options. Nat. Rev. Microbiol. 2015, 13, 269–284. [Google Scholar] [CrossRef] [PubMed]
- Medina, M.; Castillo-Pino, E. An Introduction to the Epidemiology and Burden of Urinary Tract Infections. Ther. Adv. Urol. 2019, 11, 1756287219832172. [Google Scholar] [CrossRef] [Green Version]
- Chan, C.C.Y.; Lewis, I.A. Role of Metabolism in Uropathogenic Escherichia coli. Trends Microbiol. 2022, 30, 1174–1204. [Google Scholar] [CrossRef] [PubMed]
- Alteri, C.J.; Smith, S.N.; Mobley, H.L.T. Fitness of Escherichia coli during Urinary Tract Infection Requires Gluconeogenesis and the TCA Cycle. PLoS Pathog. 2009, 5, e1000448. [Google Scholar] [CrossRef] [Green Version]
- Alteri, C.J.; Himpsl, S.D.; Shea, A.E.; Mobley, H.L.T. Flexible Metabolism and Suppression of Latent Enzymes Are Important for Escherichia coli Adaptation to Diverse Environments within the Host. J. Bacteriol. 2019, 201, e00181-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Himpsl, S.D.; Shea, A.E.; Zora, J.; Stocki, J.A.; Foreman, D.; Alteri, C.J.; Mobley, H.L.T. The Oxidative Fumarase FumC Is a Key Contributor for E. Coli Fitness under Iron-Limitation and during UTI. PLoS Pathog. 2020, 16, e1008382. [Google Scholar] [CrossRef] [Green Version]
- Mann, R.; Mediati, D.G.; Duggin, I.G.; Harry, E.J.; Bottomley, A.L. Metabolic Adaptations of Uropathogenic E. Coli in the Urinary Tract. Front. Cell. Infect. Microbiol. 2017, 7, 241. [Google Scholar] [CrossRef] [Green Version]
- Connolly, J.P.R.; Goldstone, R.J.; Burgess, K.; Cogdell, R.J.; Beatson, S.A.; Vollmer, W.; Smith, D.G.E.; Roe, A.J. The Host Metabolite D-Serine Contributes to Bacterial Niche Specificity through Gene Selection. ISME J. 2015, 9, 1052. [Google Scholar] [CrossRef] [Green Version]
- O’Boyle, N.; Connolly, J.P.R.; Tucker, N.P.; Roe, A.J. Genomic Plasticity of Pathogenic Escherichia coli Mediates D-Serine Tolerance via Multiple Adaptive Mechanisms. Proc. Natl. Acad. Sci. USA 2020, 117, 22484–22493. [Google Scholar] [CrossRef]
- Durham, N.N.; Milligan, R. A Mechanism of Growth Inhibition by D-Serine in a Flavobacterium. Biochem. Biophys. Res. Commun. 1962, 7, 342–345. [Google Scholar] [CrossRef]
- Maas, W.K.; Davis, B.D. Pantothenate Studies. I. Interference by D-Serine and L-Aspartic Acid with Pantothenate Synthesis in Escherichia coli. J. Bacteriol. 1950, 60, 733–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cosloy, S.D.; McFall, E. Metabolism of D-Serine in Escherichia coli K-12: Mechanism of Growth Inhibition. J. Bacteriol. 1973, 114, 685–694. [Google Scholar] [CrossRef] [Green Version]
- Heincz, M.C.; McFall, E. Role of the DsdC Activator in Regulation of D-Serine Deaminase Synthesis. J. Bacteriol. 1978, 136, 96–103. [Google Scholar] [CrossRef] [Green Version]
- Anfora, A.T.; Welch, R.A. DsdX Is the Second D-Serine Transporter in Uropathogenic Escherichia coli Clinical Isolate CFT073. J. Bacteriol. 2006, 188, 6622–6628. [Google Scholar] [CrossRef] [Green Version]
- McFall, E. Mapping of the D-Serine Deaminase Region in Escherichia coli K-12. Genetics 1967, 55, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Nørregaard-Madsen, M.; McFall, E.; Valentin-Hansen, P. Organization and Transcriptional Regulation of the Escherichia coli K-12 D-Serine Tolerance Locus. J. Bacteriol. 1995, 177, 6456–6461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bloom, F.R.; McFall, E.; Young, M.C.; Carothers, A.M. Positive Control in the D-Serine Deaminase System of Escherichia coli K-12. J. Bacteriol. 1975, 121, 1092–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urusova, D.V.; Isupov, M.N.; Antonyuk, S.; Kachalova, G.S.; Obmolova, G.; Vagin, A.A.; Lebedev, A.A.; Burenkov, G.P.; Dauter, Z.; Bartunik, H.D.; et al. Crystal Structure of D-Serine Dehydratase from Escherichia coli. Biochim. Biophys. Acta 2012, 1824, 422–432. [Google Scholar] [CrossRef] [PubMed]
- Schiltz, E.; Schmitt, W. Sequence of Escherichia coli D-Serine Dehydratase. Location of the Pyridoxal-Phosphate Binding Site. FEBS Lett. 1981, 134, 57–62. [Google Scholar] [CrossRef] [Green Version]
- Tong, M.; French, S.; El Zahed, S.S.; Ong, W.K.; Karp, P.D.; Brown, E.D. Gene Dispensability in Escherichia coli Grown in Thirty Different Carbon Environments. MBio 2020, 11, e02259-20. [Google Scholar] [CrossRef]
- Zlitni, S.; Ferruccio, L.F.; Brown, E.D. Metabolic Suppression Identifies New Antibacterial Inhibitors under Nutrient Limitation. Nat. Chem. Biol. 2013, 9, 796–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marrero, J.; Rhee, K.Y.; Schnappinger, D.; Pethe, K.; Ehrt, S. Gluconeogenic Carbon Flow of Tricarboxylic Acid Cycle Intermediates Is Critical for Mycobacterium Tuberculosis to Establish and Maintain Infection. Proc. Natl. Acad. Sci. USA 2010, 107, 9819–9824. [Google Scholar] [CrossRef] [Green Version]
- Okuda, J.; Nagata, S.; Yasuda, M.; Suezawa, C. Validating the Inhibitory Effects of D- and l-Serine on the Enzyme Activity of d-3-Phosphoglycerate Dehydrogenases That Are Purified from Pseudomonas aeruginosa, Escherichia coli and Human Colon. Gut Pathog. 2019, 11, 35. [Google Scholar] [CrossRef] [PubMed]
- Leonardi, R.; Jackowski, S. Biosynthesis of Pantothenic Acid and Coenzyme A. EcoSal Plus 2007, 2, e2324-6200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hryckowian, A.J.; Baisa, G.A.; Schwartz, K.J.; Welch, R.A. DsdA Does Not Affect Colonization of the Murine Urinary Tract by Escherichia coli CFT073. PLoS ONE 2015, 10, e0138121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.-M.; Frank, M.W.; Virga, K.G.; Lee, R.E.; Rock, C.O.; Jackowski, S. Acyl Carrier Protein Is a Cellular Target for the Antibacterial Action of the Pantothenamide Class of Pantothenate Antimetabolites. J. Biol. Chem. 2004, 279, 50969–50975. [Google Scholar] [CrossRef] [Green Version]
- Virga, K.G.; Zhang, Y.-M.; Leonardi, R.; Ivey, R.A.; Hevener, K.; Park, H.-W.; Jackowski, S.; Rock, C.O.; Lee, R.E. Structure-Activity Relationships and Enzyme Inhibition of Pantothenamide-Type Pantothenate Kinase Inhibitors. Bioorg. Med. Chem. 2006, 14, 1007–1020. [Google Scholar] [CrossRef]
- Arnott, Z.L.P.; Nozaki, S.; Monteiro, D.C.F.; Morgan, H.E.; Pearson, A.R.; Niki, H.; Webb, M.E. The Mechanism of Regulation of Pantothenate Biosynthesis by the PanD-PanZ·AcCoA Complex Reveals an Additional Mode of Action for the Antimetabolite N-Pentyl Pantothenamide (N5-Pan). Biochemistry 2017, 56, 4931–4939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clifton, G.; Bryant, S.R.; Skinner, C.G. N’-(Substituted) Pantothenamides, Antimetabolites of Pantothenic Acid. Arch. Biochem. Biophys. 1970, 137, 523–528. [Google Scholar] [CrossRef]
- Thomas, J.; Cronan, J.E. Antibacterial Activity of N-Pentylpantothenamide Is Due to Inhibition of Coenzyme a Synthesis. Antimicrob. Agents Chemother. 2010, 54, 1374–1377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awuah, E.; Ma, E.; Hoegl, A.; Vong, K.; Habib, E.; Auclair, K. Exploring Structural Motifs Necessary for Substrate Binding in the Active Site of Escherichia coli Pantothenate Kinase. Bioorg. Med. Chem. 2014, 22, 3083–3090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snyder, J.A.; Haugen, B.J.; Buckles, E.L.; Lockatell, C.V.; Johnson, D.E.; Donnenberg, M.S.; Welch, R.A.; Mobley, H.L.T. Transcriptome of Uropathogenic Escherichia coli during Urinary Tract Infection. Infect. Immun. 2004, 72, 6373–6381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagan, E.C.; Lloyd, A.L.; Rasko, D.A.; Faerber, G.J.; Mobley, H.L.T. Escherichia coli Global Gene Expression in Urine from Women with Urinary Tract Infection. PLoS Pathog. 2010, 6, e1001187. [Google Scholar] [CrossRef] [Green Version]
- Nogales, J.; Garmendia, J. Bacterial Metabolism and Pathogenesis Intimate Intertwining: Time for Metabolic Modelling to Come into Action. Microb. Biotechnol. 2022, 15, 95–102. [Google Scholar] [CrossRef]
- Yan, L.; Nie, W.; Lv, H. Metabolic Phenotyping of the Yersinia High-Pathogenicity Island That Regulates Central Carbon Metabolism. Analyst 2015, 140, 3356–3361. [Google Scholar] [CrossRef]
- Smith, J.M.; Vierling, R.J.; Meyers, C.F. Selective Inhibition of E. coli 1-Deoxy-D-Xylulose-5-Phosphate Synthase by Acetylphosphonates(). Medchemcomm 2012, 3, 65–67. [Google Scholar] [CrossRef]
- Strauss, E.; Begley, T.P. The Antibiotic Activity of N-Pentylpantothenamide Results from Its Conversion to Ethyldethia-Coenzyme a, a Coenzyme a Antimetabolite. J. Biol. Chem. 2002, 277, 48205–48209. [Google Scholar] [CrossRef] [Green Version]
- Jansen, P.A.M.; van der Krieken, D.A.; Botman, P.N.M.; Blaauw, R.H.; Cavina, L.; Raaijmakers, E.M.; de Heuvel, E.; Sandrock, J.; Pennings, L.J.; Hermkens, P.H.H.; et al. Stable Pantothenamide Bioisosteres: Novel Antibiotics for Gram-Positive Bacteria. J. Antibiot. 2019, 72, 682–692. [Google Scholar] [CrossRef] [Green Version]
- Patrone, J.D.; Yao, J.; Scott, N.E.; Dotson, G.D. Selective Inhibitors of Bacterial Phosphopantothenoylcysteine Synthetase. J. Am. Chem. Soc. 2009, 131, 16340–16341. [Google Scholar] [CrossRef] [Green Version]
- Smith, J.M.; Warrington, N.V.; Vierling, R.J.; Kuhn, M.L.; Anderson, W.F.; Koppisch, A.T.; Freel Meyers, C.L. Targeting DXP Synthase in Human Pathogens: Enzyme Inhibition and Antimicrobial Activity of Butylacetylphosphonate. J. Antibiot. 2014, 67, 77–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moody, J. Clinical Microbiology Procedures Handbook, 2nd ed.; American Society for Microbiology: Washington, DC, USA, 2004. [Google Scholar]
- Pillai, S.K.; Moellering, R.C.; Eliopoulos, G.M. Antimicrobial Combinations. In Antibiotics in Laboratory Medicine; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2005. [Google Scholar]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, E.C.; Freel Meyers, C.L. DXP Synthase Function in a Bacterial Metabolic Adaptation and Implications for Antibacterial Strategies. Antibiotics 2023, 12, 692. https://doi.org/10.3390/antibiotics12040692
Chen EC, Freel Meyers CL. DXP Synthase Function in a Bacterial Metabolic Adaptation and Implications for Antibacterial Strategies. Antibiotics. 2023; 12(4):692. https://doi.org/10.3390/antibiotics12040692
Chicago/Turabian StyleChen, Eric C., and Caren L. Freel Meyers. 2023. "DXP Synthase Function in a Bacterial Metabolic Adaptation and Implications for Antibacterial Strategies" Antibiotics 12, no. 4: 692. https://doi.org/10.3390/antibiotics12040692
APA StyleChen, E. C., & Freel Meyers, C. L. (2023). DXP Synthase Function in a Bacterial Metabolic Adaptation and Implications for Antibacterial Strategies. Antibiotics, 12(4), 692. https://doi.org/10.3390/antibiotics12040692