
Using Targeted Nano-Antibiotics to Improve Antibiotic Efficacy against Staphylococcus aureus Infections


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
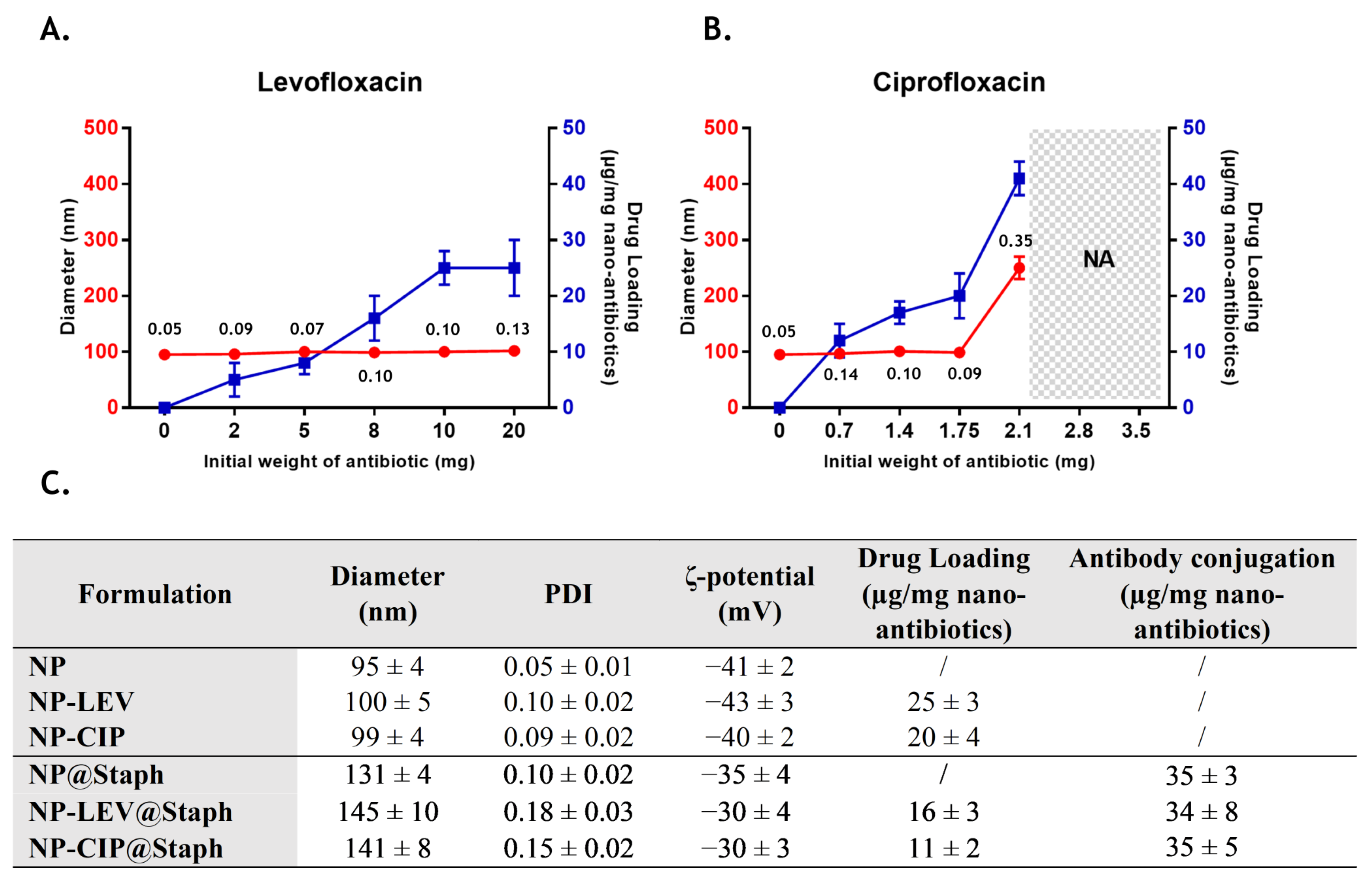
2.1. Nano-Antibiotic Preparation and Characterization
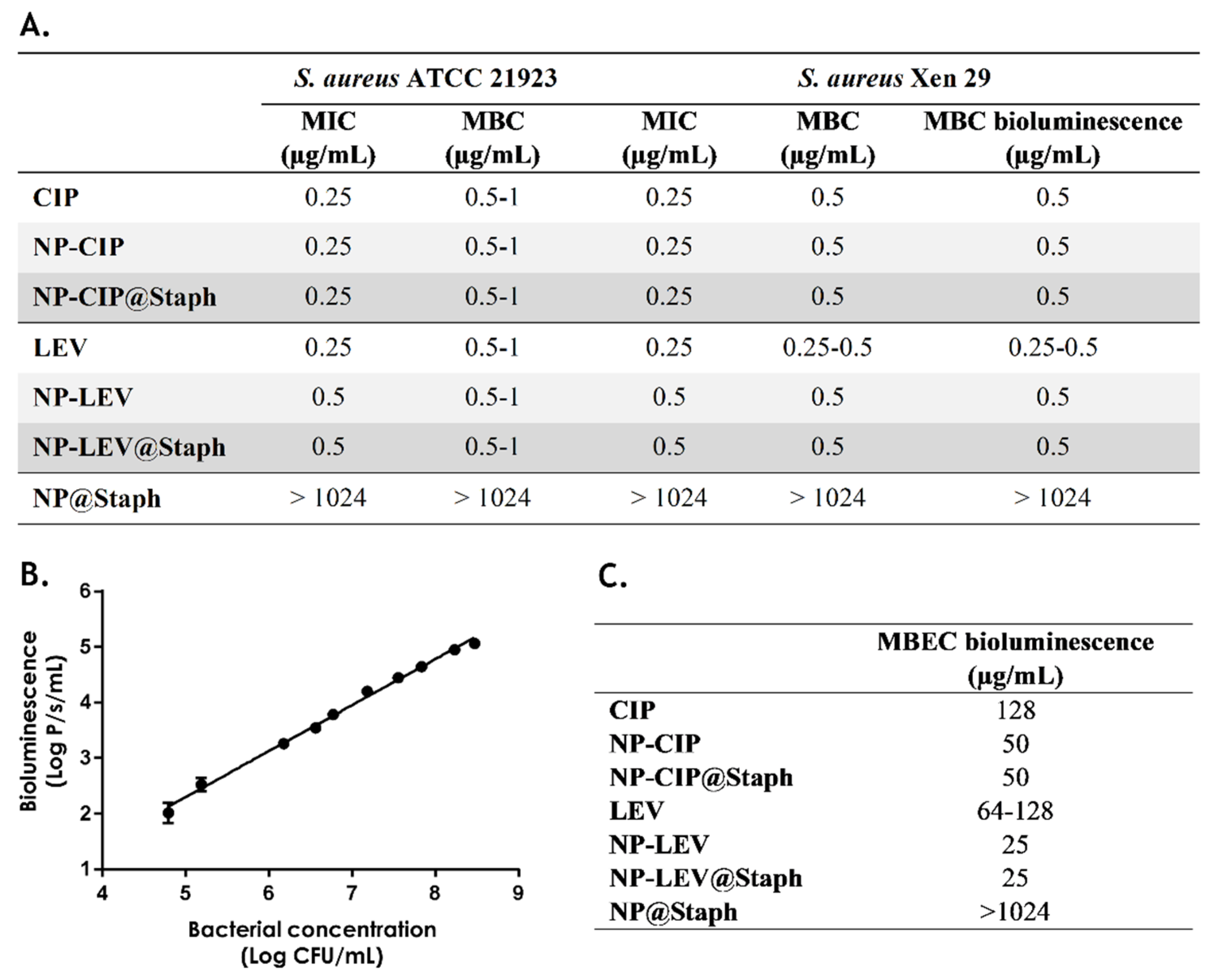
2.2. In Vitro Antimicrobial Studies of Nano-Antibiotics
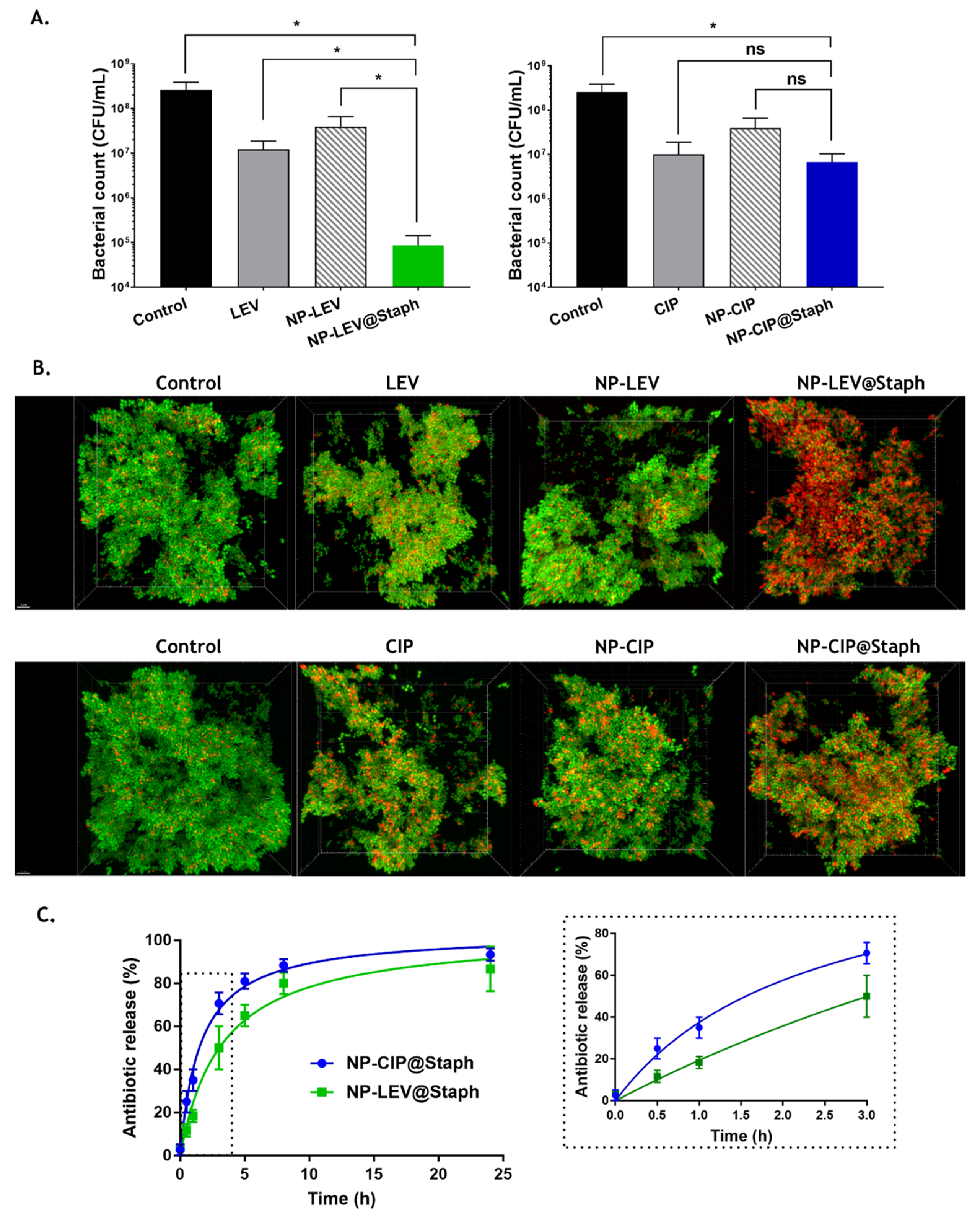
2.3. Short-Time Killing Assay
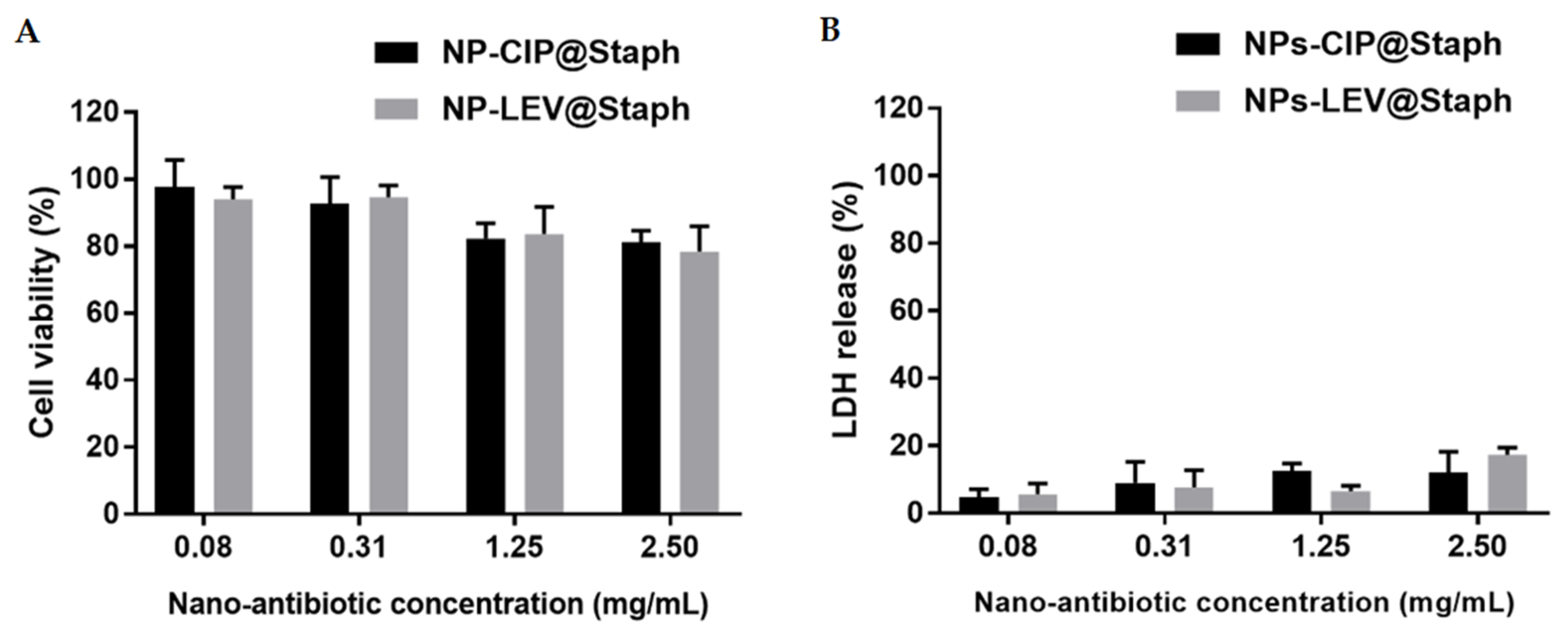
2.4. Cytotoxicity
3. Materials and Methods
3.1. Materials, Bacterial Strains, and Growth Conditions
3.2. Nano-Antibiotic Preparation
3.3. Formulation Characterization
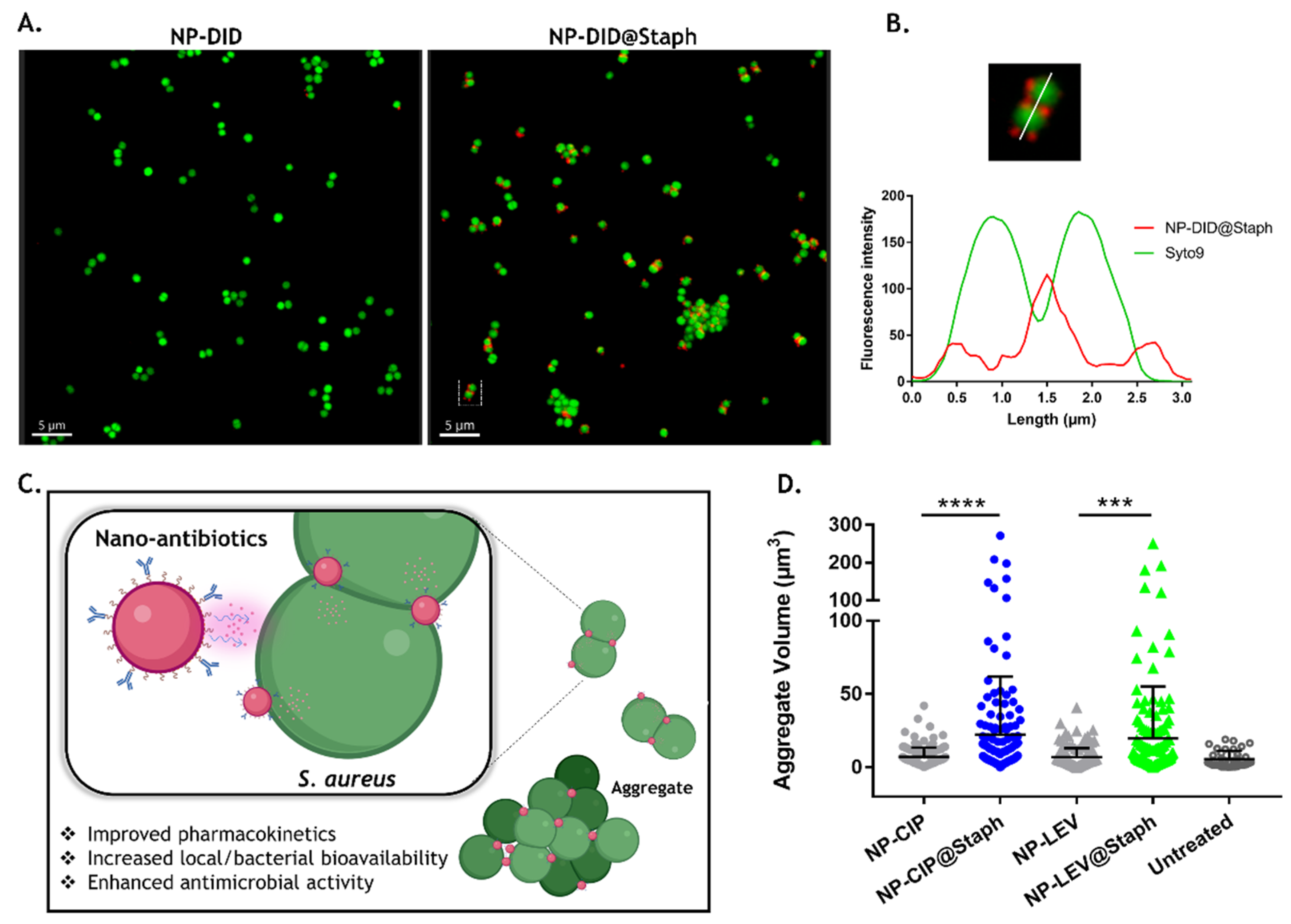
3.4. Interaction of Nano-Antibiotics with Bacterial Cells
3.5. Determination of Minimum Inhibitory Concentration (MIC) and Minimum Bactericidal Concentration (MBC)
3.6. Determination of Minimum Biofilm Eradication Concentration (MBEC) Based on Bioluminescence Measurement
3.7. Short-Time Killing Assay of Bacteria
3.8. In Vitro Cytotoxicity of Formulation
3.9. Statistical Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Howden, B.P.; Giulieri, S.G.; Wong Fok Lung, T.; Baines, S.L.; Sharkey, L.K.; Lee, J.Y.H.; Hachani, A.; Monk, I.R.; Stinear, T.P. Staphylococcus aureus host interactions and adaptation. Nat. Rev. Microbiol. 2023, 21, 380–395. [Google Scholar] [CrossRef] [PubMed]
- Klein, S.; Boutin, S.; Kocer, K.; Fiedler, M.O.; Störzinger, D.; Weigand, M.A.; Tan, B.; Richter, D.; Rupp, C.; Mieth, M.; et al. Rapid Development of Cefiderocol Resistance in Carbapenem-resistant Enterobacter cloacae During Therapy Is Associated With Heterogeneous Mutations in the Catecholate Siderophore Receptor cirA. Clin. Infect. Dis. 2021, 74, 905–908. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.; Joo, J.; Kang, J.; Kim, B.; Braun, G.B.; She, Z.-G.; Kim, D.; Mann, A.P.; Mölder, T.; Teesalu, T.; et al. Antibiotic-loaded nanoparticles targeted to the site of infection enhance antibacterial efficacy. Nat. Biomed. Eng. 2018, 2, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Aman, M.J.; Adhikari, R.P. Staphylococcal Bicomponent Pore-Forming Toxins: Targets for Prophylaxis and Immunotherapy. Toxins 2014, 6, 950–972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, H.; Karakasyan, C.; Jouenne, T.; Le Cerf, D.; Dé, E. Application of Polymeric Nanocarriers for Enhancing the Bioavailability of Antibiotics at the Target Site and Overcoming Antimicrobial Resistance. Appl. Sci. 2021, 11, 10695. [Google Scholar] [CrossRef]
- Ropponen, H.-K.; Richter, R.; Hirsch, A.K.H.; Lehr, C.-M. Mastering the Gram-negative bacterial barrier—Chemical approaches to increase bacterial bioavailability of antibiotics. Adv. Drug Deliv. Rev. 2021, 172, 339–360. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Chen, Y.; Zhang, Y.; Zhang, Q.; Zhang, L. Nanoparticle-based local antimicrobial drug delivery. Adv. Drug Deliv. Rev. 2018, 127, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Tewes, F.; Lamy, B.; Laroche, J.; Lamarche, I.; Marchand, S. PK-PD Evaluation of Inhaled Microparticles loaded with Ciprofloxacin-Copper complex in a Rat Model of Chronic Pseudomonas aeruginosa Lung Infection. Int. J. Pharm. X 2023, 5, 100178. [Google Scholar] [CrossRef]
- Radovic-Moreno, A.F.; Lu, T.K.; Puscasu, V.A.; Yoon, C.J.; Langer, R.; Farokhzad, O.C. Surface Charge-Switching Polymeric Nanoparticles for Bacterial Cell Wall-Targeted Delivery of Antibiotics. ACS Nano 2012, 6, 4279–4287. [Google Scholar] [CrossRef] [Green Version]
- Le, H.; Arnoult, C.; Dé, E.; Schapman, D.; Galas, L.; Le Cerf, D.; Karakasyan, C. Antibody-Conjugated Nanocarriers for Targeted Antibiotic Delivery: Application in the Treatment of Bacterial Biofilms. Biomacromolecules 2021, 22, 1639–1653. [Google Scholar] [CrossRef]
- Puertas, S.; Batalla, P.; Moros, M.; Polo, E.; del Pino, P.; Guisán, J.M.; Grazú, V.; de la Fuente, J.M. Taking Advantage of Unspecific Interactions to Produce Highly Active Magnetic Nanoparticle−Antibody Conjugates. ACS Nano 2011, 5, 4521–4528. [Google Scholar] [CrossRef] [PubMed]
- Günday Türeli, N.; Türeli, A.E.; Schneider, M. Counter-ion complexes for enhanced drug loading in nanocarriers: Proof-of-concept and beyond. Int. J. Pharm. 2016, 511, 994–1001. [Google Scholar] [CrossRef] [PubMed]
- Frick, A.; Möller, H.; Wirbitzki, E. Biopharmaceutical characterization of oral immediate release drug products. In vitro/in vivo comparison of phenoxymethylpenicillin potassium, glimepiride and levofloxacin. Eur. J. Pharm. Biopharm. 1998, 46, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Correia, S.; Poeta, P.; Hébraud, M.; Capelo, J.L.; Igrejas, G. Mechanisms of quinolone action and resistance: Where do we stand? J. Med. Microbiol. 2017, 66, 551–559. [Google Scholar] [CrossRef]
- Furlanut, M.; Brollo, L.; Lugatti, E.; Di Qual, E.; Dolcet, F.; Talmassons, G.; Pea, F. Pharmacokinetic aspects of levofloxacin 500 mg once daily during sequential intravenous/oral therapy in patients with lower respiratory tract infections. J. Antimicrob. Chemother. 2003, 51, 101–106. [Google Scholar] [CrossRef] [Green Version]
- Child, J.; Mortiboy, D.; Andrews, J.M.; Chow, A.T.; Wise, R. Open-label crossover study to determine pharmacokinetics and penetration of two dose regimens of levofloxacin into inflammatory fluid. Antimicrob. Agents Chemother. 1995, 39, 2749–2751. [Google Scholar] [CrossRef] [Green Version]
- Jacoby, G.A. Mechanisms of Resistance to Quinolones. Clin. Infect. Dis. 2005, 41 (Suppl. 2), S120–S126. [Google Scholar] [CrossRef] [Green Version]
- Peltola, H.; Ukkonen, P.; Saxen, H.; Stass, H. Single-dose and steady-state pharmacokinetics of a new oral suspension of ciprofloxacin in children. Pediatrics 1998, 101 Pt 1, 658–662. [Google Scholar] [CrossRef]
- Sharma, P.C.; Jain, A.; Jain, S.; Pahwa, R.; Yar, M.S. Ciprofloxacin: Review on developments in synthetic, analytical, and medicinal aspects. J. Enzym. Inhib. Med. Chem. 2010, 25, 577–589. [Google Scholar] [CrossRef] [Green Version]
- Talan, D.A.; Naber, K.G.; Palou, J.; Elkharrat, D. Extended-release ciprofloxacin (Cipro XR) for treatment of urinary tract infections. Int. J. Antimicrob. Agents 2004, 23, 54–66. [Google Scholar] [CrossRef]
- Wang, Q.; Chen, C.; Liu, W.; He, X.; Zhou, N.; Zhang, D.; Gu, H.; Li, J.; Jiang, J.; Huang, W. Levofloxacin loaded mesoporous silica microspheres/nano-hydroxyapatite/polyurethane composite scaffold for the treatment of chronic osteomyelitis with bone defects. Sci. Rep. 2017, 7, 41808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-López, M.; Fernández-Delgado, A.; Moyá, M.L.; Blanco-Arévalo, D.; Carrera, C.; de la Haba, R.R.; Ventosa, A.; Bernal, E.; López-Cornejo, P. Optimized Preparation of Levofloxacin Loaded Polymeric Nanoparticles. Pharmaceutics 2019, 11, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanco, E.; Shen, H.; Ferrari, M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 2015, 33, 941–951. [Google Scholar] [CrossRef] [PubMed]
- Gheffar, C.; Le, H.; Jouenne, T.; Schaumann, A.; Corbière, A.; Vaudry, D.; LeCerf, D.; Karakasyan, C. Antibacterial Activity of Ciprofloxacin-Loaded Poly(lactic-co-glycolic acid)-Nanoparticles Against Staphylococcus aureus. Part. Part. Syst. Charact. 2021, 38, 2000253. [Google Scholar] [CrossRef]
- Danhier, F.; Ansorena, E.; Silva, J.M.; Coco, R.; Le Breton, A.; Préat, V. PLGA-based nanoparticles: An overview of biomedical applications. J. Control. Release 2012, 161, 505–522. [Google Scholar] [CrossRef]
- Kamaly, N.; Yameen, B.; Wu, J.; Farokhzad, O.C. Degradable Controlled-Release Polymers and Polymeric Nanoparticles: Mechanisms of Controlling Drug Release. Chem. Rev. 2016, 116, 2602–2663. [Google Scholar] [CrossRef] [Green Version]
- Dillen, K.; Vandervoort, J.; Van den Mooter, G.; Ludwig, A. Evaluation of ciprofloxacin-loaded Eudragit® RS100 or RL100/PLGA nanoparticles. Int. J. Pharm. 2006, 314, 72–82. [Google Scholar] [CrossRef]
- Jeong, Y.-I.; Na, H.-S.; Seo, D.-H.; Kim, D.-G.; Lee, H.-C.; Jang, M.-K.; Na, S.-K.; Roh, S.-H.; Kim, S.-I.; Nah, J.-W. Ciprofloxacin-encapsulated poly(dl-lactide-co-glycolide) nanoparticles and its antibacterial activity. Int. J. Pharm. 2008, 352, 317–323. [Google Scholar] [CrossRef]
- Cheng, J.; Teply, B.A.; Sherifi, I.; Sung, J.; Luther, G.; Gu, F.X.; Levy-Nissenbaum, E.; Radovic-Moreno, A.F.; Langer, R.; Farokhzad, O.C. Formulation of functionalized PLGA–PEG nanoparticles for in vivo targeted drug delivery. Biomaterials 2007, 28, 869–876. [Google Scholar] [CrossRef] [Green Version]
- Fay, F.; McLaughlin, K.M.; Small, D.M.; Fennell, D.A.; Johnston, P.G.; Longley, D.B.; Scott, C.J. Conatumumab (AMG 655) coated nanoparticles for targeted pro-apoptotic drug delivery. Biomaterials 2011, 32, 8645–8653. [Google Scholar] [CrossRef]
- Krivić, H.; Himbert, S.; Sun, R.; Feigis, M.; Rheinstädter, M.C. Erythro-PmBs: A Selective Polymyxin B Delivery System Using Antibody-Conjugated Hybrid Erythrocyte Liposomes. ACS Infect. Dis. 2022, 8, 2059–2072. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-H.; Chang, D.-S. Fabrication, characterization, and biological evaluation of anti-HER2 indocyanine green-doxorubicin-encapsulated PEG-b-PLGA copolymeric nanoparticles for targeted photochemotherapy of breast cancer cells. Sci. Rep. 2017, 7, 46688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeDent, A.C.; McAdow, M.; Schneewind, O. Distribution of Protein A on the Surface of Staphylococcus aureus. J. Bacteriol. 2007, 189, 4473–4484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, S.; Frankel, M.B.; Schneewind, O.; Missiakas, D. Release of protein A from the cell wall of Staphylococcus aureus. Proc. Natl. Acad. Sci. USA 2014, 111, 1574–1579. [Google Scholar] [CrossRef] [Green Version]
- Marcelo, G.A.; Galhano, J.; Duarte, M.P.; Capelo-Martínez, J.L.; Lodeiro, C.; Oliveira, E. Validation of a Standard Luminescence Method for the Fast Determination of the Antimicrobial Activity of Nanoparticles in Escherichia coli. Nanomaterials 2022, 12, 2164. [Google Scholar] [CrossRef] [PubMed]
- Kadurugamuwa, J.L.; Sin, L.; Albert, E.; Yu, J.; Francis, K.; DeBoer, M.; Rubin, M.; Bellinger-Kawahara, C.; Parr, T.R.; Contag, P.R. Direct Continuous Method for Monitoring Biofilm Infection in a Mouse Model. Infect. Immun. 2003, 71, 882–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebeaux, D.; Ghigo, J.-M.; Beloin, C. Biofilm-related infections: Bridging the gap between clinical management and fundamental aspects of recalcitrance toward antibiotics. Microbiol. Mol. Biol. Rev. 2014, 78, 510–543. [Google Scholar] [CrossRef] [Green Version]
- Fuqua, C.; Filloux, A.; Ghigo, J.-M.; Visick, K.L. Biofilms 2018: A Diversity of Microbes and Mechanisms. J. Bacteriol. 2019, 201, e00118–e00119. [Google Scholar] [CrossRef] [Green Version]
- Anderl, J.N.; Franklin, M.J.; Stewart, P.S. Role of Antibiotic Penetration Limitation in Klebsiella pneumoniae Biofilm Resistance to Ampicillin and Ciprofloxacin. Antimicrob. Agents Chemother. 2000, 44, 1818–1824. [Google Scholar] [CrossRef] [Green Version]
- Forier, K.; Messiaen, A.-S.; Raemdonck, K.; Nelis, H.; De Smedt, S.; Demeester, J.; Coenye, T.; Braeckmans, K. Probing the size limit for nanomedicine penetration into Burkholderia multivorans and Pseudomonas aeruginosa biofilms. J. Control. Release 2014, 195, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Tian, J.; Zhu, J.; Chen, J.; Li, L.; Yang, C.; Chen, J.; Chen, D. Photodynamic and photothermal co-driven CO-enhanced multi-mode synergistic antibacterial nanoplatform to effectively fight against biofilm infections. Chem. Eng. J. 2021, 426, 131919. [Google Scholar] [CrossRef]
- Tahrioui, A.; Duchesne, R.; Bouffartigues, E.; Rodrigues, S.; Maillot, O.; Tortuel, D.; Hardouin, J.; Taupin, L.; Groleau, M.-C.; Dufour, A.; et al. Extracellular DNA release, quorum sensing, and PrrF1/F2 small RNAs are key players in Pseudomonas aeruginosa tobramycin-enhanced biofilm formation. NPJ Biofilms Microbiomes 2019, 5, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magana, M.; Sereti, C.; Ioannidis, A.; Mitchell, C.A.; Ball, A.R.; Magiorkinis, E.; Chatzipanagiotou, S.; Hamblin, M.R.; Hadjifrangiskou, M.; Tegos, G.P. Options and Limitations in Clinical Investigation of Bacterial Biofilms. Clin. Microbiol. Rev. 2018, 31, e00084-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, W.; Chen, X.; Fu, L.; Zhu, J.; Fan, M.; Chen, J.; Yang, C.; Yang, G.; Wu, L.; Mao, G.; et al. Ultra-efficient Antibacterial System Based on Photodynamic Therapy and CO Gas Therapy for Synergistic Antibacterial and Ablation Biofilms. ACS Appl. Mater. Interfaces 2020, 12, 22479–22491. [Google Scholar] [CrossRef]
- Azzopardi, E.A.; Ferguson, E.L.; Thomas, D.W. The enhanced permeability retention effect: A new paradigm for drug targeting in infection. J. Antimicrob. Chemother. 2013, 68, 257–274. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Zhang, J.; Chen, M.; Gong, H.; Thamphiwatana, S.; Eckmann, L.; Gao, W.; Zhang, L. A Bioadhesive Nanoparticle–Hydrogel Hybrid System for Localized Antimicrobial Drug Delivery. ACS Appl. Mater. Interfaces 2016, 8, 18367–18374. [Google Scholar] [CrossRef] [Green Version]
- Levison, M.E.; Levison, J.H. Pharmacokinetics and pharmacodynamics of antibacterial agents. Infect. Dis. Clin. N. Am. 2009, 23, 791–815. [Google Scholar] [CrossRef] [Green Version]
- Stiefel, P.; Schmidt-Emrich, S.; Maniura-Weber, K.; Ren, Q. Critical aspects of using bacterial cell viability assays with the fluorophores SYTO9 and propidium iodide. BMC Microbiol. 2015, 15, 36. [Google Scholar] [CrossRef] [Green Version]
- Yus, C.; Irusta, S.; Sebastian, V.; Arruebo, M. Controlling Particle Size and Release Kinetics in the Sustained Delivery of Oral Antibiotics Using pH-Independent Mucoadhesive Polymers. Mol. Pharm. 2020, 17, 3314–3327. [Google Scholar] [CrossRef]
- Fredenberg, S.; Wahlgren, M.; Reslow, M.; Axelsson, A. The mechanisms of drug release in poly(lactic-co-glycolic acid)-based drug delivery systems—A review. Int. J. Pharm. 2011, 415, 34–52. [Google Scholar] [CrossRef]
- Sung, K.C.; Han, R.-Y.; Hu, O.Y.P.; Hsu, L.-R. Controlled release of nalbuphine prodrugs from biodegradable polymeric matrices: Influence of prodrug hydrophilicity and polymer composition. Int. J. Pharm. 1998, 172, 17–25. [Google Scholar] [CrossRef]
- Baek, J.-S.; Tan, C.H.; Ng, N.K.J.; Yeo, Y.P.; Rice, S.A.; Loo, S.C.J. A programmable lipid-polymer hybrid nanoparticle system for localized, sustained antibiotic delivery to Gram-positive and Gram-negative bacterial biofilms. Nanoscale Horiz. 2018, 3, 305–311. [Google Scholar] [CrossRef]
- Khannanov, A.; Rossova, A.; Ulakhovich, N.; Evtugyn, V.; Valiullin, L.; Nabatov, A.; Kutyrev, G.; Kutyreva, M. Doxorubicin-Loaded Hybrid Micelles Based on Carboxyl-Terminated Hyperbranched Polyester Polyol. ACS Appl. Polym. Mater. 2022, 4, 2553–2561. [Google Scholar] [CrossRef]
- Kurbangaleeva, S.V.; Syromiatnikova, V.Y.; Prokopeva, A.E.; Rogov, A.M.; Khannanov, A.A.; Rizvanov, A.A.; Gomzikova, M.O. Increased Yield of Extracellular Vesicles after Cytochalasin B Treatment and Vortexing. Curr. Issues Mol. Biol. 2023, 45, 2431–2443. [Google Scholar] [CrossRef]
- Grabowski, N.; Hillaireau, H.; Vergnaud, J.; Santiago, L.A.; Kerdine-Romer, S.; Pallardy, M.; Tsapis, N.; Fattal, E. Toxicity of surface-modified PLGA nanoparticles toward lung alveolar epithelial cells. Int. J. Pharm. 2013, 454, 686–694. [Google Scholar] [CrossRef]
- Risnayanti, C.; Jang, Y.-S.; Lee, J.; Ahn, H.J. PLGA nanoparticles co-delivering MDR1 and BCL2 siRNA for overcoming resistance of paclitaxel and cisplatin in recurrent or advanced ovarian cancer. Sci. Rep. 2018, 8, 7498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Sai Lung, P.; Zhao, S.; Chu, Z.; Chrzanowski, W.; Li, Q. Shape dependent cytotoxicity of PLGA-PEG nanoparticles on human cells. Sci. Rep. 2017, 7, 7315. [Google Scholar] [CrossRef]
- Doty, A.C.; Zhang, Y.; Weinstein, D.G.; Wang, Y.; Choi, S.; Qu, W.; Mittal, S.; Schwendeman, S.P. Mechanistic analysis of triamcinolone acetonide release from PLGA microspheres as a function of varying in vitro release conditions. Eur. J. Pharm. Biopharm. 2017, 113, 24–33. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Le, H.; Dé, E.; Le Cerf, D.; Karakasyan, C. Using Targeted Nano-Antibiotics to Improve Antibiotic Efficacy against Staphylococcus aureus Infections. Antibiotics 2023, 12, 1066. https://doi.org/10.3390/antibiotics12061066
Le H, Dé E, Le Cerf D, Karakasyan C. Using Targeted Nano-Antibiotics to Improve Antibiotic Efficacy against Staphylococcus aureus Infections. Antibiotics. 2023; 12(6):1066. https://doi.org/10.3390/antibiotics12061066
Chicago/Turabian StyleLe, Hung, Emmanuelle Dé, Didier Le Cerf, and Carole Karakasyan. 2023. "Using Targeted Nano-Antibiotics to Improve Antibiotic Efficacy against Staphylococcus aureus Infections" Antibiotics 12, no. 6: 1066. https://doi.org/10.3390/antibiotics12061066
APA StyleLe, H., Dé, E., Le Cerf, D., & Karakasyan, C. (2023). Using Targeted Nano-Antibiotics to Improve Antibiotic Efficacy against Staphylococcus aureus Infections. Antibiotics, 12(6), 1066. https://doi.org/10.3390/antibiotics12061066