1. Introduction
Antimicrobial resistance (AMR) is a worldwide public health crisis: there were approximately 5 million human deaths associated with AMR in 2019 [
1]. While there are new antibiotics currently being developed, most of these compounds belong to already existing antibiotic classes [
2]. Therefore, resistance against these compounds in development will be readily acquired as resistance genes for those antimicrobial classes already exist and are ubiquitous. As a result, new drug classes are much needed [
3].
One Health is a multidisciplinary approach that addresses conjointly the well-being of humans, livestock, and the environment as a whole [
4]. In that context, AMR is not limited to human clinical settings but extends to various other environments. Indeed, wherever antibiotics are used, resistance appears, including in microbes affecting domestic animals and livestock. Furthermore, resistant bacteria and their resistance genetic determinants easily transfer from one reservoir to another [
5]. Therefore, it is paramount to address the global AMR crisis by acting in all the different sectors where antibiotics are used.
In support of the One Health approach, governments worldwide have started to establish policies limiting antibiotic use in animal production [
6,
7]. Notably, these regulations aim to protect class I antibiotics, i.e., those that are critical for clinical medicine, such as fluoroquinolones and third-generation cephalosporins [
8]. Ceftiofur is an example of the latter, which was often used to treat bovine mastitis (i.e., infectious inflammatory disease of the udder) [
9].
Staphylococcus aureus is the most prevalent bacterial pathogen causing bovine mastitis [
10], and new alternatives are immediately needed.
Reported cure rates for
S. aureus intramammary infections are usually low and depend in part on the genetic background and the virulence factors of
S. aureus isolates and their ability to produce biofilm [
11,
12], a protective layer surrounding the bacterial population that limits antibiotic and host defenses to reach and act on their targets [
13]. An important consideration with this pathogen is its capacity to infect both human and livestock, leading to an increased risk of emergence of antibiotic-resistant clones like methicillin-resistant
S. aureus (MRSA) [
14,
15]. MRSA are frequent in the hospital setting (HA-MRSA) and increasingly so in the community (CA-MRSA), as well as among livestock and animal food production (LA-MRSA). MRSA were classified as “high-priority” on the 2017 WHO’s list of bacteria for which new antibiotics are urgently needed [
16].
Identification of bacterial genes expressed during infections can allow the discovery of promising new antibiotic targets. Previously, our group reported that the
S. aureus gene
guaA was overexpressed during bovine mastitis [
17]. It encodes a guanosine monophosphate (GMP) synthetase and is part of an operon (
xpt-pbuX-guaB-guaA) controlled by a guanine riboswitch. Riboswitches are structures of noncoding RNA situated in the 5′ untranslated regions of an mRNA molecule [
18,
19]. In the absence of the guanine ligand, the riboswitch adopts a conformation enabling the transcription of the downstream operon, including
guaA. When guanine is in a sufficient concentration in the bacteria, it binds to the riboswitch and switches it “off”, stopping the transcription of
guaA and GMP synthesis. Using rational design, PC1 (Pyrimidine Compound 1; 2,5,6-triaminopyrimidine- 4-one) was found to be structurally similar to guanine and able to block
guaA expression and subsequent GMP synthesis [
18]. PC1 was characterized in vitro and was also shown to significantly reduce
S. aureus infection in experimental models of intramammary infection in mice and cows [
18,
20]. PC1 has been demonstrated to be specific toward
S. aureus and some other staphylococcal species (
e.g.,
S. epidermidis and
S. hominis). This riboswitch is also found in different species, like
Bacillus sp., but only regulates
guaA in staphylococci. Unfortunately, PC1 was found to be chemically unstable and to self-dimerize in the presence of water and oxygen, requiring the addition of an antioxidant to remain active.
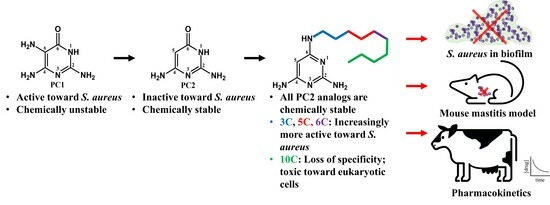
In our efforts to engineer chemically stable analogs of PC1, we have discovered PC2, a closely related analogue of PC1 that differs by the absence of the amine at position 5 on the pyrimidine cycle, which is responsible for dimerization (
Figure 1). PC2 is more chemically stable and has been shown to bind the riboswitch ex vivo, but possesses much lower antimicrobial activity against
S. aureus [
18]. Using rational design, we synthetized structural analogs of PC2 that are chemically stable and that regained activity against
S. aureus. In this study, we characterized the best candidate to date, PC206, and three other analogs (PC371, PC372 and PC383), which differ by the length of their alkyl chain bound to the amino group of position 6 on the pyrimidine cycle. The antimicrobial activity of PC206 was evaluated in vitro against
S. aureus strains from different sources (bovine and human) and the in vitro cytotoxicity was characterized. Furthermore, the efficiency of PC206 was tested in a
S. aureus mastitis mouse model and initial pharmacokinetics were measured in the udder of dairy cows. Although the mechanism of action of our new pyrimidine analogs remains to be fully elucidated, it represents the first step for the introduction of a new class of antibiotics to treat
S. aureus bovine mastitis.
3. Discussion
Treatments for
S. aureus bovine mastitis are limited or not efficient [
12,
26], and the use of third- and fourth-generation cephalosporins in dairy farms is a concern from a One Health perspective [
12,
26]. New specific treatments are clearly needed. We have previously reported PC1, a pyrimidine compound binding the guanine riboswitch of
S. aureus and inhibiting the expression of the essential gene
guaA [
18]. However, PC1 tends to self-dimerize in the presence of water and oxygen and therefore requires the addition of an antioxidant adjuvant, i.e., dithiothreitol, to maintain its antibiotic activity. By removing the amino group at position 5 on the pyrimidine cycle of PC1, we obtained PC2, which is chemically stable and able to bind the riboswitch ex vivo but loses most of its activity against
S. aureus [
18]. We hypothesized that the lack of antibacterial activity of PC2 was due to its inability to penetrate the bacterial membrane passively due to its high hydrophilicity, whereas analogs with a hydrophobic aliphatic chain could. Therefore, we set out to develop structural analogs of PC2 that would remain chemically stable and regain activity against
S. aureus.
In this article, we have characterized four analogs of PC2 with aliphatic chain of various lengths on the amino group at position 6 on the pyrimidine cycle. PC372 (5-carbon chain) and PC206 (6-carbon chain) displayed specific antibacterial activity against different staphylococci species. Accordingly, PC371, with its shorter 3-carbon chain, did not show any activity. The alkyl chain of this compound may not possess sufficient lipophilicity to significantly decrease the polarity necessary to enable its passive diffusion through bacterial membranes [
21]. Conversely, PC383 (10-carbon chain) was unspecific and targeted staphylococci and non-targeted species (i.e.,
E. coli and
E. faecalis) alike. PC383 was also shown to be cytotoxic in vitro for HepG2 and MAC-T cells, possibly due to its longer carbon chain causing non-specific cytotoxicity and cell damage through its interactions with cell membranes (i.e., very low CC
50 value in the LDH assay against HepG2 cells;
Table 3). Overall, there seems to be a balance point for the ideal length of the alkyl chain. If it is too short, the drug will not be able to cross the bacterial membrane; if it is too long, it will lose its specificity and cause damage to all cells.
PC206 was selected as the most promising of the four analogs because of its selective antimicrobial activity against staphylococci. It was also shown to remain active in biofilm, which is a challenge in antibiotic development [
13]. PC206 has low and equivalent MIC
50 and MIC
90 against a panel of
S. aureus bovine strains and MRSA strains. On the other hand, coagulase-negative staphylococci (CNS) were not equally susceptible to the drug;
S. hominis and
S. epidermidis displayed similar MIC
50 and MIC
90 values to
S. aureus, while
S. chromogenes,
S. haemolyticus, and
S. simulans were not as susceptible. A possible explanation for the reduced activity of PC206 against some CNS could be differences in their membrane composition, such that PC206 polarity would be adequate for certain species but not for others [
27]. Also, while PC206 is derived from PC1, we have not investigated his mode of action as thoroughly as for PC1. Further studies are needed to fully understand the antibiotic mode of action of these novel pyrimidine compounds. Interestingly, the lack of suitable antibiotic activity of PC206 against some CNS might be beneficial as it was previously demonstrated that
S. chromogenes, a CNS, has a protective effect against
S. aureus in a mouse co-infection model [
28].
The mode of action of our novel pyrimidine compounds was nonetheless explored to ascertain if there were indeed similarities with the antibiotic class representatives PC1 and PC2. Hence, we sought to verify if PC206 could bind to the guanine riboswitch, like its structural precursors, using a guanine riboswitch-
lacZ fusion construct integrated in
B. subtilis [
18]. Both
S. aureus and
B. subtilis belong to the order of the
Bacillales and have the same essential nucleotides for the binding of guanine to their respective riboswitch [
18]. However, the riboswitch in
B. subtilis is upstream of
xpt and
pbuX, while in
S. aureus, it also precedes essential genes
guaA and
guaB, thus tentatively explaining its differential antibiotic action. Therefore, we considered our model to accurately assess the binding of molecules to the riboswitch when β-galactosidase activity is reduced in the presence of a riboswitch ligand. In our model, PC206 and, to a lesser degree PC372, the only two compounds that showed specific antibiotic activities toward staphylococci, significantly decreased in Miller units in comparison to the untreated control (
Figure 4). PC371 did not show any detectable binding affinity as it probably could not penetrate through the
B. subtilis cytoplasmic membrane. PC383, which has unspecific antibiotic activity and displayed the highest toxicity against eukaryotic cell lines, did not impact the LacZ signal even at concentrations that were toxic for the reporter strain. Here, the binding of these novel pyrimidine compounds to the riboswitch was specifically studied because PC1 has previously been shown to bind to the guanine riboswitch and inhibit the expression of the genes under its regulation [
18]. However, the validity of the riboswitch as an antibiotic target has since been questioned in some experimental conditions [
29]. It is thus possible that there is another or an additional antibiotic target other than the guanine riboswitch, despite the correlation between MIC and the reduction in
lacZ expression in the β-galactosidase assay. Nevertheless, PC206 is a very narrow-spectrum antibiotic of a novel structural class, which is only effective against some staphylococci.
The innocuity of our novel pyrimidine compounds was verified for two eukaryotic cell lines, HepG2 and MAC-T, with two different assays, LDH and MTT. PC206 displayed little to no cytotoxicity, depending on the cell line and the assay used. Its selectivity index (S.I.), i.e., cytotoxicity compared to its antibiotic activity, is comparable to PC372, both of which are the most selective PC2 analogs. The S.I. allows us to compare antibiotics among themselves, but it does not warrant good tolerability in animals. PC206 cytotoxicity (for MAC-T cells, CC
50 of 317 and >600 μM [66 and >125 μg/mL], respectively for MTT and LDH assays;
Table 3) is comparable to antibiotics targeting
S. aureus in bovine mastitis. For example, cytotoxicity CC
50 values reported for enrofloxacin and ikarugamycin in MAC-T cells were 345 and 9.2 μg/mL, respectively (871 and 19 μM, respectively) [
30,
31]. The evaluation of cytotoxicity on cell monolayers does not always correlate well with in vivo tolerability due to many factors, including the cell line, the antibiotic contact time, and the assay conditions [
32,
33]. Therefore, the innocuity of PC206 was further verified in dairy cows. When injected into the mammary gland quarters, PC206 did not impact somatic cells count, body temperature, milk production, and appearance, nor did it cause any visible symptoms during the 69 h observation period.
The in vivo efficacy of PC206 was demonstrated using a well-established murine infectious mastitis model [
25]. The administration of PC206 reduced viable counts of
S. aureus Newbould by 1.24 log
10 CFU/g of gland, which was statistically significant. Mouse hepatic and plasmatic stability of PC206 was also assessed and was relatively stable in both environments over the course of the experiment (
Supplementary Information Figure S3). We then measured the recovery of PC206 in quarter milk of cows after a single intramammary infusion. The concentration of PC206 in the milk exceeded the MIC for
S. aureus for nearly 8 h when used at the highest dose (500 mg). In a future study, PC206 in vivo activity, pharmacokinetics, and pharmacodynamic properties could be further explored, to optimize dosing regimens, thereby improving treatment outcomes. For example, an appropriate formulation could be tested to fit dairy farm practices that often require milking every ~12 h. It should also be noted that intramammary infusion is the usual administration procedure for anti-mastitis therapies and that ceftiofur can be used at a dose of 125 mg/quarter every 24 h for up to 8 days or cephapirin at a dose of 250 mg every 12 h. Hence, our infusion procedure and dosing proposition for PC206 is already similar to dairy farm standards.
In conclusion, although its mode of action remains to be completely elucidated, this study proposes PC206 as the first stable candidate of a novel antibiotic class dedicated to the treatment of S. aureus-induced mastitis in dairy cows. Its potency and specificity were confirmed with MIC assays on large panels of S. aureus isolates from human and animal origins and against preformed biofilms. Furthermore, its impact on eukaryotic cells was thoroughly examined and deemed suitable. Finally, PC206 safety, effectiveness, and pharmacokinetics were evaluated in vivo, including in a mouse mastitis model and in lactating cows. With one-step synthesis using inexpensive, commercially available material, we expect a low-cost production, which is critical for use in food-animal production. This should help reduce broad-spectrum antibiotic overuse in farm animals, allowing clinically important antimicrobials for human health care to be preserved.
4. Materials and Methods
4.1. Synthesis of Novel Pyrimidine Analogs of PC1
This study specifically focused on novel pyrimidine analogs that have an amino group with an alkyl chain of varying lengths at position 6 (
Figure 1a). Alkylated PC2 analogs were synthesized using the same protocol with a nucleophilic aromatic substitution at position 6 from commercially available 6-chloropyrimidine-2,4-diamine with the appropriate alkylamine (
Figure 1b). The products were then purified by Prep-HPLC- MS. Syntheses are described below, and characterization data are provided in the Supplementary Information (
Supplementary Figures S5–S16).
In a 2 mL glass vial, alkylamine (0.5 mL) was added dropwise to solid 6-chloropyrimidine-2,4-diamine (50 mg, 0.34 mmol) before adding a 7 mm × 2 mm magnetic stir bar. The vial was sealed, and the reaction mixture was stirred at 110 °C for 24 h. The crude mixture was diluted with methanol to a total volume of 2 mL, filtered, and directly injected for purification. Purification was carried out on a Waters preparative HPLC-MS (column XSelectTM CSHTM Prep C18 (19 × 100 mm) packed with 5 μm particles, UV detector 2998, MS SQ Detector 2, Sample manager 2767, and binary gradient module). The eluents used were acetonitrile and water, both containing 0.1% formic acid.
A first run was conducted with the above eluents and the following method: 5% ACN to 95% ACN (0 → 10 min) in order to assess the optimal %ACN for product elution. A second run was made using the following eluent: [(%ACN) − 10 → (%ACN) + 5] (0 → 15 min). Fractions collected were analyzed on a Waters UPLC-MS system (column Acquity UPLC CSHTM C18 (2.1 mm × 50 mm) packed with 1.7 μm particles) using ACN and water + 0.1% formic acid. The method used was 0 → 0.2 min: 5% ACN; 0.2 → 1.5 min: 5% → 95% ACN; 1.5 → 1.8 min: 95% ACN; 1.8 → 2.0 min: 95% → 5% ACN; 2.0 → 2.5 min: 5% ACN. Pure fractions were pooled and lyophilized. All compounds possessed ≥95% purity as determined by UPLC-MS. HRMS spectra of the final compounds were recorded using a maXis ESI-Q-TOF with ESI in positive mode.
4.2. Characterization of Novel Pyrimidine Analogs of PC1
4.2.1. PC206
Yield: 32% (23 mg), off-white fluffy powder; UPLC-MS: retention time of 1.28 min, purity 98.8%, mass found (m/z) [M + 1]+: 210.1; 1H NMR (400 MHz, DMSO-d6) δ ppm 0.84 (t, J = 6.50 Hz, 3 H) 1.17–1.33 (m, 6 H) 1.45 (quin, J = 7.00 Hz, 2 H) 3.12 (q, J = 5.50 Hz, 2 H) 4.98 (s, 1 H) 6.77 (br. s., 2 H) 6.94 (br. s., 2 H) 7.31 (br. s., 1 H); 13C NMR (400 MHz, DMSO-d6) δ ppm 167.24, 161.78, 156.48, 72.45, 40.58, 31.06, 28.84, 26.15, 22.14, 13.95; HRMS (m/z): [M + H]+ calcd. for C10H20N5, 210.1713; found, 210.1717.
4.2.2. PC371
Yield: 15% (9 mg), off-white fluffy powder; UPLC-MS: retention time of 1.05 min, purity 98.1%, mass found (m/z) [M + 1]+: 167.9; 1H NMR (400 MHz, DMSO-d6) δ ppm 0.86 (t, J = 7.43 Hz, 3 H) 1.47 (sxt, J = 7.28 Hz, 2 H) 3.07 (q, J = 6.43 Hz, 2 H) 4.90 (s, 1 H) 6.18 (br. s., 4 H) 6.72 (br. s., 1 H); 13C NMR (400 MHz, DMSO-d6) δ ppm 165.46, 162.88, 159.20, 73.07, 42.30, 22.28, 11.48; HRMS (m/z): [M + H]+ calcd. for C7H14N5, 168.1244; found, 168.1246.
4.2.3. PC372
Yield: 28% (19 mg), off-white fluffy powder; UPLC-MS: retention time of 1.21 min, purity 97.6%, mass found (m/z) [M + 1]+: 196.0; 1H NMR (400 MHz, DMSO-d6) δ ppm 0.87 (t, J = 7.00 Hz, 3 H) 1.21–1.33 (m, 4 H) 1.46 (quin, J = 7.09 Hz, 2 H) 3.12 (q, J = 6.00 Hz, 2 H) 4.95 (s, 1 H) 6.57 (br. s., 2 H) 6.66 (br. s., 2 H) 7.11 (br. s., 1 H); 13C NMR (400 MHz, DMSO-d6) δ ppm 165.28, 161.09, 155.91, 72.39, 40.53, 28.61, 28.52, 21.90, 13.96; HRMS (m/z): [M + H]+ calcd. for C9H18N5, 196.1557; found, 196.1558.
4.2.4. PC383
Yield: 31% (28 mg), off-white fluffy powder; UPLC-MS: retention time of 1.53 min, purity 97.2%, mass found (m/z) [M + 1]+: 266.3; 1H NMR (400 MHz, DMSO-d6) δ ppm 0.85 (t, J = 7.00 Hz, 3 H) 1.18–1.32 (m, 14 H) 1.46 (quin, J = 6.50 Hz, 2 H) 3.14 (q, J = 5.00 Hz, 2 H) 4.99 (s, 1 H) 6.82 (br. s., 2 H) 6.98 (br. s., 2 H) 7.38 (br. s., 1 H); 13C NMR (400 MHz, DMSO-d6) δ ppm 165.26, 160.95, 155.51, 72.40, 40.57, 31.32, 29.06, 28.98, 28.79, 28.73, 28.44, 26.42, 22.12, 13.99; HRMS (m/z): [M + H]+ calcd. for C14H28N5, 266.2339; found, 266.2344.
4.3. Bacterial Strains
Reference strains for this study (
S. aureus ATCC 29213, ATCC 29740 [bovine mastitis strain Newbould],
S. epidermidis ATCC 12228,
E. coli ATCC 35695,
E. faecalis ATCC 29212) were obtained from the American Type Culture Collection (ATCC, Rockville, MD).
S. aureus strains CMRSA-10 and CMRSA-2 were obtained from the
Laboratoire de santé publique du Québec (LSPQ, Sainte-Anne-de-Bellevue, QC, Canada) and are Canadian CA-MRSA USA-300 and HA-MRSA USA-100 clones, respectively. The strain
E. coli imp4213 (
lptD), used as a control in MIC assays, is a derivative of
E. coli ATCC 35695 (
E. coli MC4100) with an increased membrane permeability [
34]. This strain possesses a mutation in the
lptD gene (LPS transport), resulting in an outer-membrane permeability defect and thus hyper-susceptibility to a variety of antibiotics, including vancomycin, which is normally inactive against Gram-negative bacteria due to the permeability barrier [
35]. Other MRSA strains used in this study are clinical isolates of human origin obtained from the microbiology laboratory of the
Centre Hospitalier Universitaire de Sherbrooke (CHUS, Sherbrooke, QC, Canada). Bovine mastitis strains (
S. aureus,
S. chromogenes,
S. haemolyticus,
S. hominis,
S. simulans) were obtained from the Canadian Bovine Mastitis and Milk Quality Research Network (CBRQMN, St-Hyacinthe, QC, Canada) [
10]. Among these strains,
S. aureus 2117 is a high producer of biofilm [
10] and was used in the biofilm-eradication assay.
S. epidermidis isolates were provided by
Centre de Recherche en Infectiologie (CRI) de Québec (Québec, QC, Canada).
Bacillus subtilis xpt-lacZ had a transcriptional fusion construct integrated in its genome by recombination as previously described and was used in a reporter gene assay [
18].
4.4. Antibiotic Susceptibility Testing
Susceptibility tests were performed by a broth microdilution assay in 96-well plates according to the Clinical and Laboratory Standards Institute (CLSI) guidelines [
36]. Briefly, each compound was serially diluted 2-fold and bacteria were inoculated in approximately 5 × 10
5 colony-forming units per mL (CFU/mL). Testing of oxacillin against MSSA and MRSA strains and coagulase-negative staphylococci was performed in cation-adjusted Mueller–Hinton (CAMHB) broth, supplemented with 2% NaCl. The supplemental NaCl was excluded for the testing of the other drugs. Every tested strain was incubated for 20 h at 35 °C, except when exposed to vancomycin and oxacillin; strains were then incubated for 24 h at 35 °C. The minimal inhibitory concentration (MIC) of each drug is defined as the lowest concentration of the antibiotic that prevents visible growth of a bacterial strain. It was determined by measuring the absorbance at 600 nm with an Epoch microplate reader (Agilent Technologies, Mississauga, ON, Canada), as well as by visual confirmation. Vancomycin (Sigma Aldrich, Oakville, ON, Canada) was used as a control antibiotic for Gram-positive bacteria, and oxacillin (Sigma Aldrich) was also used as a control for
S. aureus, MRSA, and the
B. subtilis xpt-lacZ reporter strain. Ceftazidime (Sigma Aldrich) was used as a control for
E. coli strains. Pirlimycin (Pfizer, Kirkland, QC, Canada) was included as the mastitis antibiotic control.
The antibacterial activity of the most promising compound, PC206, was also evaluated against larger groups of clinical isolates of human and animal origin. The MIC50 and MIC90 values represented the concentrations of drug required to inhibit the growth of 50% and 90% of the bacterial strains tested, respectively.
4.5. Kill Kinetics
Time-kill kinetics studies were performed to define the bactericidal effect of PC206. S. aureus Newbould, CMRSA-10, or S. epidermidis ATCC 12228 were inoculated at a density of approximately 5 × 105 CFU/mL in CAMHB in the absence or presence of PC206 and were incubated with agitation (225 rpm) for 24 h at 35 °C. Right before inoculation and 0.5, 1, 2, 4, 8 and 24 h thereafter, bacterial cultures were sampled, serially diluted, and plated on tryptic soya agar (TSA) plates that were incubated at 35 °C for 24 h to determine viable counts with a detection limit of 2 log10 CFU/mL.
4.6. Biofilm-Eradication Test
The viability of bacteria in preformed biofilms treated with PC206 was evaluated using the MBEC assay™ system (formerly the Calgary Biofilm Device) [
22,
37,
38]. The device consists of a plastic lid with 96 pegs and a 96-well plate. The wells were first inoculated with a suspension of
S. aureus bovine strain 2117 adjusted to a 0.5 McFarland standard in brain heart infusion (BHI) media supplemented with 0.25% glucose, and then covered with the peg lids. Plates were incubated at 35 °C for 24 h with agitation (120 rpm). The biofilm formed on the pegs was then washed three times with PBS and was further incubated in a 96-well plate containing BHI, 0.25% glucose, and antibiotics (two-fold dilutions) at 35 °C for 24 h with agitation (120 rpm). The pegs were washed three times with PBS and the biofilm was sonicated for 10 min, and then transferred into a new 96-well base plate. The 96-well plate was centrifuged for 5 min at 180 g to recover the detached bacteria. Bacteria were resuspended, serially diluted, and plated on TSA plates that were incubated at 35 °C for 24 h to determine the remaining viable counts with a detection limit of 10 CFU/mL. Gentamicin and vancomycin were used as positive and negative controls, respectively.
4.7. Riboswitch Regulation Assay
β-galactosidase gene expression in the presence of the new pyrimidine analogs was studied, using an
xpt-lacZ transcriptional fusion construct integrated in the genome of
B. subtilis as previously described [
18]. Briefly, cultures were grown for 5 h at 35 °C in CAMHB, from an initial optical density of 0.05 at 600 nm (OD
600), and then diluted 1:20 and in the presence of different concentrations of the compounds tested. Cultures were centrifuged for 10 min at 13,100 rpm, the supernatant was discarded, and pellets were suspended in a Z-buffer (NaH
2PO
4 40 mM, Na
2HPO
4 60 mM, MgSO
4 1 mM, KCl 10 mM). Cultures OD
600 were measured, and bacteria were then lysed with lysozyme (1 mg/mL) and β-mercaptoethanol 50 mM for 30 min. Lysates were centrifuged for 10 min at 13,100 rpm, and supernatants were incubated for 5 min at 30 °C with the substrate orthonitrophenyl-β-galactoside (ONPG; Sigma Aldrich). Reactions were stopped with Na
2CO
3 1M, supernatants OD
420 were measured, and Miller units were calculated [
39]. A decrease in β-galactosidase activity indicated binding of the test compound to the riboswitch. Guanine and oxacillin were used as negative and positive controls, respectively. The formula to measure Miller units is as follows:
Here, t represents the time of incubation with ONPG, and v represents the volume of the reaction.
4.8. Evaluation of Cell Viability Using the MTT Assay
A bovine mammary epithelial cell line, MAC-T, and a human hepatocellular cell line, HepG2, were used to evaluate the effect of the various pyrimidine analogs on cell viability. Both cell lines were cultured in Dulbecco’s modified Eagle medium (DMEM) with 4.5 g glucose per liter and supplemented with 10% heat-inactivated fetal bovine serum (FBS), 1% sodium pyruvate, and 1% antimycotic solution. Cell cultures were incubated at 37 °C with 5% CO2.
Twenty-four hours prior to the MTT assay, cells were seeded in a 96-well plate at a concentration of 1 × 10
5 and 2 × 10
5 cells/mL for MAC-T and HepG2, respectively. The next day, in a separate plate, pyrimidine compounds and 4-chloro-7-nitrobenzofurazan (NBD-Cl) control were serially diluted in the cell culture medium (DMEM containing 1% FSB, 1% sodium pyruvate and 1% antimycotic solution). DMSO was adjusted to 0.1%. The MTT assay was performed in triplicate according to the kit recommendations (Roche, Cell proliferation kit I (MTT)). Briefly, the cells were exposed to various concentrations of pyrimidine compounds. High-viability controls consisted of cells exposed to the cell culture medium with 0.1% DMSO. After 24 h of incubation, 10 µL of the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) solutions was added directly on the cells and incubated for four more hours. The solubilization buffer was then added and incubated overnight. Absorbances were measured at 550 nm (OD
550) to detect the formation of formazan and at 690 nm (OD
690) for reference using an Epoch microplate reader. The OD
550 was corrected (cOD
550) by subtracting the OD
690. Viability was measured using the following formula:
For each compound, the cytotoxic concentration 50% (CC50), i.e., the concentration leading to 50% cell death, was determined using a non-linear regression fit (variable slope, four parameters) of the plot of the Log10 inhibitor concentration versus the percent cell viability with the software GraphPad prism 9.4.1.
4.9. Evaluation of the Cell Viability by the LDH Release Assay
The MAC-T and HepG2 cell lines were routinely cultured as explained in the MTT assay section above.
The lactate dehydrogenase release (LDH) assay was used to verify the impact of the new pyrimidine analogs on cell membrane integrity. Damaged cell membranes will release LDH in the medium, and the enzyme activity can be quantified. Here, cells were seeded in a 96-well plate at concentrations of 5 × 10
4 and 1 × 10
5 cells/mL for MAC-T and HepG2, respectively. Antibiotics and controls were serially diluted in a second plate in the same medium as for the MTT assay. DMSO was adjusted to 0.1%. Triton 1% was used as a complete lysis control and wells that did not contain any test compounds were used as high-viability controls. The LDH assay was conducted in triplicate and according to the kit recommendations (Roche, Cytotoxicity detection kit (LDH)). The cells were exposed to various concentrations of test compounds or controls. After 24 h of incubation, 100 µL of the cell supernatant was sampled and used for the quantification of the LDH activity. Cell supernatant was incubated with the revelation buffer for 20 to 30 min in the dark and absorbances were measured at 492 nm (OD
492) and for reference at 690 nm (OD
690) using an Epoch microplate reader. The OD
492 was corrected (cOD
492) by subtracting the OD
690. The formula to measure the LDH release is as follows:
Here, L represents the mean of the low LDH release (high viability) and H the high LDH release (complete cell lysis). CC50 were measured as explained in the MTT assay section above.
4.10. Murine Mastitis Model
A well-characterized mouse mastitis model was used to evaluate the in vivo efficacy of PC206 [
25]. Briefly, CD-1 lactating mice were separated from their pups (12–14 days following birth), anesthetized using isoflurane, and infected with
S. aureus Newbould. For inoculation, the fourth pair of glands found from head to tail (L4 and R4 glands) was first disinfected with 70% ethanol. Then, 100 µL of PBS containing 75–125 CFU was slowly injected into the lactiferous duct with a 32-gauge blunt needle attached to a 1 mL syringe. One and four hours post-inoculation, mice were anesthetized again, and 500 µg of compound PC206 (in PBS) was injected directly into the previously infected mammary glands (6 mammary glands:
n = 6). Some mice were injected with PBS instead and were used as an untreated control group (8 mammary glands:
n = 8). Each infected gland was considered to be the experimental unit. Then, 14 h after bacterial inoculation, mice were anesthetized and humanely euthanized, and mammary glands were harvested and homogenized. CFU counts were obtained after plating serial dilution of mammary gland homogenates on TSA plates that were incubated at 35 °C for 24 h. The detection limit was approximately 200 CFU per gram of mammary glands.
4.11. Metabolic and Plasmatic Stability
4.11.1. Metabolic Stability
All incubations were performed minimally in triplicate. A total of 1 mL of mice liver S9 fraction (XenoTech, Kansas City, KS, USA) was fortified with 50 μL of NADPH-regenerating solution A and 10 μL of solution B (Corning BD Biosciences catalog no. 451200) and preincubated at 37 °C for 5 min prior to the addition of PC206. Immediately after fortification of PC206 into the mice liver S9 fraction containing the NADPH-regeneration system, the sampling points were taken at 2, 5, 10, 15, 30, 45 and 60 min for metabolic stability experiments (i.e., the substrate concentration was 10 μM). Samples of 50 μL were taken and mixed with 250 μL of the internal standard solution (caffeine) in microcentrifuge tubes. The samples were centrifuged at 12,000× g for 10 min and 200 μL of the supernatant was transferred into an injection vial for HPLC-MS analysis. A Q Exactive Orbitrap Mass Spectrometer was interfaced with a Vanquish Flex UHPLC system (Thermo Fisher Scientific, Rochester, NY, USA). Chromatography was achieved using a gradient mobile phase, along with a microbore column, namely Thermo BioBasic Phenyl (ThermoFisher Scientific), of 50 × 1 mm with a particle size of 5 μm. The initial mobile phase condition consisted of acetonitrile and water (both fortified with 0.1% of formic acid) at a ratio of 5:95. From 0 to 1 min, the ratio was maintained at 5:95. From 1 to 3 min, it was set to a ratio of 80:20. The mobile phase composition ratio was reverted at the initial conditions, and the column was allowed to re-equilibrate for 5 min for a total run time of 8 min. The flow rate was fixed at 75 µL/min and 2 µL of samples was injected. MS detection was performed in positive-ion mode, operating in high-resolution accurate-mass (HRAM) scan mode. Nitrogen was used for sheath and auxiliary gases and was set at 10 and 5 arbitrary units. The ESI probe was set to 4000 V and the ion transfer tube temperature was set to 200 °C. The scan range was set to m/z 200–500. Data were acquired at a resolving power of 140,000 (FWHM) using an automatic gain control target of 1.0 × 106 and maximum ion injection time of 100 msec. Targeted drug quantification was performed by MS detection using specific precursor masses based on monoisotopic masses (i.e., [M + H]+ ions). Quantification was performed by extracting specific precursor ions using a 5 ppm mass window. Instrument calibration was performed prior to all analysis and mass accuracy was notably below 1 ppm using PierceTM LTQ Velos ESI positive-ion calibration solution (ThermoFisher Scientific) and automated instrument protocol.
4.11.2. Plasmatic Stability
Blood samples were collected from CD-1 female mice by jugular vein puncture and subsequently transferred to K3-EDTA-coated tubes (Sarstedt, St-Leonard, QC, Canada). Samples were gently inverted and centrifuged at 1500× g for 10 min at 4 °C, and plasma aliquots were stored at −80 °C until analysis. All incubations were performed in triplicate. A total of 1 mL of plasma was fortified with 10 µM of PC206 and incubated at room temperature (~22 °C). Samples were taken at 2, 5, 10, 15, 30, 45, and 60 min. Plasma samples of 50 μL were taken and mixed with 250 μL of the internal standard solution (caffeine) in microcentrifuge tubes. The samples were centrifuged at 12,000× g for 10 min, and 200 μL of the supernatant was transferred into an injection vial for HPLC-MS analysis. Drug detection was performed as described in the previous paragraph.
4.12. Initial Safety and Pharmacokinetics Assessments in Cows
The innocuity of PC206 was evaluated in cows, as previously described for other test substances [
20]. Following morning milking, each of the four mammary gland quarters four 4 cows was infused with various doses of PC206 (100, 250, 500 mg) or saline as a negative control. Milk samples were collected aseptically from each quarter prior to treatment, as well as 2, 4, 8, 21, 33, 45, and 69 h after intra-mammary infusion of PC206 or saline to assess the onset of inflammation by determining the somatic cell counts through Lactanet (Saint-Anne-de-Bellevue, QC, Canada). Cows were milked using an individual quarter-milking unit in order to measure individual quarter-milk production. PC206 concentration in milk was also measured; milk samples were diluted with an equal volume of isopropanol and washed 3 times with hexane. PC206 was extracted using ethyl acetate, filtered on Na
2SO
4, and evaporated. The dried samples were diluted 80:20 in ACN:H
2O containing an internal standard. The content of PC206 was determined using single-ion monitoring (SIMS) on UPLC-MS.




 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}