A RADAR-Based Assay to Isolate Covalent DNA Complexes in Bacteria

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
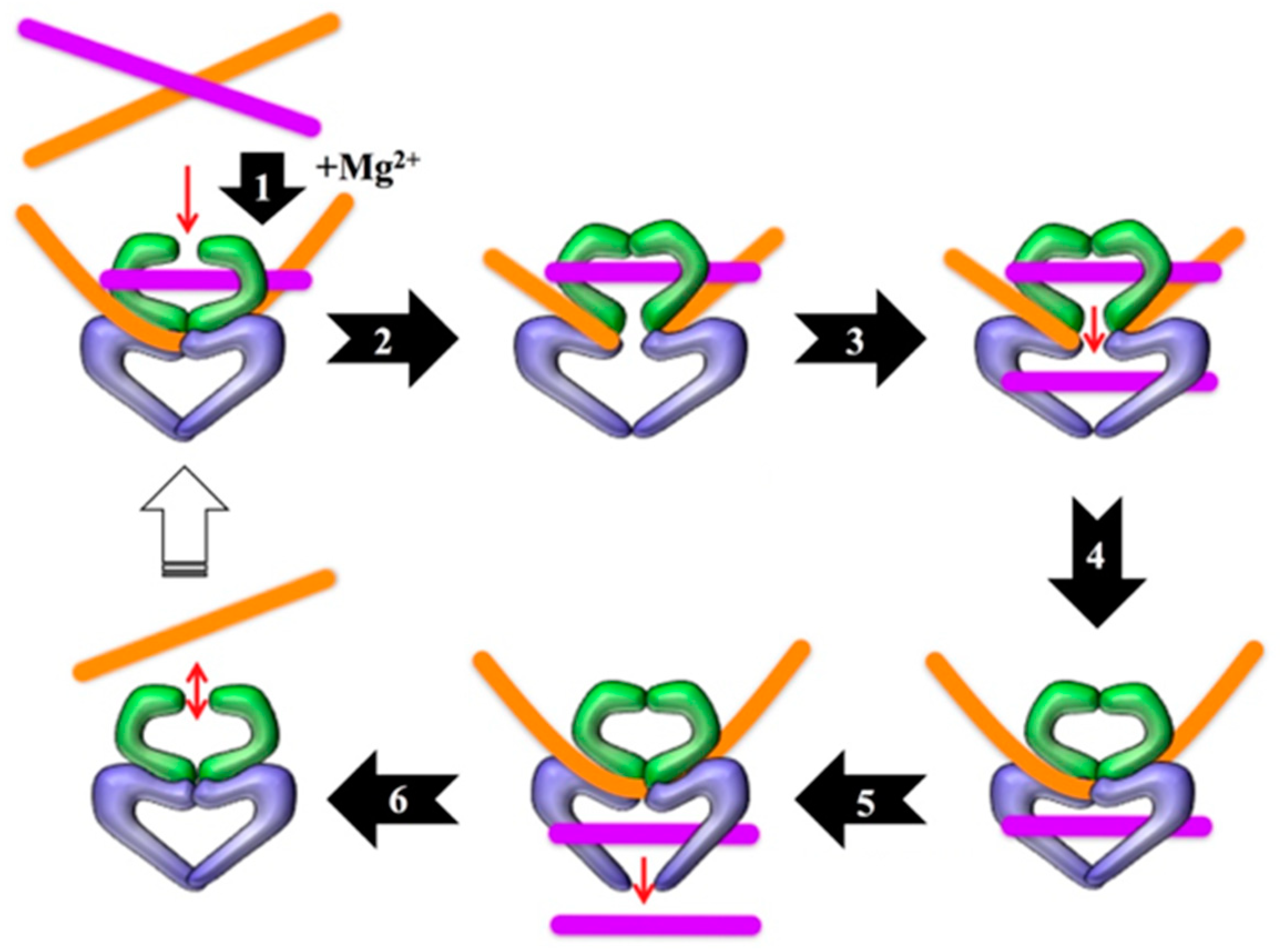
2.1. Technical Considerations
2.2. Early Development
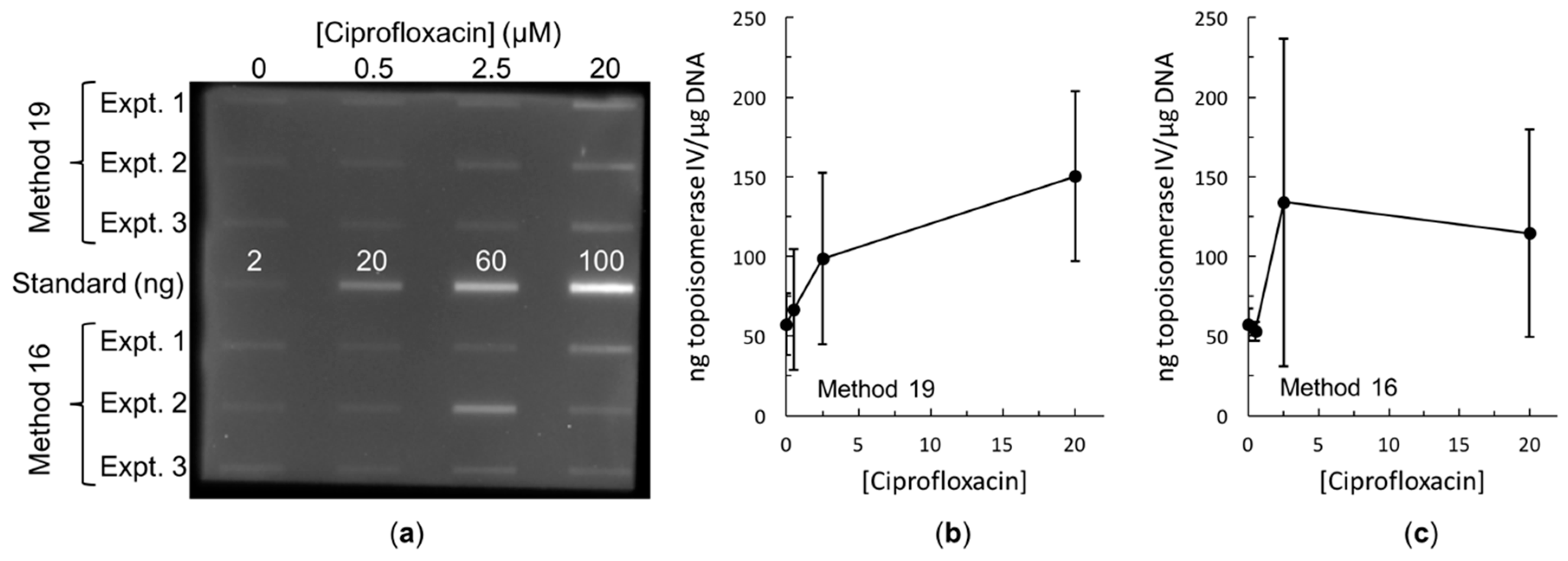
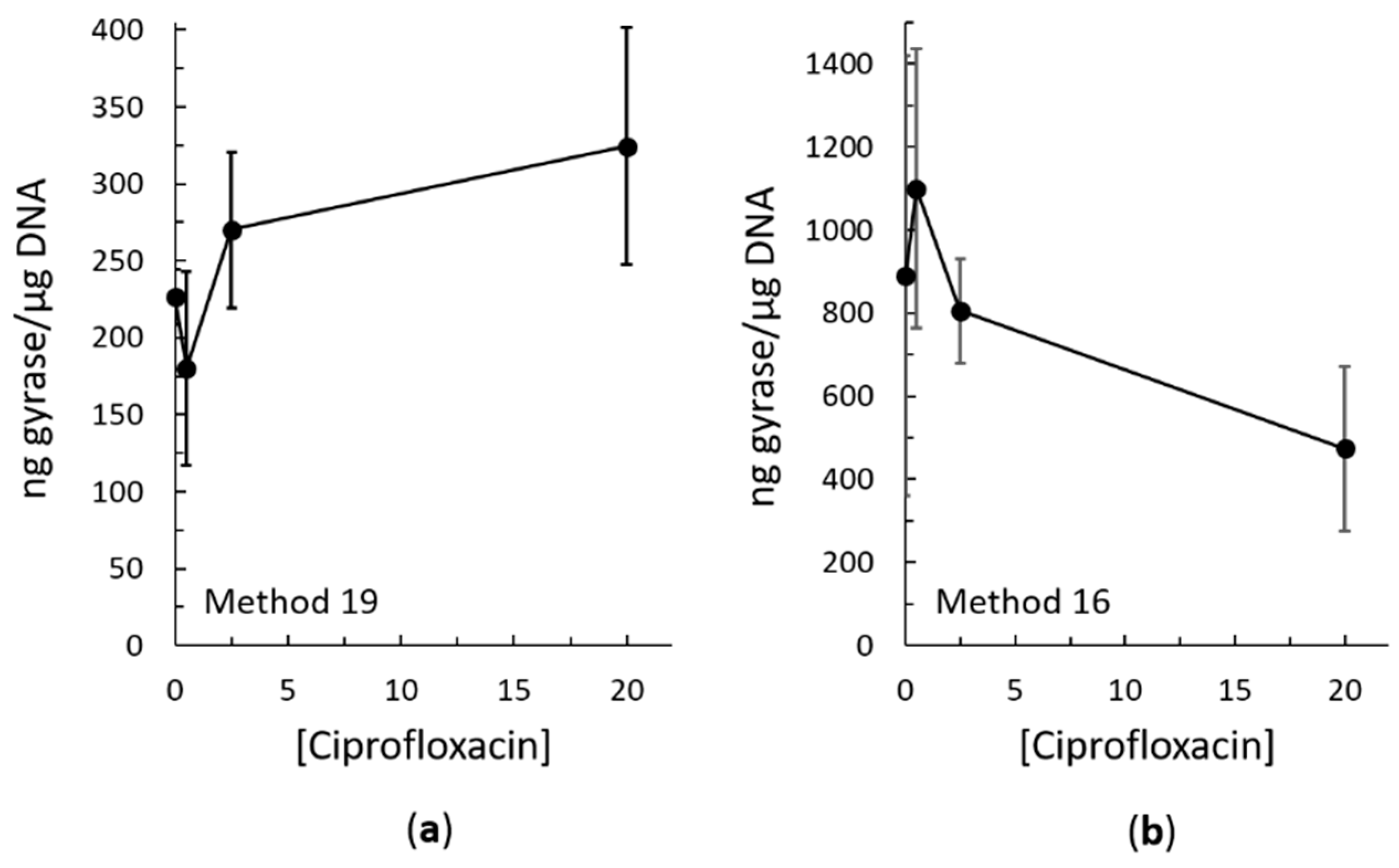
2.3. Bacterial RADAR Assay Method 16 versus 19
3. Discussion
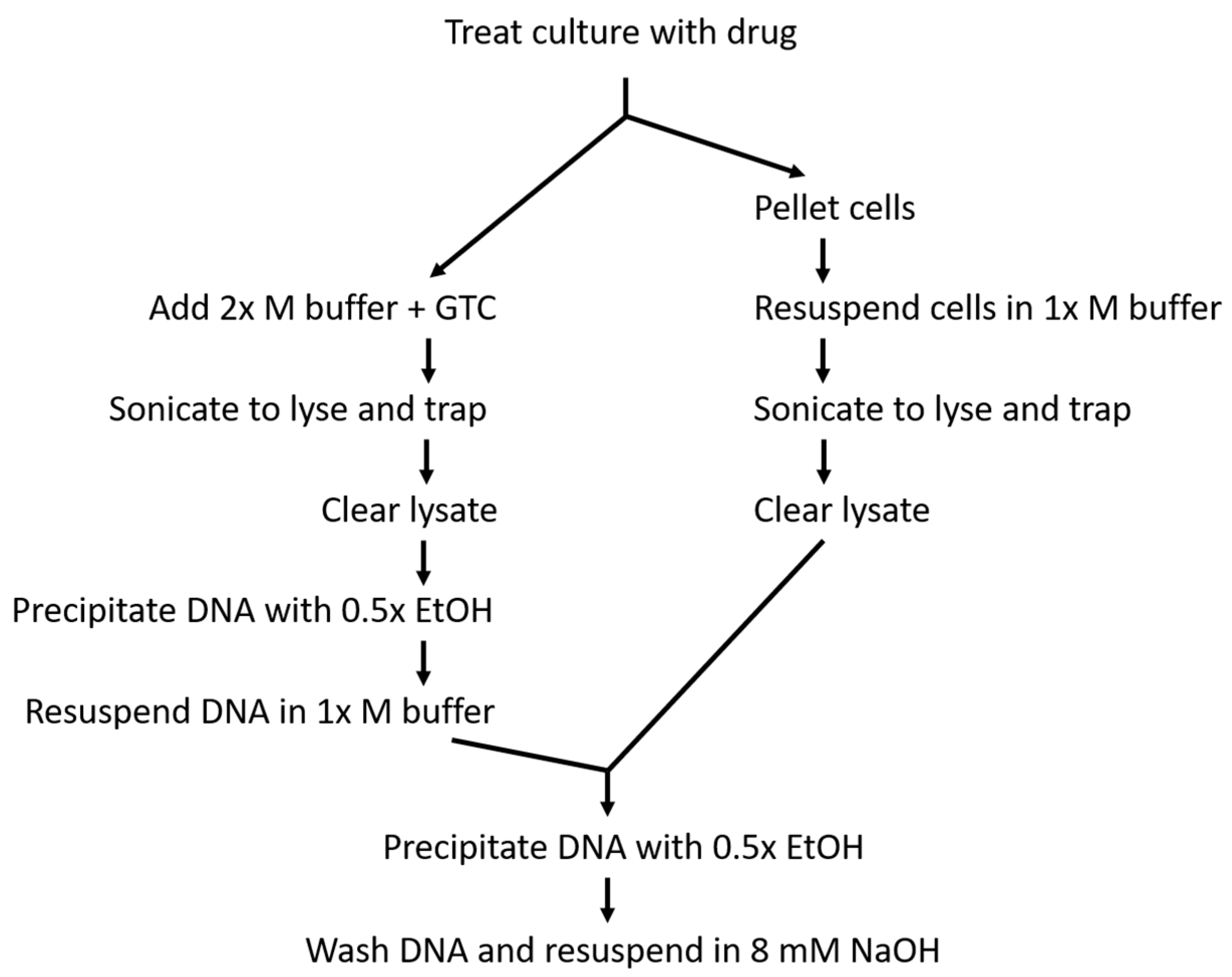
4. Materials and Methods
4.1. Strains, Antibodies, and Reagents
4.2. Culture Growth and Treatment
4.3. Cell Lysis and DNA Capture
4.4. DNA Quantification and Blotting
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Levine, C.; Hiasa, H.; Marians, K.J. DNA gyrase and topoisomerase IV: Biochemical activities, physiological roles during chromosome replication, and drug sensitivities. Biochim. Biophys. Acta 1998, 1400, 29–43. [Google Scholar] [CrossRef]
- Champoux, J.J. DNA topoisomerases: Structure, function, and mechanism. Annu. Rev. Biochem. 2001, 70, 369–413. [Google Scholar] [CrossRef] [PubMed]
- Deweese, J.E.; Osheroff, M.A.; Osheroff, N. DNA Topology and Topoisomerases: Teaching a “Knotty” Subject. Biochem. Mol. Biol. Educ. 2008, 37, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Nitiss, J.L. DNA topoisomerase II and its growing repertoire of biological functions. Nat. Rev. Cancer 2009, 9, 327–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pommier, Y.; Leo, E.; Zhang, H.; Marchand, C. DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem. Biol. 2010, 17, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Gentry, A.C.; Osheroff, N. DNA topoisomerases: Type II. In Encyclopedia of Biological Chemistry; Academic Press: Waltham, MA, USA, 2013; pp. 163–168. [Google Scholar]
- Wang, J.C. DNA topoisomerases. Annu. Rev. Biochem. 1996, 65, 635–692. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.C. Cellular roles of DNA topoisomerases: A molecular perspective. Nat. Rev. Mol. Cell Biol. 2002, 3, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Nitiss, J.L. Investigating the biological functions of DNA topoisomerases in eukaryotic cells. Biochim. Biophys. Acta 1998, 1400, 63–81. [Google Scholar] [CrossRef]
- Leppard, J.B.; Champoux, J.J. Human DNA topoisomerase I: Relaxation, roles, and damage control. Chromosoma 2005, 114, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y.; Pourquier, P.; Fan, Y.; Strumberg, D. Mechanism of action of eukaryotic DNA topoisomerase I and drugs targeted to the enzyme. Biochim. Biophys. Acta 1998, 1400, 83–105. [Google Scholar] [CrossRef]
- Stein, G.E. The 4-quinolone antibiotics: Past, present, and future. Pharmacotherapy 1988, 8, 301–314. [Google Scholar] [CrossRef] [PubMed]
- Anderson, V.E.; Osheroff, N. Type II topoisomerases as targets for quinolone antibacterials: Turning Dr. Jekyll into Mr. Hyde. Curr. Pharm. Des. 2001, 7, 337–353. [Google Scholar] [CrossRef] [PubMed]
- Emmerson, A.M.; Jones, A.M. The quinolones: Decades of development and use. J. Antimicrob. Chemother. 2003, 51 (Suppl. 1), 13–20. [Google Scholar] [CrossRef] [PubMed]
- Andersson, M.I.; MacGowan, A.P. Development of the quinolones. J. Antimicrob. Chemother. 2003, 51 (Suppl. 1), 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitscher, L.A. Bacterial topoisomerase inhibitors: Quinolone and pyridone antibacterial agents. Chem. Rev. 2005, 105, 559–592. [Google Scholar] [CrossRef] [PubMed]
- Andriole, V.T. The quinolones: Past, present, and future. Clin. Infect. Dis. 2005, 41 (Suppl. 2), S113–S119. [Google Scholar] [CrossRef] [PubMed]
- Forterre, P.; Gribaldo, S.; Gadelle, D.; Serre, M.C. Origin and evolution of DNA topoisomerases. Biochimie 2007, 89, 427–446. [Google Scholar] [CrossRef] [PubMed]
- Forterre, P.; Gadelle, D. Phylogenomics of DNA topoisomerases: Their origin and putative roles in the emergence of modern organisms. Nucleic Acids Res. 2009, 37, 679–692. [Google Scholar] [CrossRef] [PubMed]
- Aldred, K.J.; Kerns, R.J.; Osheroff, N. Mechanism of quinolone action and resistance. Biochemistry 2014, 53, 1565–1574. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y. DNA topoisomerase I inhibitors: Chemistry, biology, and interfacial inhibition. Chem. Rev. 2009, 109, 2894–2902. [Google Scholar] [CrossRef] [PubMed]
- Aldred, K.J. Mechanism of Quinolone Action and Resistance in Bacterial and Human Type II Topoisomerases. Ph.D. Thesis, Vanderbilt University, Nashville, TN, USA, 2014. [Google Scholar]
- Deweese, J.E.; Osheroff, N. The DNA cleavage reaction of topoisomerase II: Wolf in sheep’s clothing. Nucleic Acids Res. 2009, 37, 738–748. [Google Scholar] [CrossRef] [PubMed]
- Nitiss, J.L. Targeting DNA topoisomerase II in cancer chemotherapy. Nat. Rev. Cancer 2009, 9, 338–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hooper, D.C. Mode of action of fluoroquinolones. Drugs 1999, 58 (Suppl. 2), 6–10. [Google Scholar] [CrossRef] [PubMed]
- Hooper, D.C. Mechanisms of action of antimicrobials: Focus on fluoroquinolones. Clin. Infect. Dis. 2001, 32 (Suppl. 1), S9–S15. [Google Scholar] [CrossRef]
- Hooper, D.C. Bacterial topoisomerases, anti-topoisomerases, and anti-topoisomerase resistance. Clin. Infect. Dis. 1998, 27 (Suppl. 1), S54–S63. [Google Scholar] [CrossRef] [PubMed]
- Drlica, K.; Malik, M.; Kerns, R.J.; Zhao, X. Quinolone-mediated bacterial death. Antimicrob. Agents Chemother. 2008, 52, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Drlica, K.; Hiasa, H.; Kerns, R.; Malik, M.; Mustaev, A.; Zhao, X. Quinolones: Action and resistance updated. Curr. Top. Med. Chem. 2009, 9, 981–998. [Google Scholar] [CrossRef] [PubMed]
- Wohlkonig, A.; Chan, P.F.; Fosberry, A.P.; Homes, P.; Huang, J.; Kranz, M.; Leydon, V.R.; Miles, T.J.; Pearson, N.D.; Perera, R.L.; et al. Structural basis of quinolone inhibition of type IIA topoisomerases and target-mediated resistance. Nat. Struct. Mol. Biol. 2010, 17, 1152–1153. [Google Scholar] [CrossRef] [PubMed]
- Aldred, K.J.; McPherson, S.A.; Turnbough, C.L., Jr.; Kerns, R.J.; Osheroff, N. Topoisomerase IV-quinolone interactions are mediated through a water-metal ion bridge: Mechanistic basis of quinolone resistance. Nucleic Acids Res. 2013, 41, 4628–4639. [Google Scholar] [CrossRef] [PubMed]
- Jacoby, G.A. Mechanisms of resistance to quinolones. Clin. Infect. Dis. 2005, 41 (Suppl. 2), S120–S126. [Google Scholar] [CrossRef] [PubMed]
- Anderson, V.E.; Zaniewski, R.P.; Kaczmarek, F.S.; Gootz, T.D.; Osheroff, N. Quinolones inhibit DNA religation mediated by Staphylococcus aureus topoisomerase IV. Changes in drug mechanism across evolutionary boundaries. J. Biol. Chem. 1999, 274, 35927–35932. [Google Scholar] [CrossRef] [PubMed]
- Laponogov, I.; Sohi, M.K.; Veselkov, D.A.; Pan, X.S.; Sawhney, R.; Thompson, A.W.; McAuley, K.E.; Fisher, L.M.; Sanderson, M.R. Structural insight into the quinolone-DNA cleavage complex of type IIA topoisomerases. Nat. Struct. Mol. Biol. 2009, 16, 667–669. [Google Scholar] [CrossRef] [PubMed]
- Aedo, S.; Tse-Dinh, Y.C. Isolation and quantitation of topoisomerase complexes accumulated on Escherichia coli chromosomal DNA. Antimicrob. Agents Chemother. 2012, 56, 5458–5464. [Google Scholar] [CrossRef] [PubMed]
- Kiianitsa, K.; Maizels, N. A rapid and sensitive assay for DNA-protein covalent complexes in living cells. Nucleic Acids Res. 2013, 41, e104. [Google Scholar] [CrossRef] [PubMed]
- Aldred, K.J.; Schwanz, H.A.; Li, G.; Williamson, B.H.; McPherson, S.A.; Turnbough, C.L., Jr.; Kerns, R.J.; Osheroff, N. Activity of quinolone CP-115,955 against bacterial and human type II topoisomerases is mediated by different interactions. Biochemistry 2015, 54, 1278–1286. [Google Scholar] [CrossRef] [PubMed]
- Bandele, O.J.; Osheroff, N. The efficacy of topoisomerase II-targeted anticancer agents reflects the persistence of drug-induced cleavage complexes in cells. Biochemistry 2008, 47, 11900–11908. [Google Scholar] [CrossRef] [PubMed]
- Nitiss, J.L.; Soans, E.; Rogojina, A.; Seth, A.; Mishina, M. Topoisomerase assays. Curr. Protoc. Pharmacol. 2012, 57, 3. [Google Scholar]
- Aldred, K.J.; McPherson, S.A.; Wang, P.; Kerns, R.J.; Graves, D.E.; Turnbough, C.L., Jr.; Osheroff, N. Drug interactions with Bacillus anthracis topoisomerase IV: Biochemical basis for quinolone action and resistance. Biochemistry 2012, 51, 370–381. [Google Scholar] [CrossRef] [PubMed]
- Aldred, K.J.; Breland, E.J.; Vlckova, V.; Strub, M.P.; Neuman, K.C.; Kerns, R.J.; Osheroff, N. Role of the water-metal ion bridge in mediating interactions between quinolones and Escherichia coli topoisomerase IV. Biochemistry 2014, 53, 5558–5567. [Google Scholar] [CrossRef] [PubMed]
- Luan, G.; Drlica, K. Fluoroquinolone-Gyrase-DNA Cleaved Complexes. Methods Mol. Biol. 2018, 1703, 269–281. [Google Scholar] [PubMed]
- Aldred, K.J.; Blower, T.R.; Kerns, R.J.; Berger, J.M.; Osheroff, N. Fluoroquinolone interactions with Mycobacterium tuberculosis gyrase: Enhancing drug activity against wild-type and resistant gyrase. Proc. Natl. Acad. Sci. USA 2016, 113, E839–E846. [Google Scholar] [CrossRef] [PubMed]
- Dalhoff, A. Resistance surveillance studies: A multifaceted problem—The fluoroquinolone example. Infection 2012, 40, 239–262. [Google Scholar] [CrossRef] [PubMed]
- Wise, R.; Honeybourne, D. Pharmacokinetics and pharmacodynamics of fluoroquinolones in the respiratory tract. Eur. Respir. J. 1999, 14, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Lode, H.; Borner, K.; Koeppe, P. Pharmacodynamics of fluoroquinolones. Clin. Infect. Dis. 1998, 27, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.K.; Nightingale, C.H. Pharmacokinetics and pharmacodynamics of the fluoroquinolones. In The Quinolones, 3rd ed.; Andriole, V.T., Ed.; Academic Press: San Diego, CA, USA, 2000; pp. 169–202. [Google Scholar]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aldred, K.J.; Payne, A.; Voegerl, O. A RADAR-Based Assay to Isolate Covalent DNA Complexes in Bacteria. Antibiotics 2019, 8, 17. https://doi.org/10.3390/antibiotics8010017
Aldred KJ, Payne A, Voegerl O. A RADAR-Based Assay to Isolate Covalent DNA Complexes in Bacteria. Antibiotics. 2019; 8(1):17. https://doi.org/10.3390/antibiotics8010017
Chicago/Turabian StyleAldred, Katie J., Adeline Payne, and Olivia Voegerl. 2019. "A RADAR-Based Assay to Isolate Covalent DNA Complexes in Bacteria" Antibiotics 8, no. 1: 17. https://doi.org/10.3390/antibiotics8010017
APA StyleAldred, K. J., Payne, A., & Voegerl, O. (2019). A RADAR-Based Assay to Isolate Covalent DNA Complexes in Bacteria. Antibiotics, 8(1), 17. https://doi.org/10.3390/antibiotics8010017