Development of a High Throughput Screen for the Identification of Inhibitors of Peptidoglycan O-Acetyltransferases, New Potential Antibacterial Targets


Abstract
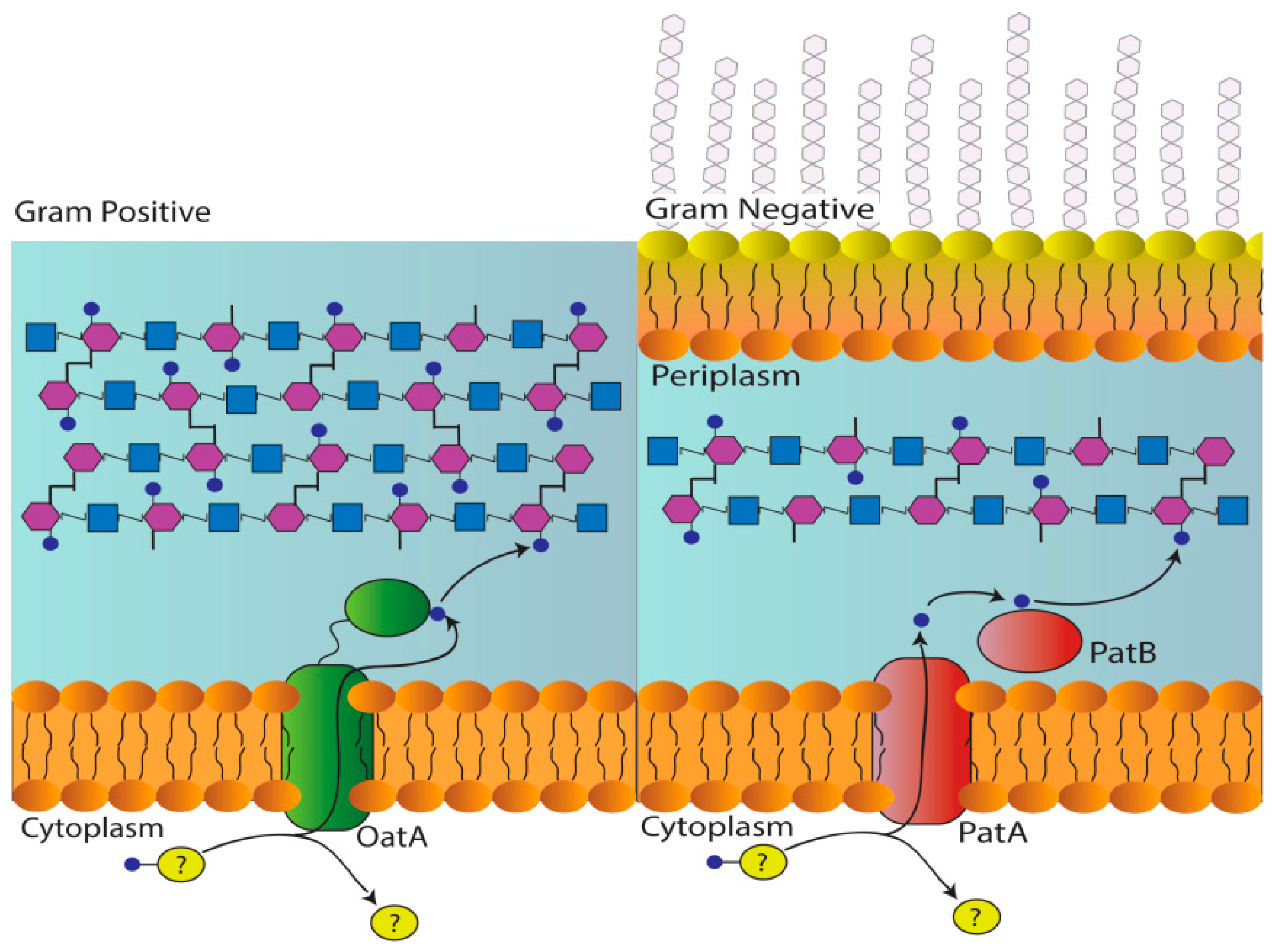
:1. Introduction
2. Results
2.1. Production, Purification and Stability of NgPatB and SaOatAC Constructs
2.2. Kinetic Parameters of NgPatBΔ100 and SaOatAΔ445
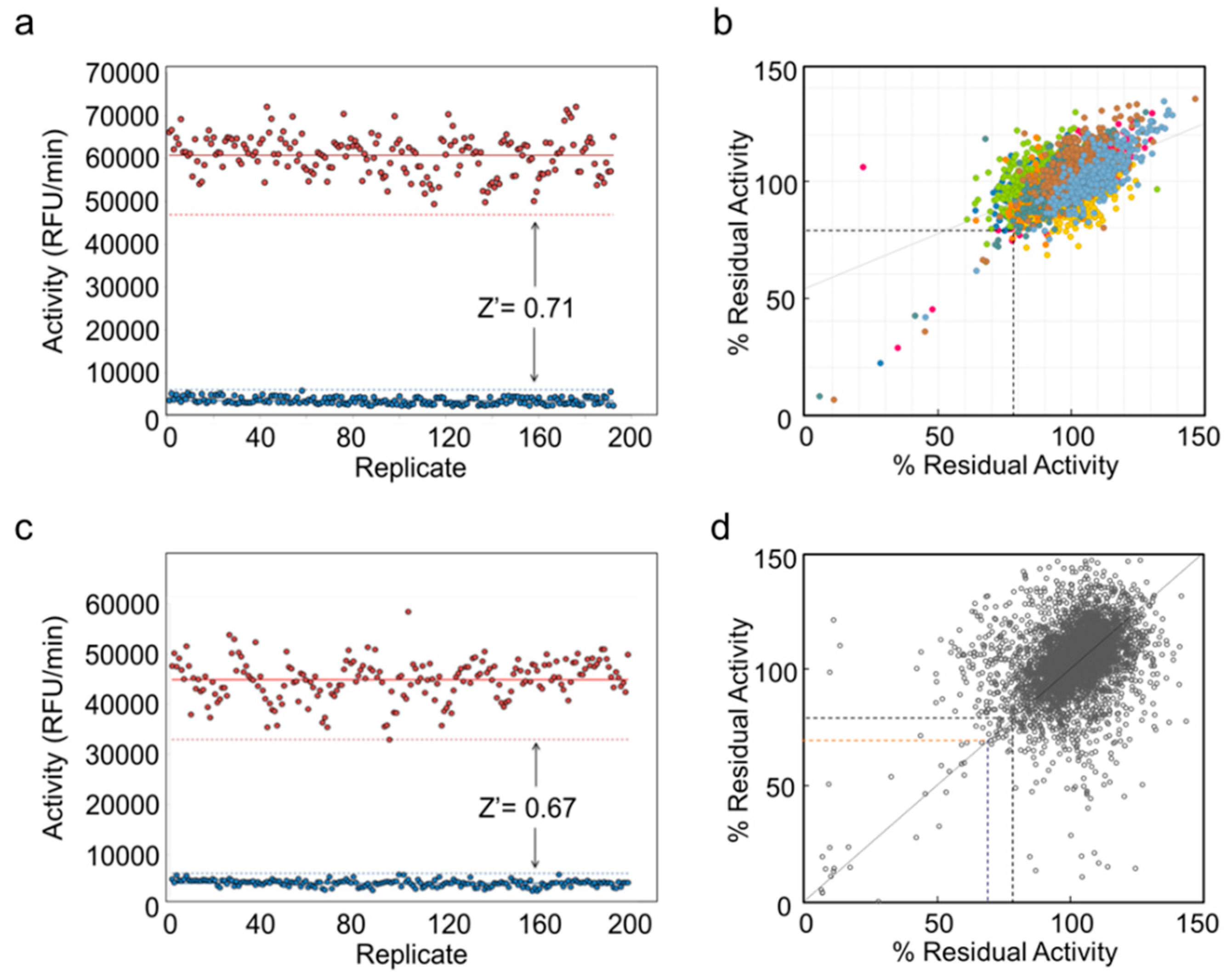
2.3. Pilot High Throughput Screen for Inhibitors of SaOatACΔ435 and NgPatBΔ69 Esterase Activity
2.4. Secondary Screening of Pilot Screen Hits
2.5. Large Scale High Throughput Screen for Inhibitors of NgPatBΔ100 and SaOatAΔ445
2.6. Secondary Screening of Potential PatB Inhibitors from Large Scale HTS
2.7. Inhibition Kinetics of Esculetin and Compound 89224 with NgPatB
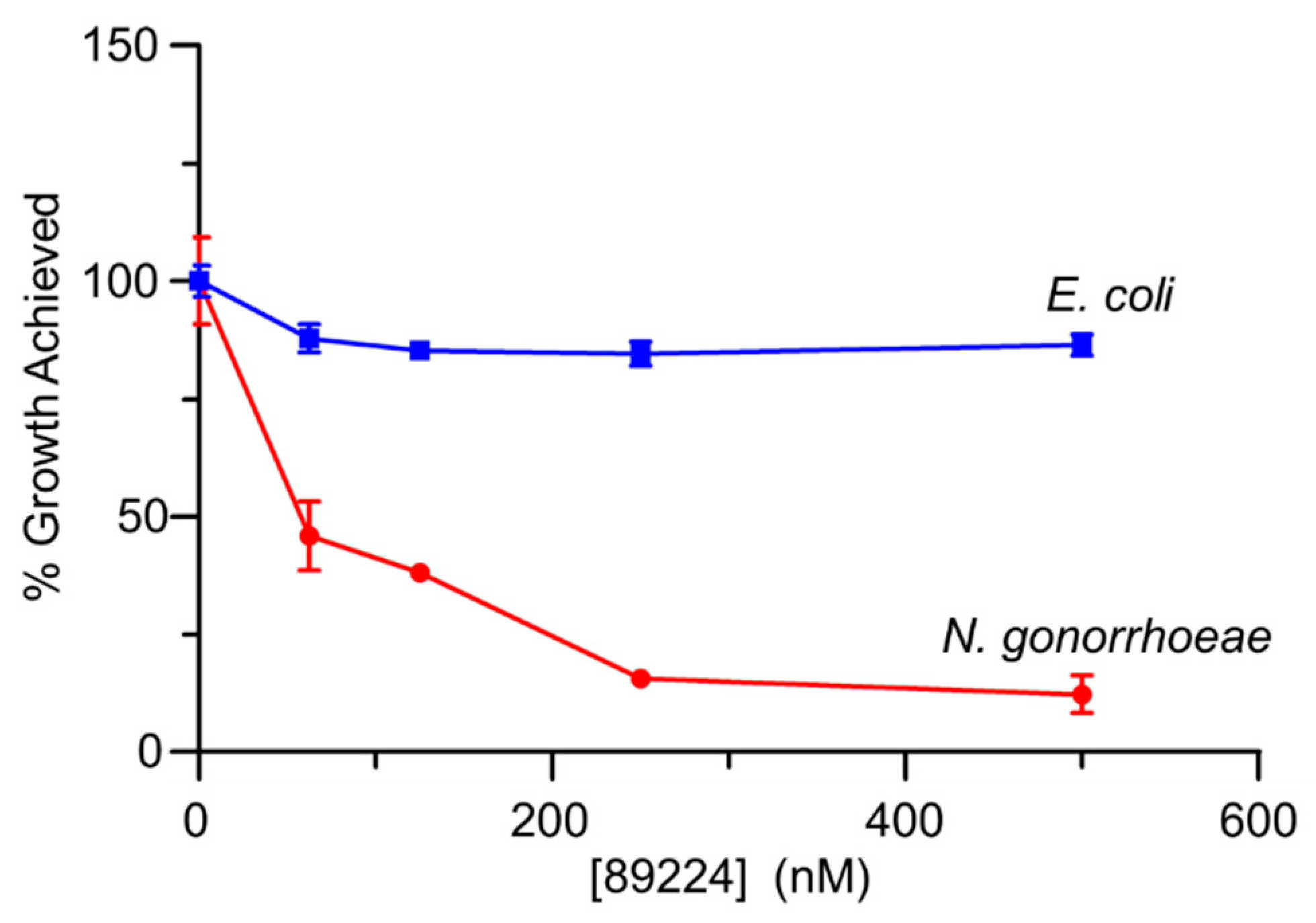
2.8. Antibacterial Activity of Inhibitors
3. Discussion
4. Materials and Methods
4.1. Engineering of NgPatB
4.2. Production and Purification of NgPatB Variants
4.3. Engineering of SaOatAΔ445
4.4. Production and Purification of SaOatAΔ445
4.5. Assays for NgPatB and SaOatA Activity and Inhibition Kinetics
4.6. Pilot HTS for Inhibitors of NgPatBΔ69 and SaOatAΔ435 and Esterase Activity
4.7. Secondary Screening of Potential NgPatB Inhibitors from Pilot HTS
4.8. Large Scale High Throughput Screen for Inhibitors of NgPatBΔ100 and SaOatAΔ445 Esterase Activity
4.9. Secondary Screening of Potential NgPatB and SaOatAΔ445 Inhibitors from Large Scale HTS
4.10. Bacterial Strains and Growth Conditions for Minimum Inhibitory Concentration Assays
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Callewaert, L.; Michiels, C.W. Lysozymes in the animal kingdom. J. Biosci. 2010, 35, 127–160. [Google Scholar] [CrossRef]
- Sorbara, M.T.; Philpott, D.J. Peptidoglycan: A critical activator of the mammalian immune system during infection and homeostasis. Immunol. Rev. 2011, 243, 40–60. [Google Scholar] [CrossRef]
- Shimada, T.; Park, B.G.; Wolf, A.J.; Briko, C.; Goodridge, H.S.; Becker, C.A.; Reyes, C.N.; Miao, E.A.; Aderem, A.; Götz, F.; et al. Staphylococcus aureus evades lysozyme-based peptidoglycan digestion that links phagocytosis, inflammasome activation, and IL-1beta secretion. Cell Host Microbe 2010, 7, 38–49. [Google Scholar] [CrossRef]
- Hancock, R.E.W.; Scott, M.G. The role of antimicrobial peptides in animal defenses. Proc. Natl. Acad. Sci. USA 2000, 97, 8856–8861. [Google Scholar] [CrossRef] [Green Version]
- Ellison, R.T., 3rd; Giehl, T.J. Killing of Gram-negative bacteria by lactoferrin and lysozyme. J. Clin. Investig. 1991, 88, 1080–1091. [Google Scholar] [CrossRef]
- Pushkaran, A.C.; Nataraj, N.; Nair, N.; Götz, F.; Biswas, R.; Mohan, C.G. Understanding the structure-function relationship of lysozyme resistance in Staphylococcus aureus by peptidoglycan O-acetylation using molecular docking, dynamics, and lysis assay. J. Chem. Inf. Model. 2015, 55, 760–770. [Google Scholar] [CrossRef]
- Dupont, C.; Clarke, A.J. Dependence of lysozyme-catalysed solubilization of Proteus mirabilis peptidoglycan on the extent of O-acetylation. Eur. J. Biochem. 1991, 195, 763–769. [Google Scholar] [CrossRef]
- Rosenthal, R.S.; Folkening, W.J.; Miller, D.R.; Swim, S.C. Resistance of O-acetylated gonococcal peptidoglycan to human peptidoglycan-degrading enzymes. Infect. Immun. 1983, 40, 903–911. [Google Scholar] [Green Version]
- Rosenthal, R.S.; Blundell, J.K.; Perkins, H.R. Strain-related differences in lysozyme sensitivity and extent of O-acetylation of gonococcal peptidoglycan. Infect. Immun. 1982, 37, 826–829. [Google Scholar] [Green Version]
- Swim, S.C.; Gfell, M.A.; Wilde, C.E., 3rd; Rosenthal, R.S. Strain distribution in extents of lysozyme resistance and O-acetylation of gonococcal peptidoglycan determined by high-performance liquid chromatography. Infect. Immun. 1983, 42, 446–452. [Google Scholar] [PubMed]
- Sukhithasri, V.; Nisha, N.; Biswas, L.; Anil Kumar, V.; Biswas, R. Innate immune recognition of microbial cell wall components and microbial strategies to evade such recognitions. Microbiol. Res. 2013, 168, 396–406. [Google Scholar] [CrossRef] [PubMed]
- Sychantha, D.; Brott, A.S.; Jones, C.S.; Clarke, A.J. Mechanistic pathways for peptidoglycan O-acetylation and de-O-acetylation. Front. Microbiol. 2018, 9, 2332. [Google Scholar] [CrossRef] [PubMed]
- Moynihan, P.J.; Sychantha, D.; Clarke, A.J. Chemical biology of peptidoglycan acetylation and deacetylation. Bioorg. Chem. 2014, 54, 44–50. [Google Scholar] [CrossRef]
- Moynihan, P.J.; Clarke, A.J. O-Acetylated peptidoglycan: Controlling the activity of bacterial autolysins and lytic enzymes of innate immune systems. Intl. J. Biochem. Cell Biol. 2011, 43, 1655–1659. [Google Scholar] [CrossRef] [PubMed]
- Clarke, A.J.; Dupont, C. O-Acetylated peptidoglycan: Its occurrence, pathobiological significance and biosynthesis. Can. J. Microbiol. 1991, 38, 85–91. [Google Scholar] [CrossRef]
- Pfeffer, J.M.; Strating, H.; Weadge, J.T.; Clarke, A.J. Peptidoglycan O-acetylation and autolysin profile of Enterococcus faecalis in the viable but nonculturable state. J. Bacteriol. 2006, 188, 902–908. [Google Scholar] [CrossRef] [PubMed]
- Strating, H.; Clarke, A.J. Changes in peptidoglycan structure and metabolism during differentiation of Proteus mirabilis into swarmer cells. Can. J. Microbiol. 2012, 58, 1183–1194. [Google Scholar] [CrossRef]
- Bera, A.; Biswas, R.; Herbert, S.; Götz, F. The presence of O-acetyltransferase in various staphylococci species correlates with lysozyme resistance and pathogenicity. Infect. Immun. 2006, 74, 4598–4604. [Google Scholar] [CrossRef] [PubMed]
- Bera, A.; Herbert, S.; Jakob, A.; Vollmer, W.; Götz, F. Why are pathogenic staphylococci so lysozyme resistant? The peptidoglycan O-acetyltransferase OatA is the major determinant for lysozyme resistance of Staphylococcus aureus. Mol. Microbiol. 2005, 55, 778–787. [Google Scholar] [CrossRef] [PubMed]
- Wichgers Schreur, P.J.; van Weeghel, C.; Rebel, J.M.; Smits, M.A.; van Putten, J.P.; Smith, H.E. Lysozyme resistance in Streptococcus suis is highly variable and multifactorial. PLoS ONE 2012, 7, e36281. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.P.; Neely, M.N. CpsY influences Streptococcus iniae cell wall adaptations important for neutrophil intracellular survival. Infect. Immun. 2012, 80, 1707–1715. [Google Scholar] [CrossRef]
- Hébert, L.; Courtin, P.; Torelli, R.; Sanguinetti, M.; Chapot-Chartier, M.P.; Auffray, Y.; Benachour, A. Enterococcus faecalis constitutes an unusual bacterial model in lysozyme resistance. Infect. Immun. 2007, 75, 5390–5398. [Google Scholar] [CrossRef]
- Burke, T.P.; Loukitcheva, A.; Zemansky, J.; Wheeler, R.; Boneca, I.G.; Portnoy, D.A. Listeria monocytogenes is resistant to lysozyme through the regulation, not the acquisition, of cell wall-modifying enzymes. J. Bacteriol. 2014, 196, 3756–3767. [Google Scholar] [CrossRef]
- Aubry, C.; Goulard, C.; Nahori, M.A.; Cayet, N.; Decalf, J.; Sachse, M.; Boneca, I.G.; Cossart, P.; Dussurget, O. OatA, a peptidoglycan O-acetyltransferase involved in Listeria monocytogenes immune escape, is critical for virulence. J. Infect. Dis. 2011, 204, 731–740. [Google Scholar] [CrossRef]
- Rae, C.S.; Geissler, A.; Adamson, P.C.; Portnoy, D.A. Mutations of the peptidoglycan N-deacetylase and O-acetylase result in enhanced lysozyme sensitivity, bacteriolysis, and hyperinduction of innate immune pathways. Infect. Immun. 2011, 79, 3596–3606. [Google Scholar] [CrossRef] [PubMed]
- Guariglia-Oropeza, V.; Helmann, J.D. Bacillus subtilis σ(V) confers lysozyme resistance by activation of two cell wall modification pathways, peptidoglycan O-acetylation and D-alanylation of teichoic acids. J. Bacteriol. 2011, 193, 6223–6232. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Lo, L.F.; Forsberg, L.S.; Maier, R.J. Helicobacter pylori peptidoglycan modifications confer lysozyme resistance and contribute to survival in the host. WMBio 2012, 3, e00409–e00412. [Google Scholar] [CrossRef]
- Fleming, T.J.; Wallsmith, D.E.; Rosenthal, R.S. Arthropathic properties of gonococcal peptidoglycan fragments: Implications for the pathogenesis of disseminated gonococcal disease. Infect. Immun. 1986, 52, 600–608. [Google Scholar]
- Veyrier, F.J.; Williams, A.H.; Mesnage, S.; Schmitt, C.; Taha, M.K.; Boneca, I.G. De-O-acetylation of peptidoglycan regulates glycan chain extension and affects in vivo survival of Neisseria meningitidis. Mol. Microbiol. 2013, 87, 1100–1112. [Google Scholar] [CrossRef]
- Crisóstomo, M.L.; Vollmer, W.; Kharat, A.S.; Inhülsen, S.; Gehre, F.; Buckenmaier, S.; Tomasz, A. Attenuation of penicillin resistance in a peptidoglycan O-acetyl transferase mutant of Streptococcus pneumoniae. Mol. Microbiol. 2006, 61, 1497–1509. [Google Scholar] [CrossRef]
- Pfeffer, J.M.; Moynihan, P.J.; Clarke, C.A.; Vandenende, C.; Clarke, A.J. Control of lytic transglycosylase activity within bacterial cell walls. In Glycomicrobiology; Reid, C.W., Twin, S.M., Reid, A.N., Eds.; Caister Academic Press: Norfolk, UK, 2012; pp. 55–68. [Google Scholar]
- Bernard, E.; Rolain, T.; David, B.; André, G.; Dupres, V.; Dufrêne, Y.F.; Hallet, B.; Chapot-Chartier, M.P.; Hols, P. Dual role for the O-acetyltransferase OatA in peptidoglycan modification and control of cell septation in Lactobacillus plantarum. PLoS ONE 2012, 7, e47893. [Google Scholar] [CrossRef]
- Laaberki, M.-H.; Pfeffer, J.; Clarke, A.J.; Dworkin, J. O-Acetylation of peptidoglycan is required for proper cell separation and S-layer anchoring in Bacillus anthracis. J. Biol. Chem. 2011, 286, 5278–5288. [Google Scholar] [CrossRef] [PubMed]
- Emirian, A.; Fromentin, S.; Eckert, C.; Chau, F.; Dubost, L.; Delepierre, M.; Gutmann, L.; Arthur, M.; Mesnage, S. Impact of peptidoglycan O-acetylation on autolytic activities of the Enterococcus faecalis N-acetylglucosaminidase AtlA and N-acetylmuramidase AtlB. FEBS Lett. 2009, 583, 3033–3038. [Google Scholar] [CrossRef]
- Strating, H.; Clarke, A.J. Differentiation of bacterial autolysins by zymogram analysis. Anal. Biochem. 2001, 290, 388–393. [Google Scholar] [CrossRef]
- Scheurwater, E.M.; Reid, C.W.; Clarke, A.J. Lytic transglycosylases: Bacterial space-making autolysins. Int. J. Biochem. Cell Biol. 2008, 40, 586–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Höltje, J.-V.; Mirelman, D.; Sharon, N.; Schwarz, U. Novel type of murein transglycosylase in Escherichia coli. J. Bacteriol. 1975, 124, 1067–1076. [Google Scholar] [Green Version]
- Clarke, A.J.; Strating, H.; Blackburn, N. Pathways for the O-acetylation of bacterial cell wall polysaccharides. In Glycomicrobiology; Doyle, R.J., Ed.; Plenum: New York, NY, USA, 1999; pp. 187–223. [Google Scholar]
- Moynihan, P.F.; Clarke, A.J. O-Acetylation of peptidoglycan in Gram-negative bacteria: Identification and characterization of PG O-acetyltransferase in Neisseria gonorrhoeae. J. Biol. Chem. 2010, 285, 13264–13273. [Google Scholar] [CrossRef]
- Dillard, J.P.; Hackett, K.T. Mutations affecting peptidoglycan acetylation in Neisseria gonorrhoeae and Neisseria meningitidis. Infect. Immun. 2005, 73, 5697–5705. [Google Scholar] [CrossRef]
- Sychantha, D.; Jones, C.S.; Little, D.J.; Moynihan, P.J.; Robinson, H.; Galley, N.F.; Roper, D.I.; Dowson, C.G.; Howell, P.L.; Clarke, A.J. In vitro characterization of the antivirulence target of Gram-positive pathogens, peptidoglycan O-acetyltransferase A (OatA). PLoS Pathog. 2017, 13, e1006667. [Google Scholar] [CrossRef]
- Bernard, E.; Rolain, T.; Courtin, P.; Guillot, A.; Langella, P.; Hols, P.; Chapot-Chartier, M.P. Characterization of O-acetylation of N-acetylglucosamine: A novel structural variation of bacterial peptidoglycan. J. Biol. Chem. 2011, 286, 23950–23958. [Google Scholar] [CrossRef]
- Sychantha, D.; Clarke, A.J. Peptidoglycan modification by the catalytic domain of Streptococcus pneumoniae OatA follows a ping-pong bi-bi mechanism of action. Biochemistry 2018, 57, 2394–2401. [Google Scholar] [CrossRef] [PubMed]
- Moynihan, P.J.; Clarke, A.J. Substrate specificity and kinetic characterization of PatB from N. gonorrhoeae. J. Biol. Chem. 2014, 289, 16748–16760. [Google Scholar] [CrossRef]
- Moynihan, P.J.; Clarke, A.J. The mechanism of peptidoglycan O-acetyltransferase involves an Asp-His-Ser catalytic triad. Biochemistry 2014, 53, 6243–6251. [Google Scholar] [CrossRef] [PubMed]
- Moynihan, P.J.; Clarke, A.J. Assay for peptidoglycan O-acetyltransferase: A potential new antibacterial target. Anal. Biochem. 2013, 439, 73–79. [Google Scholar] [CrossRef]
- Zhang, J.H.; Chung, T.D.; Oldenburg, K.R. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 1999, 4, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Brown, E.D. Screening in academe: A perspective on implementation of university-based small molecule screening. J. Biomol. Screen. 2003, 8, 377–379. [Google Scholar] [CrossRef] [PubMed]
- Kell, L.F. Investigation of novel inhibitory compounds of O-acetyltransferase A (OatA) from Staphylococcus aureus. Ph.D. Thesis, University of Guelph, Guelph, ON, Canada, December 2015. [Google Scholar]
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, J.M.; Clarke, A.J. Identification of the first known inhibitors of O-acetylpeptidoglycan esterase: A potential new antibacterial target. Chembiochem 2012, 13, 722–731. [Google Scholar] [CrossRef]
- Moynihan, P.J. Biochemical characterization of peptidoglycan O-acetyltransferases. Ph.D. Thesis, University of Guelph, Guelph, ON, Canada, December 2013. [Google Scholar]
- Song, H.; Inaka, K.; Maenaka, K.; Matsushima, M. Structural changes of active site cleft and different saccharide binding modes in human lysozyme co-crystallized with hexa-N-acetyl-chitohexose at pH 4.0. J. Mol. Biol. 1994, 224, 522–540. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.H.; Veyrier, F.J.; Bonis, M.; Michaud, Y.; Raynal, B.; Taha, M.K.; White, S.W.; Haouz, A.; Boneca, I.G. Visualization of a substrate-induced productive conformation of the catalytic triad of the Neisseria meningitidis peptidoglycan O-acetylesterase reveals mechanistic conservation in SGNH esterase family members. Acta Crystallogr. D Biol. Crystallogr. 2014, 70, 2631–2639. [Google Scholar] [CrossRef]
- Brott, A.S. Inhibition and structural insights into the mechanism of peptidoglycan O-acetylation in Neisseria gonorrhoeae. Ph.D. Thesis, University of Guelph, Guelph, ON, Canada. In preparation.
- Weeke, P.; Roden, D.M. Applied pharmacogenomics in cardiovascular medicine. Ann. Rev. Med. 2014, 65, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Emami, S.; Dadashpour, S. Current developments of coumarin-based anti-cancer agents in medicinal chemistry. Eur. J. Med. Chem. 2015, 102, 611–630. [Google Scholar] [CrossRef] [PubMed]
- Pochet, L.; Doucet, C.; Dive, G.; Wouters, J.; Masereel, B.; Reboud-Ravaux, M.; Pirotte, B. Coumarinic derivatives as mechanism-based inhibitors of alpha-chymotrypsin and human leukocyte elastase. Bioorg. Med. Chem. 2000, 8, 1489–1501. [Google Scholar] [CrossRef]
- Harper, J.W.; Powers, J.C. Reaction of serine proteases with substituted 3-alkoxy-4-chloroisocoumarins and 3-alkoxy-7-amino-4-chloroisocoumarins: New reactive mechanism-based inhibitors. Biochemistry 1985, 24, 7200–7213. [Google Scholar] [CrossRef] [PubMed]
- Heynekamp, J.J.; Hunsaker, L.A.; Vander Jagt, T.A.; Royer, R.E.; Deck, L.M.; Vander Jagt, D.L. Isocoumarin-based inhibitors of pancreatic cholesterol esterase. Bioorg. Med. Chem. 2008, 16, 5285–5294. [Google Scholar] [CrossRef] [Green Version]
- De Souza, S.M.; Delle Monache, F.; Smânia, A., Jr. Antibacterial activity of coumarins. Z. Fuer Naturforsch. C 2005, 60, 693–700. [Google Scholar] [CrossRef]
- Haggerty, C.L.; Ness, R.B. Epidemiolgy, pathogenesis and treatment of pelvic inflammatory disease. Expert Rev. Anti. Infect. Ther. 2006, 4, 235–247. [Google Scholar] [CrossRef]
- Baranwal, F.; Mohammad, M.; Jarneborn, A.; Reddy, B.R.; Golla, A.; Chakravarty, S.; Biswas, L.; Götz, F.; Shankarappa, S.; Jin, T.; et al. Impact of cell wall peptidoglycan O-acetylation on the pathogenesis of Staphylococcus aureus in septic arthritis. Int. J. Med. Microbiol. 2017, 307, 388–397. [Google Scholar] [CrossRef]
- Sanchez, M.; Kolar, S.L.; Müller, S.; Reyes, C.N.; Wolf, A.J.; Ogawa, C.; Singhania, R.; De Carvalho, D.D.; Arditi, M.; Underhill, D.M.; et al. O-Acetylation of peptidoglycan limits helper T cell priming and permits Staphylococcus aureus reinfection. Cell Host Microbe 2017, 22, 1–9. [Google Scholar] [CrossRef]
- Reverter, D.; Lima, C.D. Preparation of SUMO proteases and kinetic analysis using endogenous substrates. In Methods in Molecular Biology: SUMO Protocols; Ulrich, H.D., Ed.; Humana Press, Springer Science + Business Media: New York, NY, USA, 2009. [Google Scholar]
- Dillard, J.P. Genetic manipulation of Neisseria gonorrhoeae. In Current Protocols in Microbiology: Beta Proteobacteria; John Wiley & Sons. Inc.: Hoboken, NJ, USA, 2005; pp. 4A. 2.1–4A. 2.19. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | NgPatB | SaOatA | |||||
|---|---|---|---|---|---|---|---|
| 4MU-Ac | pNP-Ac | 4MU-Ac | pNP-Ac | ||||
| 1° Screen (% RA) 1 | IC50 (µM) | Ki (µM) | IC50 (µM) | 1° Screen (% RA) 1 | IC50 (µM) | IC50 (µM) | |
| Coumarin | 82.5 | n.d. | n.d. | n.d. | 106 | n.d. | n.d. |
| 7-Hydroxy- (Umbelliferone) | n.d. | 530 ± 70 | n.d. | >3000 | n.d. | 98.5 ± 1.12 | >3000 |
| 7-Hydroxy-4-methyl- (Hymercrome) | 42.9 | 615 ± 10 | n.d. | >3000 | 67.8 | >3000 | >3000 |
| 6,7-Dihydroxy- (Esculetin) | 55.7 | 53.1 ± 1.0 | 86.7 ± 4.7 | 853 ± 79 | 71.0 | 106±1.22 | 600 ± 140 |
| 6,7-Dihyroxy-4-methyl- (4-Me-esculetin) | 92.0 | n.d. | n.d. | n.d. | 98.3 | n.d. | n.d. |
| 6-Glucopyranosyl-7- hydroxy-(Esculin) | 38.4 | 49.7 ± 3.0 | n.d. | 923±9.0 | 47.8 | 75.8 ± 1.02 | 1350 ± 150 |
| 6-Hydroxy-7-methoxy- (Isoscopoletin) | 53.5 | n.d. | n.d. | n.d. | 115 | n.d. | n.d. |
| 7-Hydroxyl-6-methoxy- (Scopoletin) | 30.0 | 47.9 ± 4.0 | n.d. | 792 ± 7.0 | 21.4 | 103 ± 1.12 | 1170 ± 100 |
| Compound 89224 | 78.8 | 22.1 ± 1.2 | 126 ± 19 | 623 ± 64 | 99.8 | n.d. | n.d. |
| Compound 18420 | 77.1 | n.d. | n.d. | n.d. | 78.2 | 86.6 ± 8.7 | >3000 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brott, A.S.; Jones, C.S.; Clarke, A.J. Development of a High Throughput Screen for the Identification of Inhibitors of Peptidoglycan O-Acetyltransferases, New Potential Antibacterial Targets. Antibiotics 2019, 8, 65. https://doi.org/10.3390/antibiotics8020065
Brott AS, Jones CS, Clarke AJ. Development of a High Throughput Screen for the Identification of Inhibitors of Peptidoglycan O-Acetyltransferases, New Potential Antibacterial Targets. Antibiotics. 2019; 8(2):65. https://doi.org/10.3390/antibiotics8020065
Chicago/Turabian StyleBrott, Ashley S., Carys S. Jones, and Anthony J. Clarke. 2019. "Development of a High Throughput Screen for the Identification of Inhibitors of Peptidoglycan O-Acetyltransferases, New Potential Antibacterial Targets" Antibiotics 8, no. 2: 65. https://doi.org/10.3390/antibiotics8020065
APA StyleBrott, A. S., Jones, C. S., & Clarke, A. J. (2019). Development of a High Throughput Screen for the Identification of Inhibitors of Peptidoglycan O-Acetyltransferases, New Potential Antibacterial Targets. Antibiotics, 8(2), 65. https://doi.org/10.3390/antibiotics8020065