Countering Gram-Negative Antibiotic Resistance: Recent Progress in Disrupting the Outer Membrane with Novel Therapeutics


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
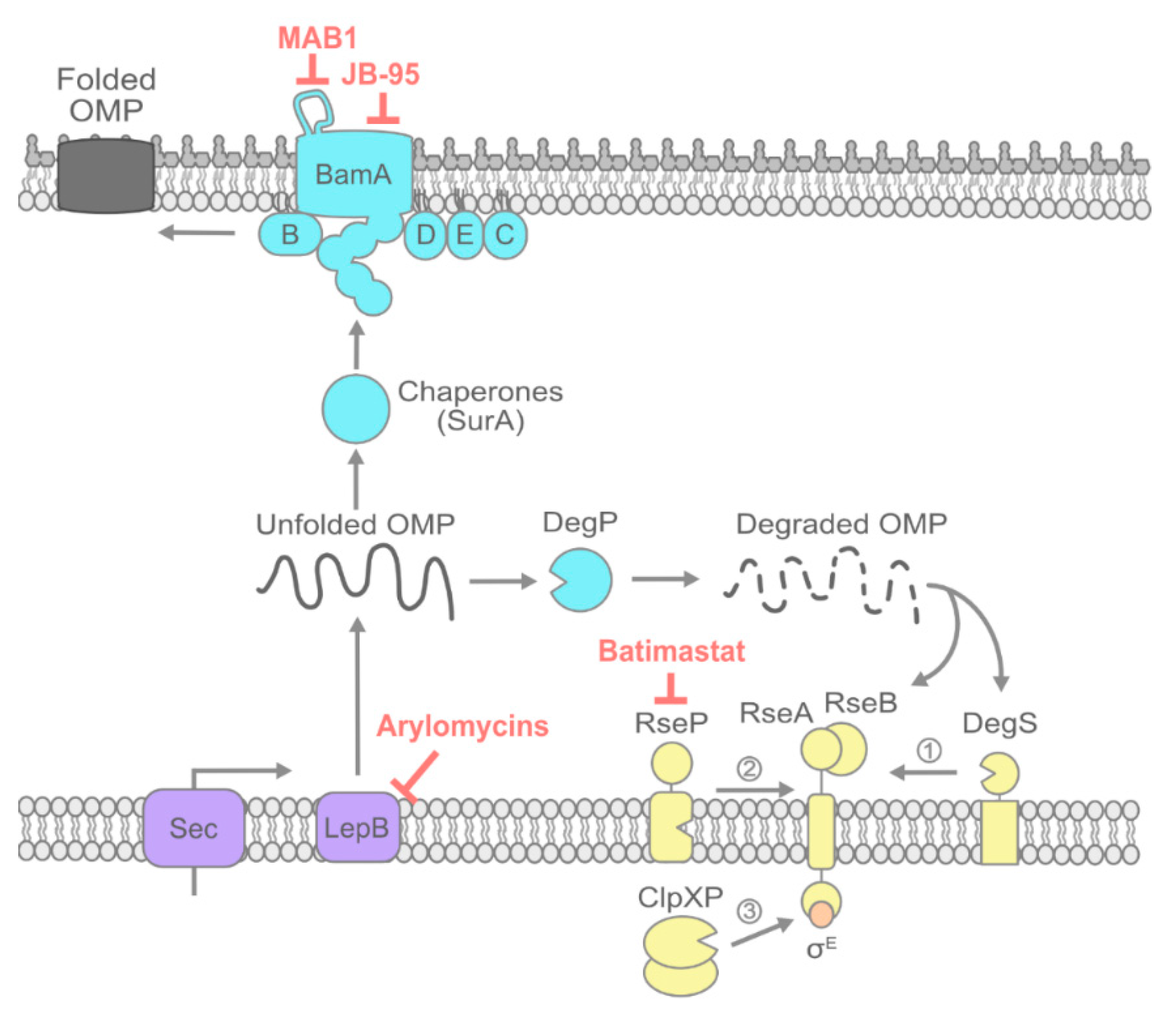
2. OMP Transport and Folding
2.1. Signal Peptide Processing by LepB
2.2. Inhibiting OMP Chaperones
2.3. Inhibiting the Bam Machine
2.4. Potentiating Bam Sensitivity by Inhibiting the σE Envelope Stress Response:
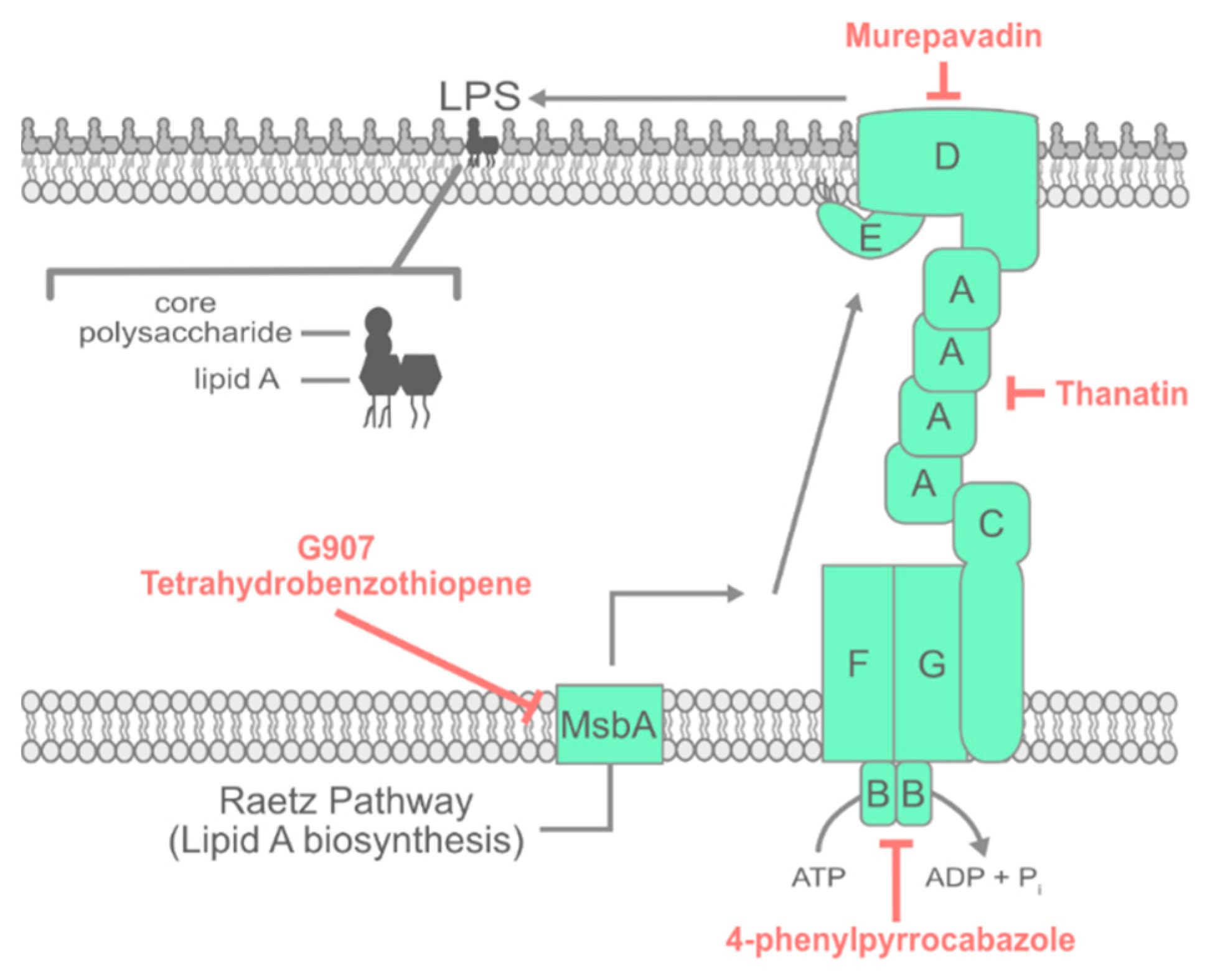
3. LPS Transport
3.1. Inhibiting Early Steps in LPS Assembly
3.2. Inhibiting the Lpt Complex
3.3. Activating Lpt Sensitizes Bacteria to Polymyxin
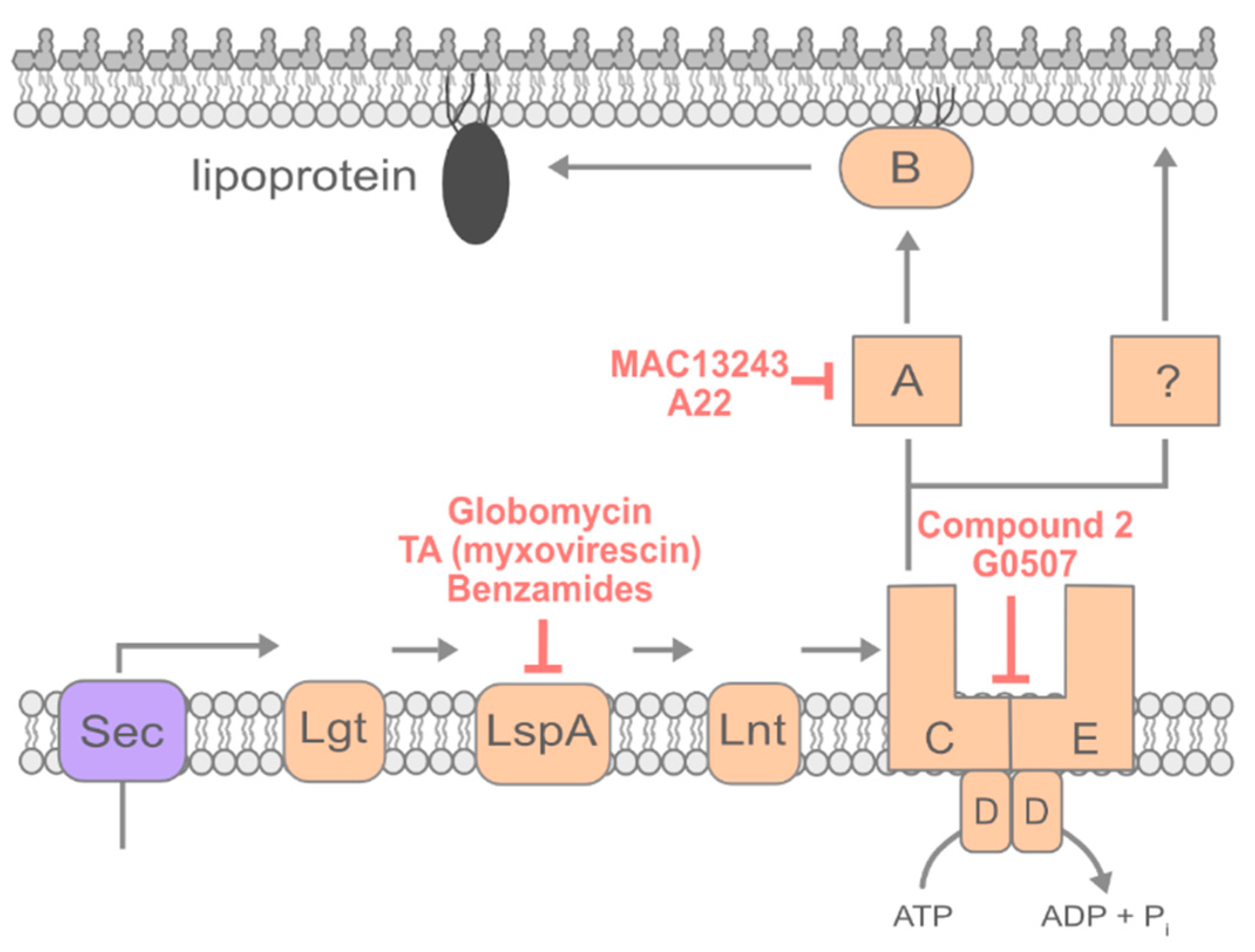
4. Lipoprotein Transport
4.1. Inhibiting Early Steps of Lipoprotein Maturation
4.2. Inhibiting Lipoprotein Transport
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- O’Neill, J. Review on Antimicrobial Resistance: Tackling a Crisis for the Health and Wealth of Nations; HM Government: London, UK, 2014.
- CDC. Antibiotic Resistance Threats in the United States, 2013; Centers of Disease Control and Prevention: Atlanta, Georgia, 2013.
- Nikaido, H. Molecular Basis of Bacterial Outer Membrane Permeability Revisited. Microbiol. Mol. Biol. Rev. 2003, 67, 593–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamio, Y.; Nikaido, H. Outer membrane of Salmonella typhimurium: Accessibility of phospholipid head groups to phospholipase C and cyanogen bromide activated dextran in the external medium. Biochemistry 1976, 15, 2561–2570. [Google Scholar] [CrossRef] [PubMed]
- Tokuda, H. Biogenesis of Outer Membranes in Gram-Negative Bacteria. Biosci. Biotechnol. Biochem. 2009, 73, 465–473. [Google Scholar] [CrossRef] [PubMed]
- Zgurskaya, H.I.; Rybenkov, V. Permeability barriers of Gram-negative pathogens. Ann. N. Y. Acad. Sci. 2019. [Google Scholar] [CrossRef] [PubMed]
- Silhavy, T.J.; Kahne, D.; Walker, S. The Bacterial Cell Envelope. Cold Spring Harb. Perspect. Biol. 2010, 2, a000414. [Google Scholar] [CrossRef]
- Okuda, S.; Sherman, D.J.; Silhavy, T.J.; Ruiz, N.; Kahne, D. Lipopolysaccharide transport and assembly at the outer membrane: The PEZ model. Nat. Rev. Microbiol. 2016, 14, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Konovalova, A.; Kahne, D.E.; Silhavy, T.J. Outer Membrane Biogenesis. Annu. Rev. Microbiol. 2017, 71, 539–556. [Google Scholar] [CrossRef]
- Grabowicz, M. Lipoprotein Transport: Greasing the machines of outer membrane biogenesis. BioEssays 2018, 40, e1700187. [Google Scholar] [CrossRef]
- Powers, M.J.; Trent, M.S. Intermembrane transport: Glycerophospholipid homeostasis of the Gram-negative cell envelope. Proc. Natl. Acad. Sci. USA 2019, 116, 17147–17155. [Google Scholar] [CrossRef]
- Henderson, I.R.; Nataro, J.P. Virulence Functions of Autotransporter Proteins. Infect. Immun. 2001, 69, 1231–1243. [Google Scholar] [CrossRef] [Green Version]
- Werner, J.; Misra, R. YaeT (Omp85) affects the assembly of lipid-dependent and lipid-independent outer membrane proteins of Escherichia coli. Mol. Microbiol. 2005, 57, 1450–1459. [Google Scholar] [CrossRef] [PubMed]
- Rapoport, T.A. Protein translocation across the eukaryotic endoplasmic reticulum and bacterial plasma membranes. Nature 2007, 450, 663–669. [Google Scholar] [CrossRef] [PubMed]
- Paetzel, M.; Karla, A.; Strynadka, N.C.J.; Dalbey, R.E. Signal peptidases. Chem. Rev. 2002, 102, 4549–4579. [Google Scholar] [CrossRef] [PubMed]
- Ollinger, J.; Ahn, J.; Odingo, J.; Parish, T. Inhibition of the sole type I signal peptidase of Mycobacterium tuberculosis is bactericidal under replicating and nonreplicating conditions. J. Bacteriol. 2012, 2614–2619. [Google Scholar] [CrossRef] [PubMed]
- Kulanthaivel, P.; Kreuzman, A.J.; Strege, M.A.; Belvo, M.D.; Smitka, T.A.; Clemens, M.; Swartling, J.B.; Minton, K.L.; Zheng, F.; Angleton, E.L.; et al. Novel lipoglycopeptides as inhibitors of bacterial signal peptidase I. J. Biol. Chem. 2004, 279, 36250–36258. [Google Scholar] [CrossRef]
- Paetzel, M.; Goodall, J.J.; Kania, M.; Dalbey, R.E.; Page, M.G.P. Crystallographic and Biophysical Analysis of a Bacterial Signal Peptidase in Complex with a Lipopeptide-based Inhibitor. J. Biol. Chem. 2004, 279, 30781–30790. [Google Scholar] [CrossRef] [Green Version]
- Roberts, T.C.; Smith, P.A.; Cirz, R.T.; Romesberg, F.E. Structural and initial biological analysis of synthetic arylomycin A2. J. Am. Chem. Soc. 2007, 129, 15830–15838. [Google Scholar] [CrossRef]
- Smith, P.A.; Koehler, M.F.T.; Girgis, H.S.; Yan, D.; Chen, Y.; Chen, Y.; Crawford, J.J.; Durk, M.R.; Higuchi, R.I.; Kang, J.; et al. Optimized arylomycins are a new class of Gram-negative antibiotics. Nature 2018, 561, 189–194. [Google Scholar] [CrossRef]
- Konovalova, A.; Grabowicz, M.; Balibar, C.J.; Malinverni, J.C.; Painter, R.E.; Riley, D. Inhibitor of intramembrane protease RseP blocks the σE response causing lethal accumulation of unfolded outer membrane proteins. Proc. Natl. Acad. Sci. USA 2018, 115, E6614–E6621. [Google Scholar] [CrossRef]
- Rizzitello, A.M.Y.E.; Harper, J.R.; Silhavy, T.J. Genetic Evidence for Parallel Pathways of Chaperone Activity in the Periplasm of Escherichia coli. J. Bacteriol. 2001, 183, 6794–6800. [Google Scholar] [CrossRef]
- Schwalm, J.; Mahoney, T.F.; Soltes, G.R.; Silhavy, T.J. Role for Skp in LptD assembly in Escherichia coli. J. Bacteriol. 2013, 195, 3734–3742. [Google Scholar] [CrossRef] [PubMed]
- Sklar, J.G.; Wu, T.; Kahne, D.; Silhavy, T.J. Defining the roles of the periplasmic chaperones SurA, Skp, and DegP in Escherichia coli. Genes Dev. 2007, 21, 2473–2484. [Google Scholar] [CrossRef] [PubMed]
- Weirich, J.; Cornelia, B.; Mühlenkamp, M.; Franz-Wachtel, M.; Macek, B.; Meuskens, I.; Skurnik, M.; Leskinen, K.; Bohn, E.; Autenrieth, I.; et al. Identifying components required for OMP biogenesis as novel targets for antiinfective drugs. Virulence 2017, 8, 1170–1188. [Google Scholar] [CrossRef] [PubMed]
- Sydenham, M.; Douce, G.; Bowe, F.; Ahmed, S.; Chatfield, S.; Dougan, G. Salmonella enterica serovar typhimurium surA mutants are attenuated and effective live oral vaccines. Infect. Immun. 2000, 68, 1109–1115. [Google Scholar] [CrossRef] [PubMed]
- Justice, S.S.; Hunstad, D.A.; Harper, J.R.; Duguay, A.R.; Pinkner, J.S.; Bann, J.; Frieden, C.; Silhavy, T.J.; Hultgren, S.J. Periplasmic peptidyl prolyl cis-trans isomerases are not essential for viability, but SurA is required for pilus biogenesis in Escherichia coli. J. Bacteriol. 2005, 187, 7680–7686. [Google Scholar] [CrossRef] [PubMed]
- Justice, S.S.; Lauer, S.R.; Hultgren, S.J.; Hunstad, D.A. Maturation of intracellular Escherichia coli communities requires SurA. Infect. Immun. 2006, 74, 4793–4800. [Google Scholar] [CrossRef] [PubMed]
- Watts, K.M.; Hunstad, D.A. Components of SurA required for outer membrane biogenesis in uropathogenic Escherichia coli. PLoS ONE 2008, 3, e3359. [Google Scholar] [CrossRef]
- Bell, E.W.; Zheng, E.J.; Ryno, L.M. Identification of inhibitors of the E. coli chaperone SurA using in silico and in vitro techniques. Bioorgan. Med. Chem. Lett. 2018, 28, 3540–3548. [Google Scholar] [CrossRef]
- Malinverni, J.C.; Werner, J.; Kim, S.; Sklar, J.G.; Kahne, D.; Misra, R.; Silhavy, T.J. YfiO stabilizes the YaeT complex and is essential for outer membrane protein assembly in Escherichia coli. Mol. Microbiol. 2006, 61, 151–164. [Google Scholar] [CrossRef]
- Hagan, C.L.; Wzorek, J.S.; Kahne, D. Inhibition of the β-barrel assembly machine by a peptide that binds BamD. Proc. Natl. Acad. Sci. USA 2015, 112, 2011–2016. [Google Scholar] [CrossRef]
- Lee, J.; Xue, M.; Wzorek, J.S.; Wu, T.; Grabowicz, M.; Gronenberg, L.S.; Sutterlin, H.A.; Davis, R.M.; Ruiz, N.; Silhavy, T.J.; et al. Characterization of a stalled complex on the β-barrel assembly machine. Proc. Natl. Acad. Sci. USA 2016, 113, 8717–8722. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Malinverni, J.C.; Sliz, P.; Silhavy, T.J.; Harrison, S.C.; Kahne, D. Structure and Function of an Essential Component of the Outer Membrane Protein Assembly Machine. Science 2007, 317, 961–964. [Google Scholar] [CrossRef] [Green Version]
- Ieva, R.; Tian, P.; Peterson, J.H.; Bernstein, H.D. Sequential and spatially restricted interactions of assembly factors with an autotransporter β domain. Proc. Natl. Acad. Sci. USA 2011, 108, E383–E391. [Google Scholar] [CrossRef] [PubMed]
- Noinaj, N.; Kuszak, A.J.; Gumbart, J.C.; Lukacik, P.; Chang, H.; Easley, N.C.; Lithgow, T.; Buchanan, S.K. Structural insight into the biogenesis of β-barrel membrane proteins. Nature 2013, 501, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Ni, D.; Wang, Y.; Yang, X.; Zhou, H.; Hou, X.; Cao, B.; Lu, Z.; Zhao, X.; Yang, K.; Huang, Y. Structural and functional analysis of the β-barrel domain of BamA from Escherichia coli. FASEB J. 2014, 28, 2677–2685. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, R.; Schütz, M.; Oberhettinger, P.; Faulstich, M.; Bermejo, I.; Rudel, T.; Diederichs, K.; Zeth, K. Structure of BamA, an essential factor in outer membrane protein biogenesis. Acta Crystallogr. D Biol. Crystallogr. 2014, 70, 1779–1789. [Google Scholar] [CrossRef] [PubMed]
- Rossiter, A.E.; Leyton, D.L.; Tveen-Jensen, K.; Browning, D.F.; Sevastsyanovich, Y.; Knowles, T.J.; Nichols, K.B.; Cunninghman, A.F.; Overduin, M.; Schembri, M.A.; et al. The Essential β-Barrel Assembly Machinery Complex Components BamD and BamA Are Required for Autotransporter Biogenesis. FEBS Lett. 1981, 135, 145–147. [Google Scholar] [CrossRef]
- Charlson, E.S.; Werner, J.N.; Misra, R. Differential effects of yfgL mutation on Escherichia coli outer membrane proteins and lipopolysaccharide. J. Bacteriol. 2006, 188, 7186–7194. [Google Scholar] [CrossRef]
- Mahoney, T.F.; Ricci, D.P.; Silhavy, T.J. Classifying β-barrel assembly substrates by manipulating essential Bam complex members. J. Bacteriol. 2016, 198, 1984–1992. [Google Scholar] [CrossRef]
- Konovalova, A.; Mitchell, A.M.; Silhavy, T.J. A lipoprotein/β-barrel complex monitors lipopolysaccharide integrity transducing information across the outer membrane. Elife 2016, 5, e15276. [Google Scholar] [CrossRef]
- Jansen, K.B.; Baker, S.L.; Sousa, M.C. Crystal structure of BamB bound to a periplasmic domain fragment of BamA, the central component of the β-barrel assembly machine. J. Biol. Chem. 2015, 290, 2126–2136. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Li, H.; Dong, H.; Zeng, Y.; Zhang, Z.; Paterson, N.G.; Stansfeld, P.J.; Wang, Z.; Zhang, Y.; Wang, W.; et al. Structural basis of outer membrane protein insertion by the BAM complex. Nature 2016, 531, 64–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, L.; Zheng, J.; Wang, Y.; Yang, X.; Liu, Y.; Sun, C.; Cao, B.; Zhou, H.; Ni, D.; Lou, J.; et al. Structure of the BAM complex and its implications for biogenesis of outer-membrane proteins. Nat. Struct. Mol. Biol. 2016, 23, 192–196. [Google Scholar] [CrossRef]
- Iadanza, M.G.; Higgins, A.J.; Schiffrin, B.; Calabrese, A.N.; Brockwell, D.J.; Ashcroft, A.E.; Radford, S.E.; Ranson, N.A. Lateral opening in the intact β-barrel assembly machinery captured by cryo-EM. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.Y.; Kwon, E.; Choi, J.; Hwang, H.; Kim, K.K. Structural basis for the negative regulation of bacterial stress response by RseB. Protein Sci. 2010, 19, 1258–1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.H.; Paetzel, M. Crystal structure of Escherichia coli BamB, a lipoprotein component of the β-barrel assembly machinery complex. J. Mol. Biol. 2011, 406, 667–678. [Google Scholar] [CrossRef] [PubMed]
- Sandoval, C.M.; Baker, S.L.; Jansen, K.; Metzner, S.I.; Sousa, M.C. Crystal Structure of BamD. An Essential Component of the β-Barrel Assembly Machinery of Gram Negative Bacteria. J. Mol. Biol. 2011, 409, 348–357. [Google Scholar] [CrossRef] [PubMed]
- Knowles, T.J.; Browning, D.F.; Jeeves, M.; Maderbocus, R.; Rajesh, S.; Sridhar, P.; Manoli, E.; Emery, D.; Sommer, U.; Spencer, A.; et al. Structure and function of BamE within the outer membrane and the β-barrel assembly machine. EMBO Rep. 2011, 12, 123–128. [Google Scholar] [CrossRef]
- Endo, T.; Kawano, S.; Yamano, K. BamE structure: The assembly of β-barrel proteins in the outer membranes of bacteria and mitochondria. EMBO Rep. 2011, 12, 94–95. [Google Scholar] [CrossRef]
- Dong, C.; Yang, X.; Hou, H.F.; Shen, Y.Q.; Dong, Y.H. Structure of Escherichia coli BamB and its interaction with POTRA domains of BamA. Acta Crystallogr. D Biol. Crystallogr. 2012, 68, 1134–1139. [Google Scholar] [CrossRef]
- Storek, K.M.; Auerbach, M.R.; Shi, H.; Garcia, N.K.; Sun, D.; Nickerson, N.N.; Vij, R.; Lin, Z.; Chiang, N.; Schneider, K.; et al. Monoclonal antibody targeting the β-barrel assembly machine of Escherichia coli is bactericidal. Proc. Natl. Acad. Sci. USA 2018, 115, 3692–3697. [Google Scholar] [CrossRef] [PubMed]
- Leonard-Rivera, M.; Misra, R. Conserved residues of the putative L6 loop of Escherichia coli BamA play a critical role in the assembly of β-barrel outer membrane proteins, including that of BamA itself. J. Bacteriol. 2012, 194, 4662–4668. [Google Scholar] [CrossRef] [PubMed]
- Rigel, N.W.; Ricci, D.P.; Silhavy, T.J. Conformation-specific labeling of BamA and suppressor analysis suggest a cyclic mechanism for β-barrel assembly in Escherichia coli. Proc. Natl. Acad. Sci. USA 2013, 110, 5151–5156. [Google Scholar] [CrossRef] [PubMed]
- Storek, K.M.; Chan, J.; Vij, R.; Chiang, N.; Lin, Z.; Iii, J.B.; Koth, C.M.; Vernes, J.-M.; Meng, Y.G.; Yin, J.; et al. Massive antibody discovery used to probe structure–function relationships of the essential outer membrane protein LptD. Elife 2019, 8, e46258. [Google Scholar] [CrossRef] [PubMed]
- Kutik, S.; Stojanovski, D.; Becker, L.; Becker, T.; Meinecke, M.; Krüger, V.; Prinz, C.; Meisinger, C.; Guiard, B.; Wagner, R.; et al. Dissecting membrane insertion of mitochondrial β-barrel proteins. Cell 2008, 132, 1011–1024. [Google Scholar] [CrossRef] [PubMed]
- Urfer, M.; Bogdanovic, J.; Lo Monte, F.; Moehle, K.; Zerbe, K.; Omasits, U.; Ahrens, C.H.; Pessi, G.; Eberl, L.; Robinson, J.A. A Peptidomimetic Antibiotic Targets Outer Membrane Proteins and Disrupts Selectively the Outer Membrane in Escherichia coli. J. Biol. Chem. 2016, 291, 1921–1932. [Google Scholar] [CrossRef] [PubMed]
- Grabowicz, M.; Silhavy, T.J. Envelope Stress Responses: An Interconnected Safety Net. Trends Biochem. Sci. 2017, 42, 232–242. [Google Scholar] [CrossRef] [PubMed]
- Ades, S.E.; Connolly, L.E.; Alba, B.M.; Gross, C.A. The Escherichia coli σE-dependent extracytoplasmic stress response is controlled by the regulated proteolysis of an anti-σ factor. Genes Dev. 1999, 13, 2449–2461. [Google Scholar] [CrossRef]
- Cezairliyan, B.O.; Sauer, R.T. Inhibition of regulated proteolysis by RseB. Proc. Natl. Acad. Sci. USA 2007, 104, 3771–3776. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.Y.; Jin, K.S.; Kwon, E.; Ree, M.; Kim, K.K. Crystal structure of RseB and a model of its binding mode to RseA. Proc. Natl. Acad. Sci. USA 2007, 104, 8779–8784. [Google Scholar] [CrossRef] [Green Version]
- Chaba, R.; Alba, B.M.; Guo, M.S.; Sohn, J.; Ahuja, N.; Sauer, R.T. Signal integration by DegS and RseB governs the σE-mediated envelope stress response in Escherichia coli. Proc. Natl. Acad. Sci. USA 2010, 108, 2106–2111. [Google Scholar] [CrossRef] [PubMed]
- Alba, B.M.; Zhong, H.J.; Pelayo, J.C.; Gross, C.A. degS (hhoB) is an essential Escherichia coli gene whose indispensable function is to provide σE activity. Mol. Microbiol. 2001, 40, 1323–1333. [Google Scholar] [CrossRef]
- Walsh, N.P.; Alba, B.M.; Bose, B.; Gross, C.A.; Sauer, R.T. OMP peptide signals initiate the envelope-stress response by activating DegS protease via relief of inhibition mediated by its PDZ domain. Cell 2003, 113, 61–71. [Google Scholar] [CrossRef]
- Wilken, C.; Kitzing, K.; Kurzbauer, R.; Ehrmann, M.; Clausen, T. Crystal structure of the DegS stress sensor: How a PDZ domain recognizes misfolded protein and activates a protease. Cell 2004, 117, 483–494. [Google Scholar] [CrossRef]
- Grigorova, I.L.; Chaba, R.; Zhong, H.J.; Alba, B.M.; Rhodius, V.; Herman, C.; Gross, C.A. Fine-tuning of the Escherichia coli σE envelope stress response relies on multiple mechanisms to inhibit signal-independent proteolysis of the transmembrane anti-sigma factor, RseA. Genes Dev. 2004, 18, 2686–2697. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, Y.; Kanehara, K.; Ito, K. RseP (YaeL), an Escherichia coli RIP protease, cleaves transmembrane sequences. EMBO J. 2004, 23, 4434–4442. [Google Scholar] [CrossRef] [PubMed]
- Kanehara, K.; Ito, K.; Akiyama, Y. YaeL (EcfE) activates the σE pathway of stress response through a site-2 cleavage of anti-σE, RseA. Genes Dev. 2002, 16, 2147–2155. [Google Scholar] [CrossRef]
- Flynn, J.M.; Levchenko, I.; Sauer, R.T.; Baker, T.A. Modulating substrate choice: The SspB adaptor delivers a regulator of the extracytoplasmic-stress response to the AAA+ protease ClpXP for degradation. Genes Dev. 2004, 18, 2292–2301. [Google Scholar] [CrossRef]
- Konovalova, A.; Schwalm, J.A.; Silhavy, T.J. A suppressor mutation that creates a faster and more robust σE envelope stress response. J. Bacteriol. 2016, 198, 2345–2351. [Google Scholar] [CrossRef]
- Davies, B.; Brown, P.D.; East, N.; Crimmin, M.J.; Balkwill, F.R. A synthetic matrix metalloproteinase inhibitor decreases tumor burden and prolongs survival of mice bearing human ovarian carcinoma xenografts. Cancer Res. 1993, 53, 2087–2091, Erratum in 1933, 53, 3652. [Google Scholar]
- Mosyak, L.; Georgiadis, K.; Shane, T.; Svenson, K.; Hebert, T.; McDonagh, T.; Mackie, S.; Olland, S.; Lin, L.; Zhong, X.; et al. Crystal structures of the two major aggrecan degrading enzymes, ADAMTS4 and ADAMTS5. Protein Sci. 2008, 17, 16–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Las Peñas, A.; Connolly, L.; Gross, C.A. σE is an essential sigma factor in Escherichia coli. J. Bacteriol. 1997, 179, 6862–6864. [Google Scholar] [CrossRef]
- Kelesidis, T.; Falagas, M.E. The Safety of Polymyxin Antibiotics. Expert Opin. Drug Saf. 2015, 14, 1687–1701. [Google Scholar] [CrossRef] [PubMed]
- Olaitan, A.O.; Morand, S.; Rolain, J.-M. Mechanisms of polymyxin resistance: Acquired and intrinsic resistance in bacteria. Front. Microbiol. 2014, 5, 643. [Google Scholar] [CrossRef]
- Vaara, M. Polymyxins and their novel derivatives. Curr. Opin. Microbiol. 2010, 13, 574–581. [Google Scholar] [CrossRef] [PubMed]
- Bialvaei, A.Z.; Samadi Kafil, H. Colistin, mechanisms and prevalence of resistance. Curr. Med. Res. Opin. 2015, 31, 707–721. [Google Scholar] [CrossRef] [PubMed]
- Simpson, B.W.; Trent, M.S. Pushing the envelope: LPS modifications and their consequences. Nat. Rev. Microbiol. 2019, 17, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Bertani, B.; Ruiz, N. Function and biogenesis of lipopolysaccharide. EcoSal Plus 2018. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; White, K.A.; Polissi, A.; Georgopoulos, C.; Raetz, C.R.H. Function of Escherichia coli MsbA, an essential ABC family transporter, in lipid A and phospholipid biosynthesis. J. Biol. Chem. 1998, 273, 12466–12475. [Google Scholar] [CrossRef] [PubMed]
- Doerrler, W.T.; Gibbons, H.S.; Christian, R.; Raetz, H. MsbA-dependent translocation of lipids across the inner membrane of Escherichia coli. J. Biol. Chem. 2004, 279, 45102–45109. [Google Scholar] [CrossRef]
- Ho, H.; Miu, A.; Alexander, M.K.; Garcia, N.K.; Oh, A.; Zilberleyb, I.; Reichelt, M.; Austin, C.D.; Tam, C.; Shriver, S.; et al. Structural basis for dual-mode inhibition of the ABC transporter MsbA. Nature 2018, 557, 196–204. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Baidin, V.; Pahil, K.S.; Moison, E.; Tomasek, D.; Ramadoss, N.S.; Chatterjee, A.K.; McNamara, C.W.; Young, T.S.; Schultz, P.G.; et al. Cell-based screen for discovering lipopolysaccharide biogenesis inhibitors. Proc. Natl. Acad. Sci. USA 2018, 115, 6834–6839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richie, D.L.; Wang, L.; Chan, H.; Pascale, G.D.; Six, D.A.; Wei, J.-R.; Dean, C.R. A pathway-directed positive growth restoration assay to facilitate the discovery of lipid A and fatty acid biosynthesis inhibitors in Acinetobacter baumannii. PLoS ONE 2018, 13, e0193851. [Google Scholar] [CrossRef] [PubMed]
- Sherman, D.J.; Lazarus, M.B.; Murphy, L.; Liu, C.; Walker, S.; Ruiz, N.; Kahne, D. Decoupling catalytic activity from biological function of the ATPase that powers lipopolysaccharide transport. Am. J. Vet. Res. 2014, 111, 4982–4987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz, N.; Gronenberg, L.S.; Kahne, D.; Silhavy, T.J. Identification of two inner-membrane proteins required for the transport of lipopolysaccharide to the outer membrane of Escherichia coli. Proc. Natl. Acad. Sci. USA 2008, 105, 5537–5542. [Google Scholar] [CrossRef] [PubMed]
- Okuda, S.; Freinkman, E.; Kahne, D. Cytoplasmic ATP Hydrolysis Powers Transport of Lipopolysaccharide Across the Periplasm in E. coli. Science 2012, 338, 1214–1217. [Google Scholar] [CrossRef] [PubMed]
- Suits, M.D.L.; Sperandeo, P.; Dehò, G.; Polissi, A.; Jia, Z. Novel Structure of the Conserved Gram-Negative Lipopolysaccharide Transport Protein A and Mutagenesis Analysis. J. Mol. Biol. 2008, 380, 476–488. [Google Scholar] [CrossRef]
- Sperandeo, P.; Cescutti, R.; Villa, R.; Di Benedetto, C.; Candia, D.; Dehò, G.; Polissi, A. Characterization of lptA and lptB, two essential genes implicated in lipopolysaccharide transport to the outer membrane of Escherichia coli. J. Bacteriol. 2007, 189, 244–253. [Google Scholar] [CrossRef] [PubMed]
- Chng, S.S.; Gronenberg, L.S.; Kahne, D. Proteins required for lipopolysaccharide assembly in Escherichia coli form a transenvelope complex. Biochemistry 2010, 49, 4565–4567. [Google Scholar] [CrossRef]
- Ishidate, K.; Creeger, E.S.; Zrike, J.; Deb, S.; Glauner, B.; MacAlister, T.J.; Rothfield, L.I. Isolation of differentiated membrane domains from Escherichia coli and Salmonella typhimurium, including a fraction containing attachment sites between the inner and outer membranes and the murein skeleton of the cell envelope. J. Biol. Chem. 1986, 261, 428–443. [Google Scholar]
- Bos, M.P.; Tefsen, B.; Geurtsen, J.; Tommassen, J. Identification of an outer membrane protein required for the transport of lipopolysaccharide to the bacterial cell surface. Proc. Natl. Acad. Sci. USA 2004, 101, 9417–9422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braun, M.; Silhavy, T.J. Imp/OstA is required for cell envelope biogenesis in Escherichia coli. Mol. Microbiol. 2002, 45, 1289–1302. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; McCandlish, A.C.; Gronenberg, L.S.; Chng, S.-S.; Silhavy, T.J.; Kahne, D. Identification of a protein complex that assembles lipopolysaccharide in the outer membrane of Escherichia coli. Proc. Natl. Acad. Sci. USA 2006, 103, 11754–11759. [Google Scholar] [CrossRef] [PubMed]
- Freinkman, E.; Chng, S.; Kahne, D. The complex that inserts lipopolysaccharide into the bacterial outer membrane forms a two-protein plug-and-barrel. Proc. Natl. Acad. Sci. USA 2011, 108, 2486–2491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiao, S.; Luo, Q.; Zhao, Y.; Zhang, X.C.; Huang, Y. Structural basis for lipopolysaccharide insertion in the bacterial outer membrane. Nature 2014, 511, 108–111. [Google Scholar] [CrossRef]
- Dong, H.; Xiang, Q.; Gu, Y.; Wang, Z.; Paterson, N.G.; Stansfeld, P.J.; He, C.; Zhang, Y.; Wang, W.; Dong, C. Structural basis for outer membrane lipopolysaccharide insertion. Nature 2014, 511, 52–56. [Google Scholar] [CrossRef] [Green Version]
- Sperandeo, P.; Lau, F.K.; Carpentieri, A.; De Castro, C.; Molinaro, A.; Dehò, G.; Silhavy, T.J.; Polissi, A. Functional analysis of the protein machinery required for transport of lipopolysaccharide to the outer membrane of Escherichia coli. J. Bacteriol. 2008, 190, 4460–4469. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, N.; Jetter, P.; Ueberbacher, B.J.; Werneburg, M.; Zerbe, K.; Steinmann, J.; Van der Meijden, B.; Bernardini, F.; Lederer, A.; Dias, R.L.A.; et al. Peptidomimetic Antibiotics Target Outer-Membrane Biogenesis in Pseudomonas aeruginosa. Science 2010, 327, 1010–1013. [Google Scholar] [CrossRef]
- Andolina, G.; Bencze, L.-C.; Zerbe, K.; Muller, M.; Steinmann, J.; Kocherla, H.; Mondal, M.; Sobek, J.; Moehle, K.; Malojcic, G.; et al. A Peptidomimetic Antibiotic Interacts with the Periplasmic Domain of LptD from Pseudomonas aeruginosa. ACS Chem. Biol. 2018, 13, 666–675. [Google Scholar] [CrossRef]
- Vetterli, S.U.; Zerbe, K.; Müller, M.; Urfer, M.; Mondal, M.; Wang, S.; Moehle, K.; Zerbe, O.; Vitale, A.; Pessi, G.; et al. Thanatin targets the intermembrane protein complex required for lipopolysaccharide transport in Escherichia coli. Sci. Adv. 2018, 4, eaau2634. [Google Scholar] [CrossRef]
- Hagan, C.L.; Kim, S.; Kahne, D. Reconstitution of outer membrane protein assembly from purified components. Sci. Rep. 2010, 328, 890–892. [Google Scholar] [CrossRef] [PubMed]
- Sherman, D.J.; Xie, R.; Taylor, R.J.; George, A.H.; Okuda, S.; Foster, P.J.; Needleman, D.J.; Kahne, D. Lipopolysaccharide is transported to the cell surface by a membrane-to-membrane protein bridge. Science 2018, 359, 798–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gronenberg, L.S.; Kahne, D.E. Development of an Activity Assay for Discovery of Inhibitors of Lipopolysaccharide Assembly. J. Am. Chem. Soc. 2010, 132, 2518–2519. [Google Scholar] [CrossRef] [PubMed]
- Sherman, D.J.; Okuda, S.; Denny, W.A.; Kahne, D. Validation of inhibitors of an ABC transporter required to transport lipopolysaccharide to the cell surface in Escherichia coli. Bioorgan. Med. Chem. 2013, 21, 4846–4851. [Google Scholar] [CrossRef] [PubMed]
- Mandler, M.D.; Baidin, V.; Lee, J.; Pahil, K.S.; Owens, T.W.; Kahne, D. Novobiocin Enhances Polymyxin Activity by Stimulating Lipopolysaccharide Transport. J. Am. Chem. Soc. 2018, 140, 6749–6753. [Google Scholar] [CrossRef] [PubMed]
- May, J.M.; Owens, T.W.; Mandler, M.D.; Simpson, B.W.; Lazarus, M.B.; Sherman, D.J.; Davis, R.M.; Okuda, S.; Massefski, W.; Ruiz, N.; et al. The antibiotic novobiocin binds and activates the ATPase that powers lipopolysaccharide transport. J. Am. Chem. Soc. 2017, 139, 17221–17224. [Google Scholar] [CrossRef] [PubMed]
- Okuda, S.; Tokuda, H. Lipoprotein Sorting in Bacteria. Annu. Rev. Microbiol. 2011, 65, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Tokunaga, M.; Tokunaga, H.; Wu, H.C. Post-translational modification and processing of Escherichia coli prolipoprotein in vitro. Proc. Natl. Acad. Sci. USA 1982, 79, 2255–2259. [Google Scholar] [CrossRef]
- Tokunaga, M.; Loranger, J.M.; Wu, H.C. Prolipoprotein modification and processing enzymes in Escherichia coli. J. Biol. Chem. 1984, 259, 3825–3830. [Google Scholar]
- Gupta, S.D.; Gan, K.; Schmid, M.B.; Wu, H.C. Characterization of a temperature-sensitive mutant of Salmonella typhimurium defective in apolipoprotein N-acyltransferase. J. Biol. Chem. 1993, 268, 16551–16556. [Google Scholar]
- Vogeley, L.; El Arnaout, T.; Bailey, J.; Stansfeld, P.J.; Boland, C.; Caffrey, M. Structural basis of lipoprotein signal peptidase II action and inhibition by the antibiotic globomycin. Science 2016, 351, 876–880. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Y.; Gerth, K.; Wall, D. Myxobacterium-Produced Antibiotic TA (Myxovirescin) Inhibits Type II Signal Peptidase. Antimicrob. Agents Chemother. 2012, 56, 2014–2021. [Google Scholar] [CrossRef] [Green Version]
- Yakushi, T.; Tajima, T.; Matsuyama, S.; Tokuda, H. Lethality of the Covalent Linkage between Mislocalized Major Outer Membrane Lipoprotein and the Peptidoglycan of Escherichia coli. J. Bacteriol. 1997, 179, 2857–2862. [Google Scholar] [CrossRef] [PubMed]
- Buddelmeijer, N. The molecular mechanism of bacterial lipoprotein modification—How, when and why? FEMS Microbiol. Rev. 2015, 39, 246–261. [Google Scholar] [CrossRef] [PubMed]
- Banaiee, N.; Jacobs, W.R.; Ernst, J.D. LspA-independent action of globomycin on Mycobacterium tuberculosis. J. Antimicrob. Chemother. 2007, 60, 414–416. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, S.; Owensby, A.; Wall, D.; Wolan, D.W. Lipoprotein signal peptidase inhibitors with antibiotic properties identified through design of a robust in vitro HT platform. Cell Chem. Biol. 2018, 25, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, K.; Yu, F.; Inouye, M. A single amino acid determinant of the membrane localization of lipoproteins in E. coli. Cell 1988, 53, 423–432. [Google Scholar] [CrossRef]
- Yakushi, T.; Masuda, K.; Narita, S.; Matsuyama, S.; Tokuda, H. A new ABC transporter mediating the detachment of lipid-modified proteins from membranes. Nat. Cell Biol. 2000, 2, 212–218. [Google Scholar] [CrossRef]
- Matsuyama, S.; Tajima, T.; Tokuda, H. A novel periplasmic carrier protein involved in the sorting and transport of Escherichia coli lipoproteins destined for the outer membrane. EMBO J. 1995, 14, 3365–3372. [Google Scholar] [CrossRef]
- Matsuyama, S.I.; Yokota, N.; Tokuda, H. A novel outer membrane lipoprotein, LolB (HemM), involved in the LolA (p20)-dependent localization of lipoproteins to the outer membrane of Escherichia coli. EMBO J. 1997, 16, 6947–6955. [Google Scholar] [CrossRef]
- Grabowicz, M.; Silhavy, T.J. Redefining the essential trafficking pathway for outer membrane lipoproteins. Proc. Natl. Acad. Sci. USA 2017, 114, 4769–4774. [Google Scholar] [CrossRef] [Green Version]
- Mcleod, S.M.; Fleming, P.R.; MacCormack, K.; Mclaughlin, R.E.; Whiteaker, J.D.; Narita, S.; Mori, M.; Tokuda, H.; Miller, A. Small-Molecule Inhibitors of Gram-Negative Lipoprotein Trafficking Discovered by Phenotypic Screening. J. Bacteriol. 2015, 197, 1075–1082. [Google Scholar] [CrossRef] [Green Version]
- Nayar, A.S.; Dougherty, T.J.; Ferguson, K.E.; Granger, B.A.; Mcwilliams, L.; Stacey, C.; Leach, L.J.; Narita, S.; Tokuda, H.; Miller, A.A.; et al. Novel Antibacterial Targets and Compounds Revealed by a High- Throughput Cell Wall Reporter Assay. J. Bacteriol. 2015, 197, 1726–1734. [Google Scholar] [CrossRef]
- Paradis-bleau, C.; Markovski, M.; Uehara, T.; Lupoli, T.J.; Walker, S.; Kahne, D.E.; Bernhardt, T.G. Lipoprotein Cofactors Located in the Outer Membrane Activate Bacterial Cell Wall Polymerases. Cell 2010, 143, 1110–1120. [Google Scholar] [CrossRef] [Green Version]
- Typas, A.; Banzhaf, M.; Van Saparoea, B.V.D.B.; Biboy, J.; Nichols, R.J.; Zietek, M.; Beilharz, K.; Von Rechenberg, M.; Breukink, E.; Blaauwen, T.D.; et al. Regulation of peptidoglycan synthesis by outer membrane proteins. Cell 2010, 143, 1097–1109. [Google Scholar] [CrossRef]
- Nickerson, N.N.; Jao, C.C.; Xu, Y.; Quinn, J.; Skippington, E.; Alexander, K.; Miu, A.; Skelton, N.; Hankins, J.V.; Lopez, M.S.; et al. A Novel Inhibitor of the LolCDE ABC Transporter Essential for Lipoprotein Trafficking in Gram-Negative Bacteria. Antimicrob. Agents Chemother. 2018, 62, e02151-17. [Google Scholar] [CrossRef]
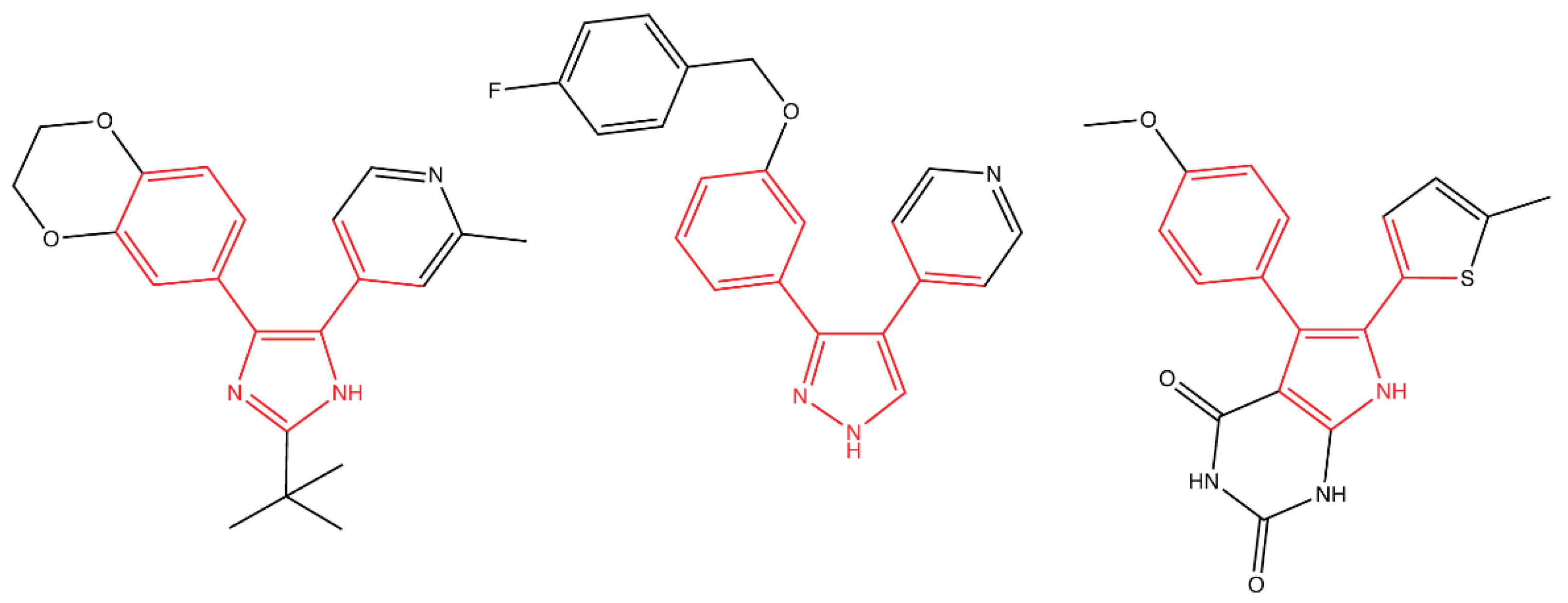
- Pathania, R.; Zlitni, S.; Barker, C.; Das, R.; Gerritsma, D.A.; Lebert, J.; Awuah, E.; Melacini, G.; Capretta, F.A.; Brown, E.D. Chemical genomics in Escherichia coli identifies an inhibitor of bacterial lipoprotein targeting. Nat. Chem. Biol. 2009, 5, 849–856. [Google Scholar] [CrossRef]
- Barker, C.A.; Allison, S.E.; Zlitni, S.; Nguyen, N.D.; Das, R.; Melacini, G.; Capretta, A.A.; Brown, E.D. Degradation of MAC13243 and studies of the interaction of resulting thiourea compounds with the lipoprotein targeting chaperone LolA. Bioorganic Med. Chem. Lett. 2013, 23, 2426–2431. [Google Scholar] [CrossRef] [Green Version]
- Muheim, C.; Götzke, H.; Eriksson, A.U.; Lindberg, S.; Lauritsen, I.; Nørholm, M.H.H.; Daley, D.O. Increasing the permeability of Escherichia coli using MAC13243. Sci. Rep. 2017, 7, 17629. [Google Scholar] [CrossRef]
- Buss, J.A.; Baidin, V.; Welsh, M.A.; Flores-Kim, J.; Cho, H.; Wood, B.M.; Uehara, T.; Walker, S.; Kahne, D.; Bernhardt, T.G. Pathway-Directed Screen for Inhibitors of the Bacterial Cell Elongation Machinery. Antimicrob. Agents Chemother. 2019, 63, e01530-18. [Google Scholar] [CrossRef]
- Richter, M.F.; Drown, B.S.; Riley, A.P.; Garcia, A.; Shirai, T.; Svec, R.L.; Hergenrother, P.J. Predictive compound accumulation rules yield a broad-spectrum antibiotic. Nature 2017, 545, 299–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lehman, K.M.; Grabowicz, M. Countering Gram-Negative Antibiotic Resistance: Recent Progress in Disrupting the Outer Membrane with Novel Therapeutics. Antibiotics 2019, 8, 163. https://doi.org/10.3390/antibiotics8040163
Lehman KM, Grabowicz M. Countering Gram-Negative Antibiotic Resistance: Recent Progress in Disrupting the Outer Membrane with Novel Therapeutics. Antibiotics. 2019; 8(4):163. https://doi.org/10.3390/antibiotics8040163
Chicago/Turabian StyleLehman, Kelly M., and Marcin Grabowicz. 2019. "Countering Gram-Negative Antibiotic Resistance: Recent Progress in Disrupting the Outer Membrane with Novel Therapeutics" Antibiotics 8, no. 4: 163. https://doi.org/10.3390/antibiotics8040163
APA StyleLehman, K. M., & Grabowicz, M. (2019). Countering Gram-Negative Antibiotic Resistance: Recent Progress in Disrupting the Outer Membrane with Novel Therapeutics. Antibiotics, 8(4), 163. https://doi.org/10.3390/antibiotics8040163