The Onset of Tacrolimus Biosynthesis in Streptomyces tsukubaensis Is Dependent on the Intracellular Redox Status

 , and
, and
Abstract
:1. Introduction
2. Results
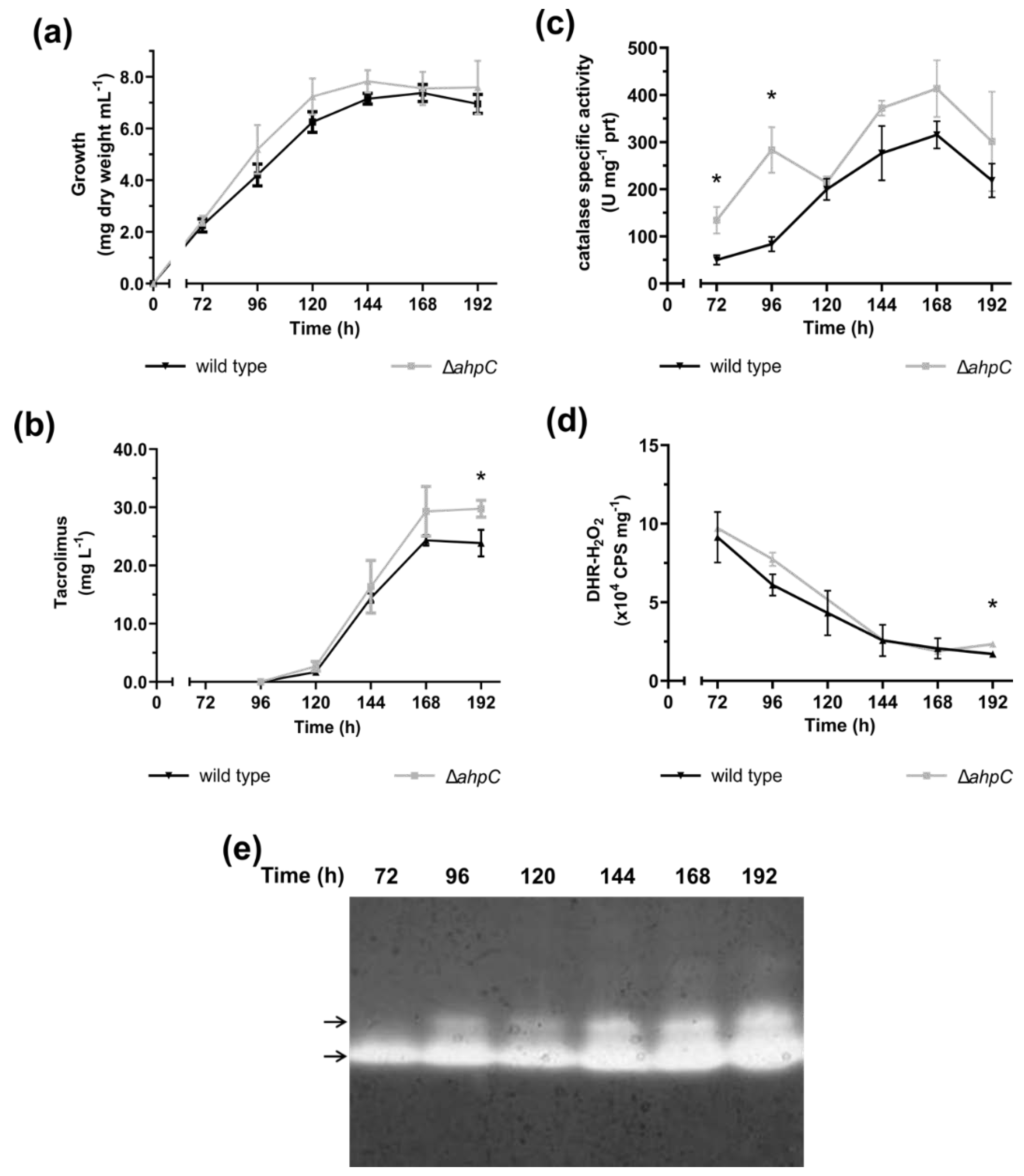
2.1. The Onset of Tacrolimus Biosynthesis Overlaps with the Induction of Catalase Activity
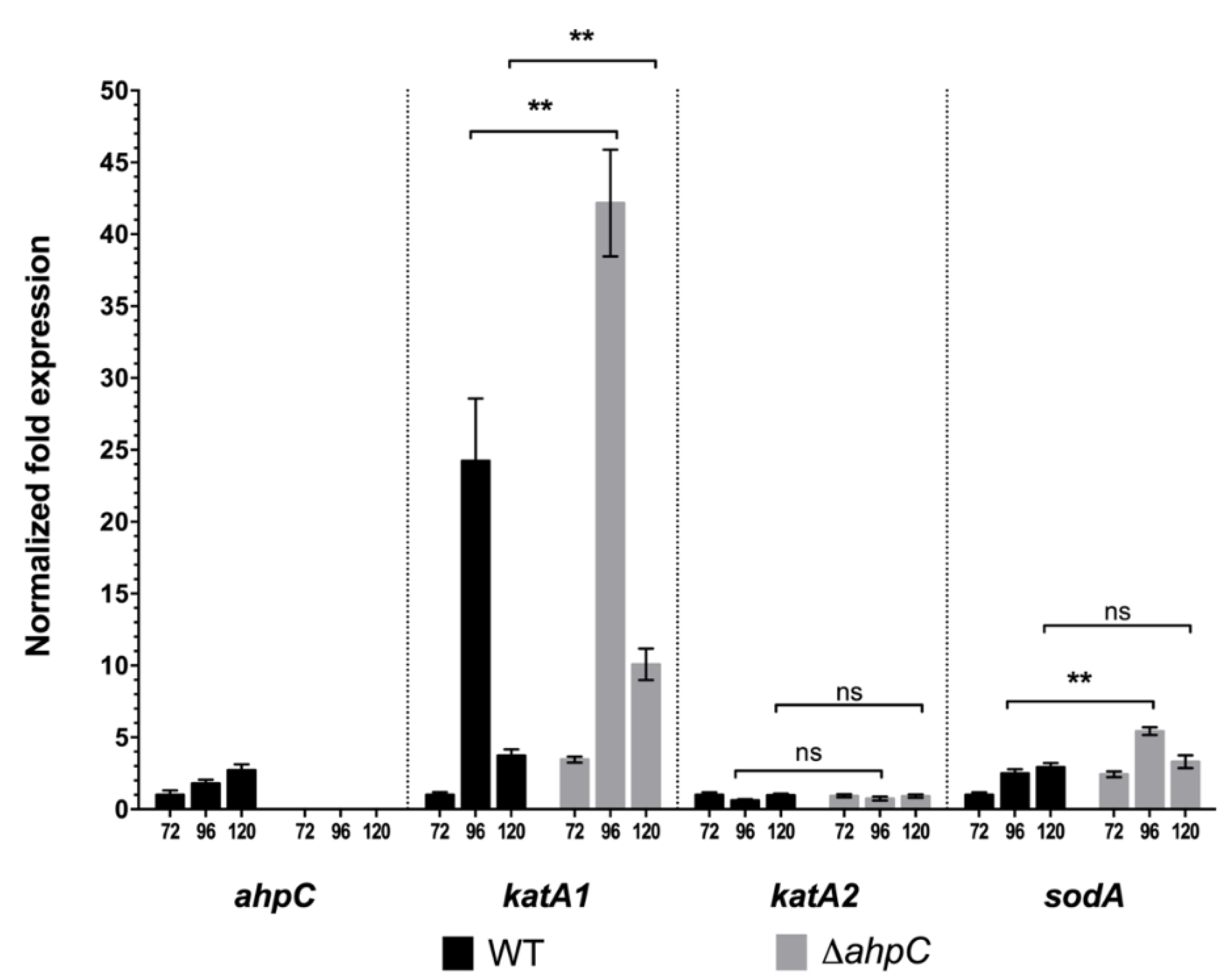
2.2. An Altered Oxidative Stress Response Leads to Tacrolimus Overproduction
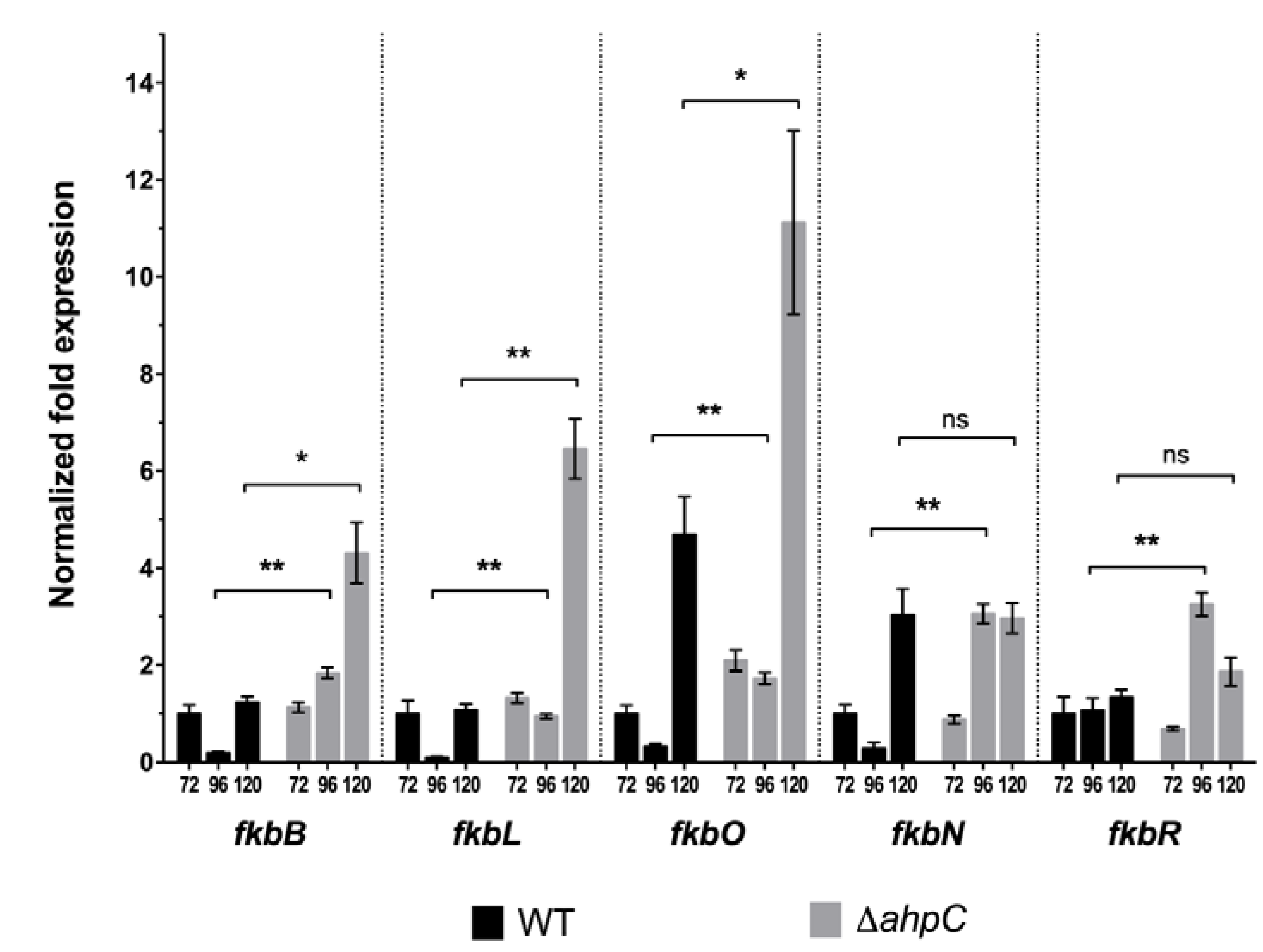
2.3. S. tsukubaensis ∆ahpC Displays a Redirection of the Metabolic Flux towards Tacrolimus Production
2.4. Tacrolimus Displays Antioxidant Activity
2.5. The Onset of Tacrolimus Biosynthesis Is Preceded by a Repression of the Oxidative Metabolism
3. Discussion
4. Materials and Methods
4.1. Bacterial Strains and Growth Conditions
4.2. Generation of Streptomyces tsukubaensis ∆ahpC Strain
4.3. Bioassays
4.4. Protein Crude Extracts and Quantification
4.5. Catalase Activity Determination
4.6. SOD Activity Determination
4.7. Quantification of Intracellular ROS Levels
4.8. Two-Dimensional Electrophoresis (2-DE) and Protein Identification
4.9. Tacrolimus Quantification
4.10. RNA Isolation and RT-qPCR
4.11. Microarray Data Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Berdy, J. Thoughts and facts about antibiotics: Where we are now and where we are heading. J. Antibiot. 2012, 65, 385–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krentz, A.J.; Dmitrewski, J.; Mayer, D.; McMaster, P.; Buckels, J.; Smith, J.M.; Nattrass, M. Tacrolimus (FK506) versus cyclosporin in prevention of liver allograft rejection. Lancet 1994, 344, 948–949. [Google Scholar] [CrossRef] [PubMed]
- Kino, T.; Hatanaka, H.; Hashimoto, M.; Nishiyama, M.; Goto, T.; Okuhara, M.; Kohsaka, M.; Aoki, H.; Imanaka, H. FK-506, a novel immunosuppressant isolated from a Streptomyces. I. Fermentation, isolation, and physico-chemical and biological characteristics. J. Antibiot. 1987, 40, 1249–1255. [Google Scholar] [CrossRef] [Green Version]
- Barreiro, C.; Martinez-Castro, M. Trends in the biosynthesis and production of the immunosuppressant tacrolimus (FK506). Appl. Microbiol. Biotechnol. 2014, 98, 497–507. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Castro, M.; Salehi-Najafabadi, Z.; Romero, F.; Perez-Sanchiz, R.; Fernandez-Chimeno, R.I.; Martin, J.F.; Barreiro, C. Taxonomy and chemically semi-defined media for the analysis of the tacrolimus producer Streptomyces tsukubaensis. Appl. Microbiol. Biotechnol. 2013, 97, 2139–2152. [Google Scholar] [CrossRef]
- Ordonez-Robles, M.; Santos-Beneit, F.; Albillos, S.M.; Liras, P.; Martin, J.F.; Rodriguez-Garcia, A. Streptomyces tsukubaensis as a new model for carbon repression: Transcriptomic response to tacrolimus repressing carbon sources. Appl. Microbiol. Biotechnol. 2017, 101, 8181–8195. [Google Scholar] [CrossRef]
- Xia, M.; Huang, D.; Li, S.; Wen, J.; Jia, X.; Chen, Y. Enhanced FK506 production in Streptomyces tsukubaensis by rational feeding strategies based on comparative metabolic profiling analysis. Biotechnol. Bioeng. 2013, 110, 2717–2730. [Google Scholar] [CrossRef]
- Wang, C.; Liu, J.; Liu, H.; Liang, S.; Wen, J. Combining metabolomics and network analysis to improve tacrolimus production in Streptomyces tsukubaensis using different exogenous feedings. J. Ind. Microbiol. Biotechnol. 2017. [Google Scholar] [CrossRef]
- Huang, D.; Xia, M.; Li, S.; Wen, J.; Jia, X. Enhancement of FK506 production by engineering secondary pathways of Streptomyces tsukubaensis and exogenous feeding strategies. J. Ind. Microbiol. Biotechnol. 2013, 40, 1023–1037. [Google Scholar] [CrossRef]
- Nielsen, J.B.; Hsu, M.J.; Byrne, K.M.; Kaplan, L. Biosynthesis of the immunosuppressant immunomycin: The enzymology of pipecolate incorporation. Biochemistry 1991, 30, 5789–5796. [Google Scholar] [CrossRef]
- Wang, C.; Huang, D.; Liang, S. Identification and metabolomic analysis of chemical elicitors for tacrolimus accumulation in Streptomyces tsukubaensis. Appl. Microbiol. Biotechnol. 2018, 102, 7541–7553. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Li, S.; Xia, M.; Wen, J.; Jia, X. Genome-scale metabolic network guided engineering of Streptomyces tsukubaensis for FK506 production improvement. Microb. Cell Fact. 2013, 12, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ordonez-Robles, M.; Rodriguez-Garcia, A.; Martin, J.F. Target genes of the Streptomyces tsukubaensis FkbN regulator include most of the tacrolimus biosynthesis genes, a phosphopantetheinyl transferase and other PKS genes. Appl. Microbiol. Biotechnol. 2016, 100, 8091–8103. [Google Scholar] [CrossRef] [PubMed]
- Goranovic, D.; Blazic, M.; Magdevska, V.; Horvat, J.; Kuscer, E.; Polak, T.; Santos-Aberturas, J.; Martinez-Castro, M.; Barreiro, C.; Mrak, P.; et al. FK506 biosynthesis is regulated by two positive regulatory elements in Streptomyces tsukubaensis. BMC Microbiol. 2012, 12, 238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.S.; Luo, H.D.; Tao, Y.; Wang, Y.Y.; Jiang, X.H.; Jiang, H.; Li, Y.Q. FkbN and Tcs7 are pathway-specific regulators of the FK506 biosynthetic gene cluster in Streptomyces tsukubaensis L19. J. Ind. Microbiol. Biotechnol. 2016, 43, 1693–1703. [Google Scholar] [CrossRef] [PubMed]
- Mo, S.; Yoo, Y.J.; Ban, Y.H.; Lee, S.K.; Kim, E.; Suh, J.W.; Yoon, Y.J. Roles of fkbN in positive regulation and tcs7 in negative regulation of FK506 biosynthesis in Streptomyces sp. strain KCTC 11604BP. Appl. Environ. Microbiol. 2012, 78, 2249–2255. [Google Scholar] [CrossRef] [Green Version]
- Mo, S.; Ban, Y.H.; Park, J.W.; Yoo, Y.J.; Yoon, Y.J. Enhanced FK506 production in Streptomyces clavuligerus CKD1119 by engineering the supply of methylmalonyl-CoA precursor. J. Ind. Microbiol. Biotechnol. 2009, 36, 1473–1482. [Google Scholar] [CrossRef]
- Poshekhontseva, V.Y.; Fokina, V.V.; Sukhodolskaya, G.V.; Shutov, A.A.; Donova, M.V. Effect of starch composition on the biosynthesis of immunosuppressant tacrolimus (FK-506) by Streptomyces tsukubaensis VKM Ac-2618D strain. Appl. Biochem. Microbiol. 2019, 55, 534–543. [Google Scholar] [CrossRef]
- van Wezel, G.P.; McDowall, K.J. The regulation of the secondary metabolism of Streptomyces: New links and experimental advances. Nat. Prod. Rep. 2011, 28, 1311–1333. [Google Scholar] [CrossRef]
- Xia, H.; Zhan, X.; Mao, X.M.; Li, Y.Q. The regulatory cascades of antibiotic production in Streptomyces. World J. Microbiol. Biotechnol. 2020, 36, 13. [Google Scholar] [CrossRef]
- Romero-Rodriguez, A.; Maldonado-Carmona, N.; Ruiz-Villafan, B.; Koirala, N.; Rocha, D.; Sanchez, S. Interplay between carbon, nitrogen and phosphate utilization in the control of secondary metabolite production in Streptomyces. Antonie Leeuwenhoek 2018, 111, 761–781. [Google Scholar] [CrossRef] [PubMed]
- Esnault, C.; Dulermo, T.; Smirnov, A.; Askora, A.; David, M.; Deniset-Besseau, A.; Holland, I.B.; Virolle, M.J. Strong antibiotic production is correlated with highly active oxidative metabolism in Streptomyces coelicolor M145. Sci. Rep. 2017, 7, 200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Millan-Oropeza, A.; Henry, C.; Blein-Nicolas, M.; Aubert-Frambourg, A.; Moussa, F.; Bleton, J.; Virolle, M.J. Quantitative proteomics analysis confirmed oxidative metabolism predominates in Streptomyces coelicolor versus glycolytic metabolism in Streptomyces lividans. J. Proteome Res. 2017, 16, 2597–2613. [Google Scholar] [CrossRef] [PubMed]
- Rui, B.; Shen, T.; Zhou, H.; Liu, J.; Chen, J.; Pan, X.; Liu, H.; Wu, J.; Zheng, H.; Shi, Y. A systematic investigation of Escherichia coli central carbon metabolism in response to superoxide stress. BMC Syst. Biol. 2010, 4, 122. [Google Scholar] [CrossRef] [Green Version]
- Beites, T.; Pires, S.D.S.; Santos, C.L.; Osorio, H.; Moradas-Ferreira, P.; Mendes, M.V. Crosstalk between ROS homeostasis and secondary metabolism in S. natalensis ATCC 27448: Modulation of pimaricin production by intracellular ROS. PLoS ONE 2011, 6, e27472. [Google Scholar] [CrossRef]
- Beites, T.; Rodriguez-Garcia, A.; Santos-Beneit, F.; Moradas-Ferreira, P.; Aparicio, J.F.; Mendes, M.V. Genome-wide analysis of the regulation of pimaricin production in Streptomyces natalensis by reactive oxygen species. Appl. Microbiol. Biotechnol. 2014, 98, 2231–2241. [Google Scholar] [CrossRef]
- Cheng, Y.; Yang, R.; Lyu, M.; Wang, S.; Liu, X.; Wen, Y.; Song, Y.; Li, J.; Chen, Z. IdeR, a DtxR family iron response regulator, controls iron homeostasis, morphological differentiation, secondary metabolism, and the oxidative stress response in Streptomyces avermitilis. Appl. Environ. Microbiol. 2018, 84. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Liu, H.; Huang, D.; Jin, L.; Wang, C.; Wen, J. Comparative proteomic and metabolomic analysis of Streptomyces tsukubaensis reveals the metabolic mechanism of FK506 overproduction by feeding soybean oil. Appl. Microbiol. Biotechnol. 2017, 101, 2447–2465. [Google Scholar] [CrossRef]
- Seaver, L.C.; Imlay, J.A. Alkyl hydroperoxide reductase is the primary scavenger of endogenous hydrogen peroxide in Escherichia coli. J. Bacteriol. 2001, 183, 7173–7181. [Google Scholar] [CrossRef] [Green Version]
- Walker, G.E.; Dunbar, B.; Hunter, I.S.; Nimmo, H.G.; Coggins, J.R. A catalase from Streptomyces coelicolor A3(2). Microbiology 1995, 141, 1377–1383. [Google Scholar] [CrossRef] [Green Version]
- Feng, J.; Jiang, J.; Liu, Y.; Li, W.; Azat, R.; Zheng, X.; Zhou, W.W. Significance of oxygen carriers and role of liquid paraffin in improving validamycin A production. J. Ind. Microbiol. Biotechnol. 2016, 43, 1365–1372. [Google Scholar] [CrossRef] [PubMed]
- Brekasis, D.; Paget, M.S. A novel sensor of NADH/NAD+ redox poise in Streptomyces coelicolor A3(2). EMBO J. 2003, 22, 4856–4865. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Cheng, Y.; Lyu, M.; Wen, Y.; Song, Y.; Chen, Z.; Li, J. Redox-sensing regulator Rex regulates aerobic metabolism, morphological differentiation, and avermectin production in Streptomyces avermitilis. Sci. Rep. 2017, 7, 44567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawers, R.G.; Falke, D.; Fischer, M. Oxygen and nitrate respiration in Streptomyces coelicolor A3(2). Adv. Microb. Physiol. 2016, 68, 1–40. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.S.; Kim, M.; Kim, E.J.; Kim, B.G. Production of pikromycin using branched chain amino acid catabolism in Streptomyces venezuelae ATCC 15439. J. Ind. Microbiol. Biotechnol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Liu, J.; Liu, H.; Wang, J.; Wen, J. A genome-scale dynamic flux balance analysis model of Streptomyces tsukubaensis NRRL18488 to predict the targets for increasing FK506 production. Biochem. Eng. J. 2017, 2017. [Google Scholar] [CrossRef]
- Begley, T.P.; Kinsland, C.; Strauss, E. The biosynthesis of coenzyme A in bacteria. Vitam. Horm. 2001, 61, 157–171. [Google Scholar] [PubMed]
- Li, L.; Jiang, W.; Lu, Y. A novel two-component system, GluR-GluK, involved in glutamate sensing and uptake in Streptomyces coelicolor. J. Bacteriol. 2017. [Google Scholar] [CrossRef] [Green Version]
- Gubbens, J.; Janus, M.M.; Florea, B.I.; Overkleeft, H.S.; van Wezel, G.P. Identification of glucose kinase-dependent and -independent pathways for carbon control of primary metabolism, development and antibiotic production in Streptomyces coelicolor by quantitative proteomics. Mol. Microbiol. 2012, 86, 1490–1507. [Google Scholar] [CrossRef]
- Ordonez-Robles, M.; Santos-Beneit, F.; Martin, J.F. Unraveling nutritional regulation of tacrolimus biosynthesis in Streptomyces tsukubaensis through omic approaches. Antibiotics 2018, 7, 39. [Google Scholar] [CrossRef] [Green Version]
- Ordonez-Robles, M.; Santos-Beneit, F.; Rodriguez-Garcia, A.; Martin, J.F. Analysis of the Pho regulon in Streptomyces tsukubaensis. Microbiol. Res. 2017, 205, 80–87. [Google Scholar] [CrossRef]
- Novotna, J.; Vohradsky, J.; Berndt, P.; Gramajo, H.; Langen, H.; Li, X.M.; Minas, W.; Orsaria, L.; Roeder, D.; Thompson, C.J. Proteomic studies of diauxic lag in the differentiating prokaryote Streptomyces coelicolor reveal a regulatory network of stress-induced proteins and central metabolic enzymes. Mol. Microbiol. 2003, 48, 1289–1303. [Google Scholar] [CrossRef] [PubMed]
- Thomas, L.; Hodgson, D.A.; Wentzel, A.; Nieselt, K.; Ellingsen, T.E.; Moore, J.; Morrissey, E.R.; Legaie, R.; Consortium, S.; Wohlleben, W.; et al. Metabolic switches and adaptations deduced from the proteomes of Streptomyces coelicolor wild type and phoP mutant grown in batch culture. Mol. Cell Proteom. 2012, 11, M111 013797. [Google Scholar] [CrossRef] [Green Version]
- Yin, P.; Li, Y.Y.; Zhou, J.; Wang, Y.H.; Zhang, S.L.; Ye, B.C.; Ge, W.F.; Xia, Y.L. Direct proteomic mapping of Streptomyces avermitilis wild and industrial strain and insights into avermectin production. J. Proteom. 2013, 79, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Beites, T.; Oliveira, P.; Rioseras, B.; Pires, S.D.S.; Oliveira, R.; Tamagnini, P.; Moradas-Ferreira, P.; Manteca, A.; Mendes, M.V. Streptomyces natalensis programmed cell death and morphological differentiation are dependent on oxidative stress. Sci. Rep. 2015, 5, 12887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seaver, L.C.; Imlay, J.A. Are respiratory enzymes the primary sources of intracellular hydrogen peroxide? J. Biol. Chem. 2004, 279, 48742–48750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, A.C.; Gust, B.; Kulik, A.; Heide, L.; Buttner, M.J.; Bibb, M.J. Phage p1-derived artificial chromosomes facilitate heterologous expression of the FK506 gene cluster. PLoS ONE 2013, 8, e69319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kieser, T.; Bibb, M.; Buttner, M.; Chater, K.; Hopwood, D.A. Practical Streptomyces Genetics; John Innes Centre: Norwich, UK, 2000. [Google Scholar]
- Gust, B.; Challis, G.L.; Fowler, K.; Kieser, T.; Chater, K.F. PCR-targeted Streptomyces gene replacement identifies a protein domain needed for biosynthesis of the sesquiterpene soil odor geosmin. Proc. Natl. Acad. Sci. USA 2003, 100, 1541–1546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beers, R.F., Jr.; Sizer, I.W. A spectrophotometric method for measuring the breakdown of hydrogen peroxide by catalase. J. Biol. Chem. 1952, 195, 133–140. [Google Scholar]
- Clare, D.A.; Duong, M.N.; Darr, D.; Archibald, F.; Fridovich, I. Effects of molecular oxygen on detection of superoxide radical with nitroblue tetrazolium and on activity stains for catalase. Anal. Biochem. 1984, 140, 532–537. [Google Scholar] [CrossRef]
- Beauchamp, C.; Fridovich, I. Superoxide dismutase: Improved assays and an assay applicable to acrylamide gels. Anal. Biochem. 1971, 44, 276–287. [Google Scholar] [CrossRef]
- Gorg, A.; Obermaier, C.; Boguth, G.; Harder, A.; Scheibe, B.; Wildgruber, R.; Weiss, W. The current state of two-dimensional electrophoresis with immobilized pH gradients. Electrophoresis 2000, 21, 1037–1053. [Google Scholar] [CrossRef]
- Goromova, I.; Celis, J.E. Protein detection in gels by silver staining: A procedure compatible with mass-spectrometry. In Cell Biology: A Laboratory Handbook, 3rd ed.; Celis, J.E., Carter, N., Hunter, T., Simons, K., Small, J.V., Shotton, D., Eds.; Elsevier Academic Press: Amsterdam, The Netherlands, 2006. [Google Scholar]
- Perkins, D.N.; Pappin, D.J.; Creasy, D.M.; Cottrell, J.S. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 1999, 20, 3551–3567. [Google Scholar] [CrossRef]
- The UniProt Consortium. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2018, 46, 2699. [Google Scholar] [CrossRef] [Green Version]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef] [PubMed]
- Vandesompele, J.; De Preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3, 34. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | SCO Orthologue | Predicted Product | ∆ahpC vs. wt Fold Variation |
|---|---|---|---|
| Oxidative Stress Response | |||
| STSU_11585 | SCO5032 | alkyl hydroperoxide reductase | only in wt |
| Protein metabolism, translation and modification | |||
| STSU_08394 | SCO5699 | Prolyl-tRNA synthetase | 0.21 1 |
| STSU_13455 | SCO4662 | Elongation factor Tu-1 | 0.37 |
| STSU_17678 | SCO3906 | 30S ribosomal protein S6 (RpsF) | 0.32 |
| STSU_28812 | SCO1648 | AAA ATPase central domain-containing protein | 0.59 |
| Energy and carbon metabolism | |||
| STSU_10154 | SCO5374 | ATP synthase subunit epsilon (AtpC) | 0.42 |
| STSU_11515 | SCO5044 | Fumarate hydratase (FumB) | 0.38 |
| STSU_12400 | SCO4921 | putative acyl-CoA carboxylase complex A subunit | 0.09 |
| STSU_12680 | SCO4855 | succinate dehydrogenase iron-sulfur subunit (SdhB) | 0.50 1 |
| STSU_30056 | SCO1391 | Phosphoenolpyruvate-protein phosphotransferase (EI component) | 0.45 |
| Amino acid metabolism | |||
| STSU_14552 | SCO3345 | Dihydroxy-acid dehydratase (IlvD) | 0.29 |
| STSU_24776 | SCO2528 | 2-isopropylmalate synthase (LeuA) | 0.21 1 |
| STSU_26189 | SCO2198 | Glutamine synthetase I (GlnA) | 0.23 |
| Hypothetical/uncharacterized proteins/not classified | |||
| STSU_10084 | SCO5389 | Hypothetical protein | 2.00 |
| STSU_13630 | SCO4637 | Hypothetical protein | 0.44 |
| STSU_30145 | SCO1374 | Putative secreted protein | 3.08 |
| STSU_31495 | SCO1116 | Hypothetical protein | 2.01 |
| STSU_33250 | SCO0167 | UspA domain-containing protein | 0.50 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pires, S.D.S.; Oliveira, R.; Moradas-Ferreira, P.; V. Mendes, M. The Onset of Tacrolimus Biosynthesis in Streptomyces tsukubaensis Is Dependent on the Intracellular Redox Status. Antibiotics 2020, 9, 703. https://doi.org/10.3390/antibiotics9100703
Pires SDS, Oliveira R, Moradas-Ferreira P, V. Mendes M. The Onset of Tacrolimus Biosynthesis in Streptomyces tsukubaensis Is Dependent on the Intracellular Redox Status. Antibiotics. 2020; 9(10):703. https://doi.org/10.3390/antibiotics9100703
Chicago/Turabian StylePires, Sílvia D. S., Rute Oliveira, Pedro Moradas-Ferreira, and Marta V. Mendes. 2020. "The Onset of Tacrolimus Biosynthesis in Streptomyces tsukubaensis Is Dependent on the Intracellular Redox Status" Antibiotics 9, no. 10: 703. https://doi.org/10.3390/antibiotics9100703
APA StylePires, S. D. S., Oliveira, R., Moradas-Ferreira, P., & V. Mendes, M. (2020). The Onset of Tacrolimus Biosynthesis in Streptomyces tsukubaensis Is Dependent on the Intracellular Redox Status. Antibiotics, 9(10), 703. https://doi.org/10.3390/antibiotics9100703