λ Recombineering Used to Engineer the Genome of Phage T7


Abstract
:1. Introduction
2. Results and Discussion
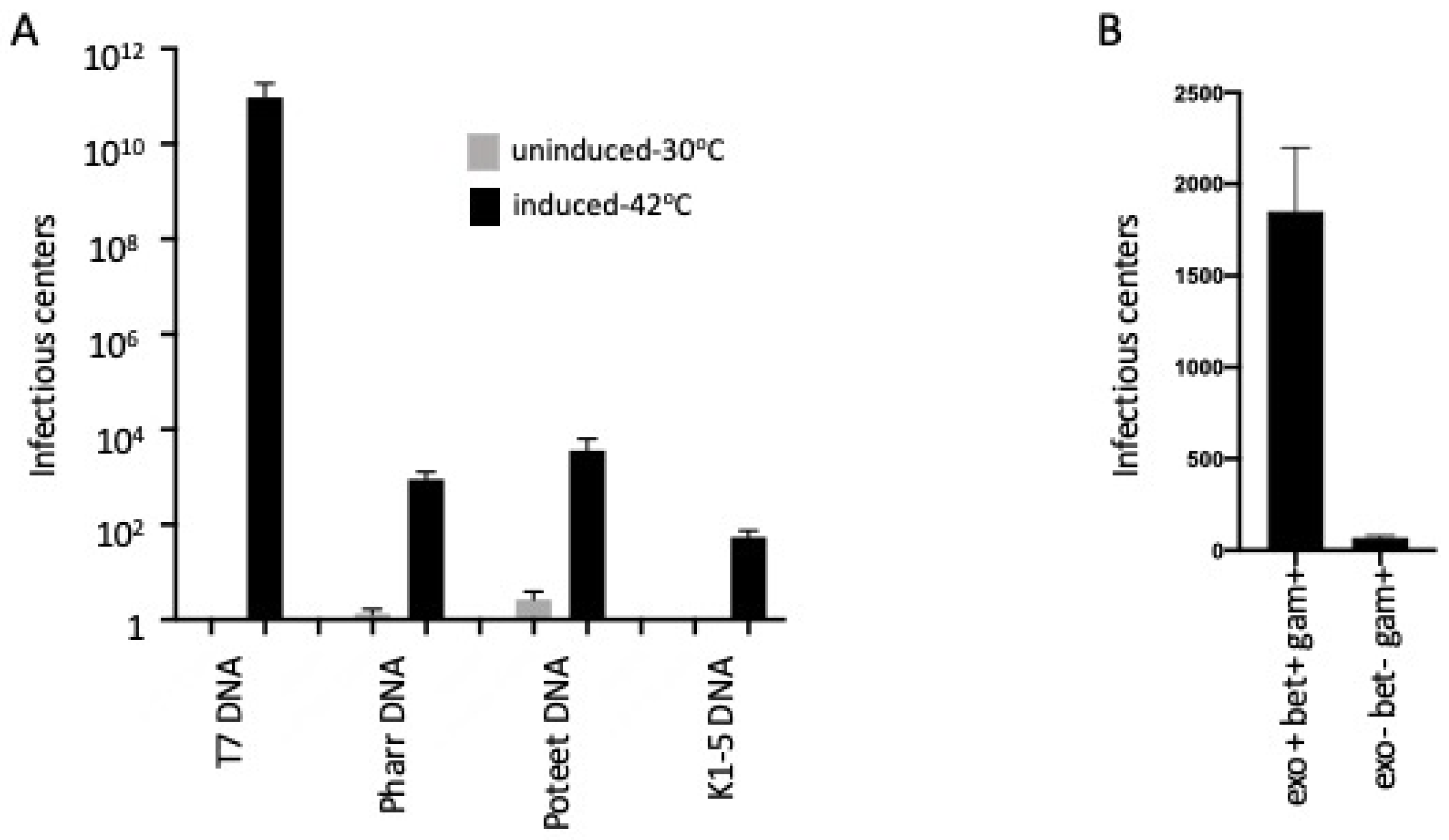
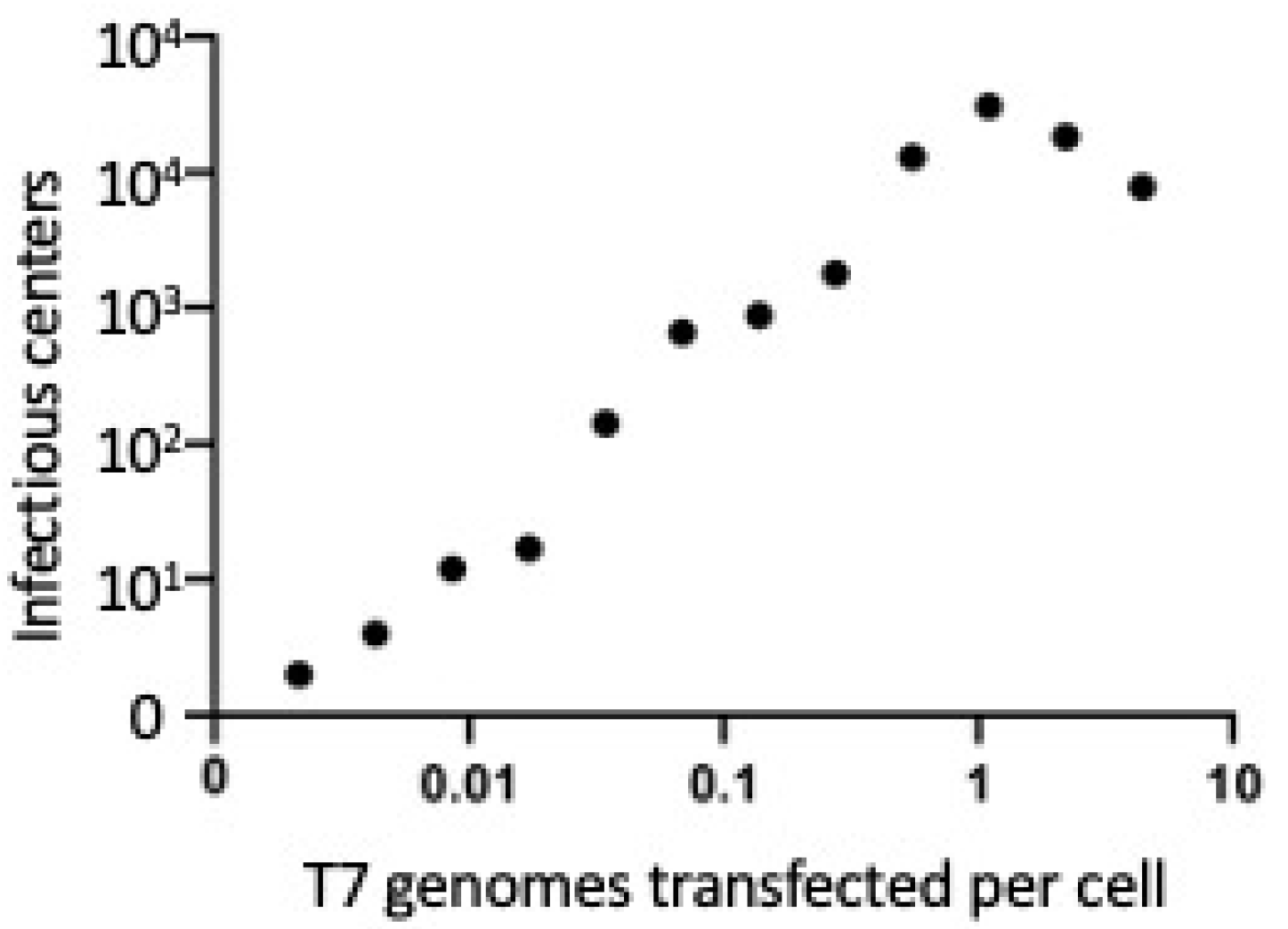
2.1. λ Red-Mediated Recombination Enhances Transfection of T7 and T7-Like Phages
2.2. T7 Transfection Requires the Exo and Bet Gene Functions and Is Independent of the Host RecA Function
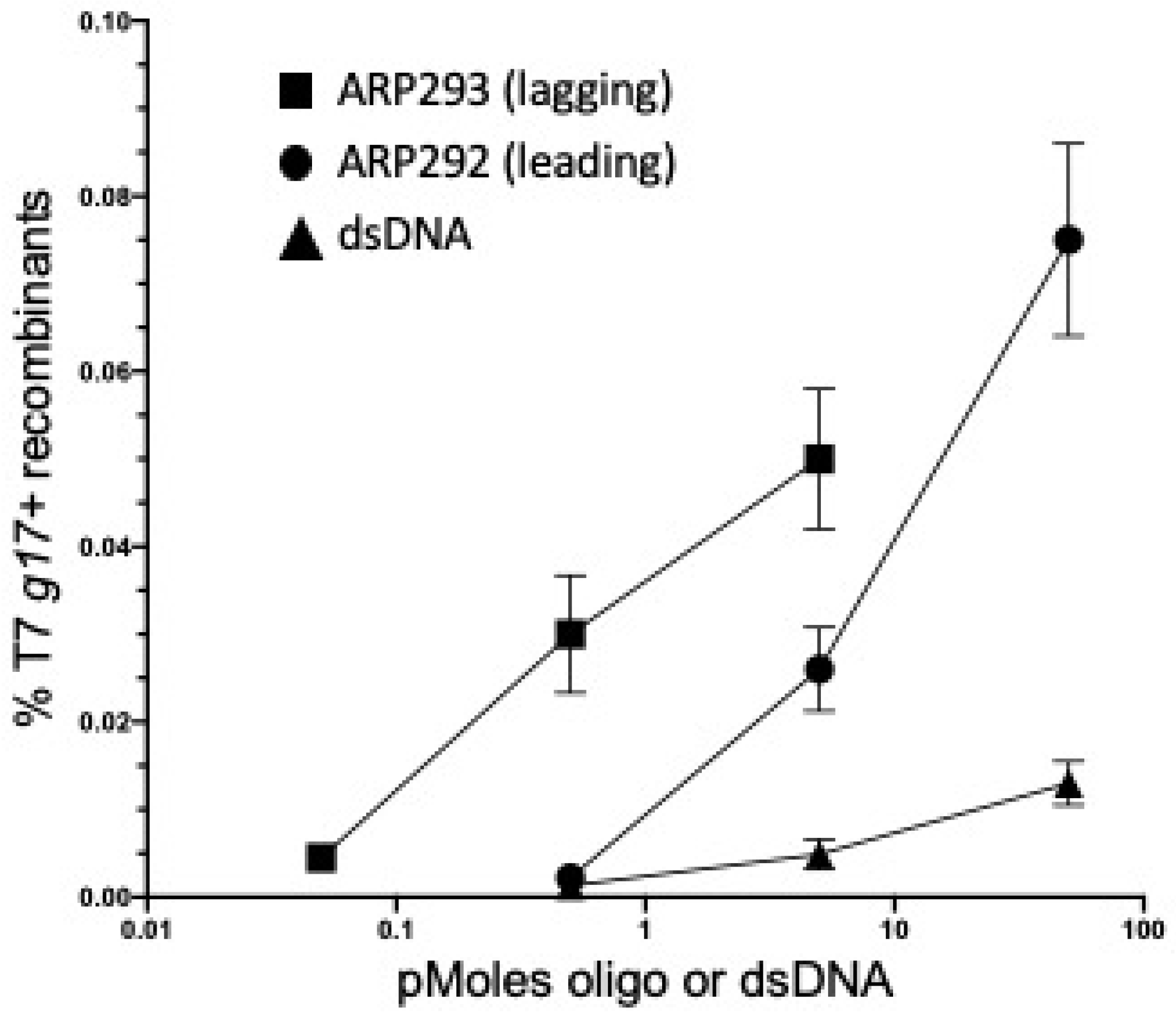
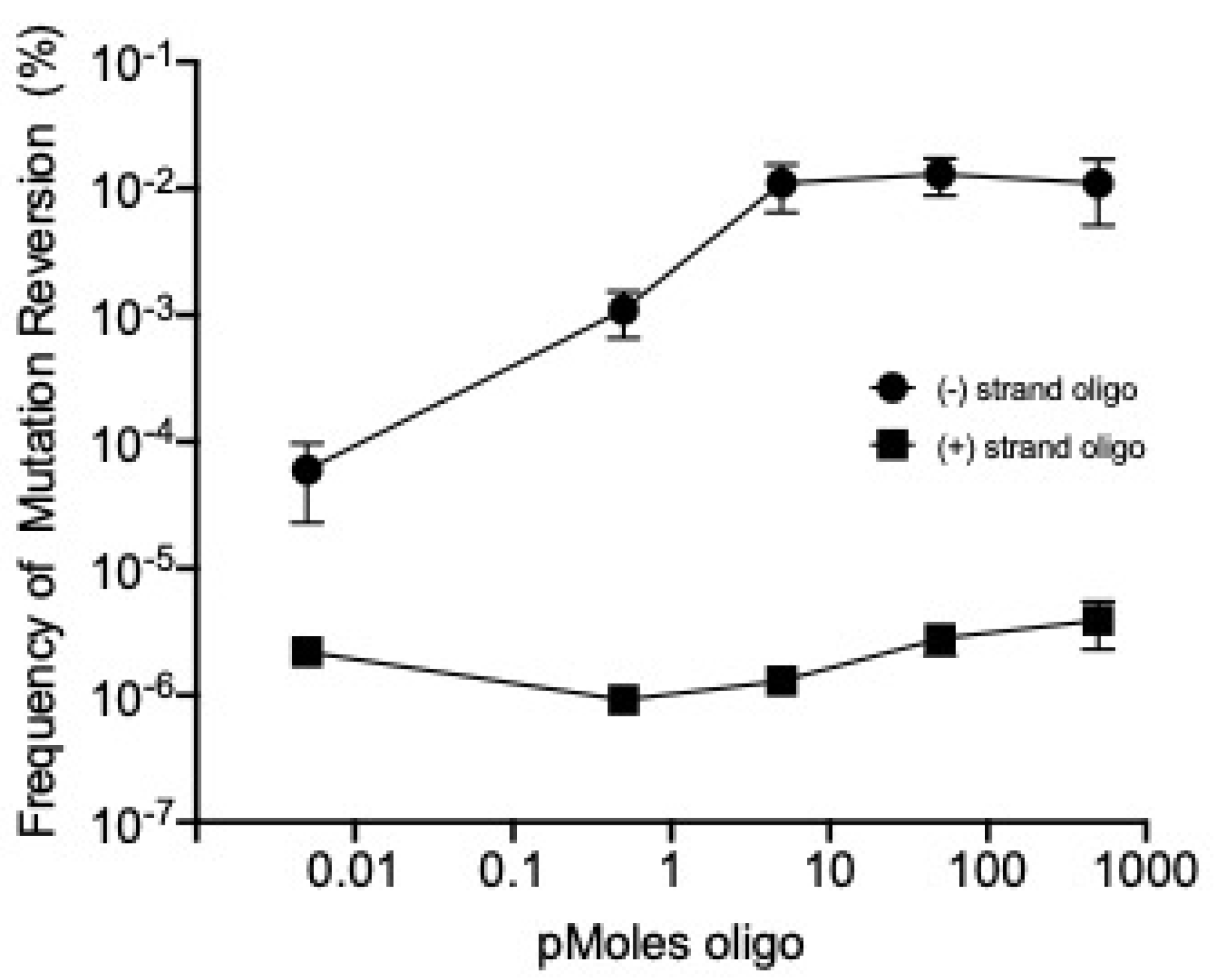
2.3. λ Red-Mediated Oligo Recombination Enables Directed Nucleotide Changes within the Phage T7 Genome
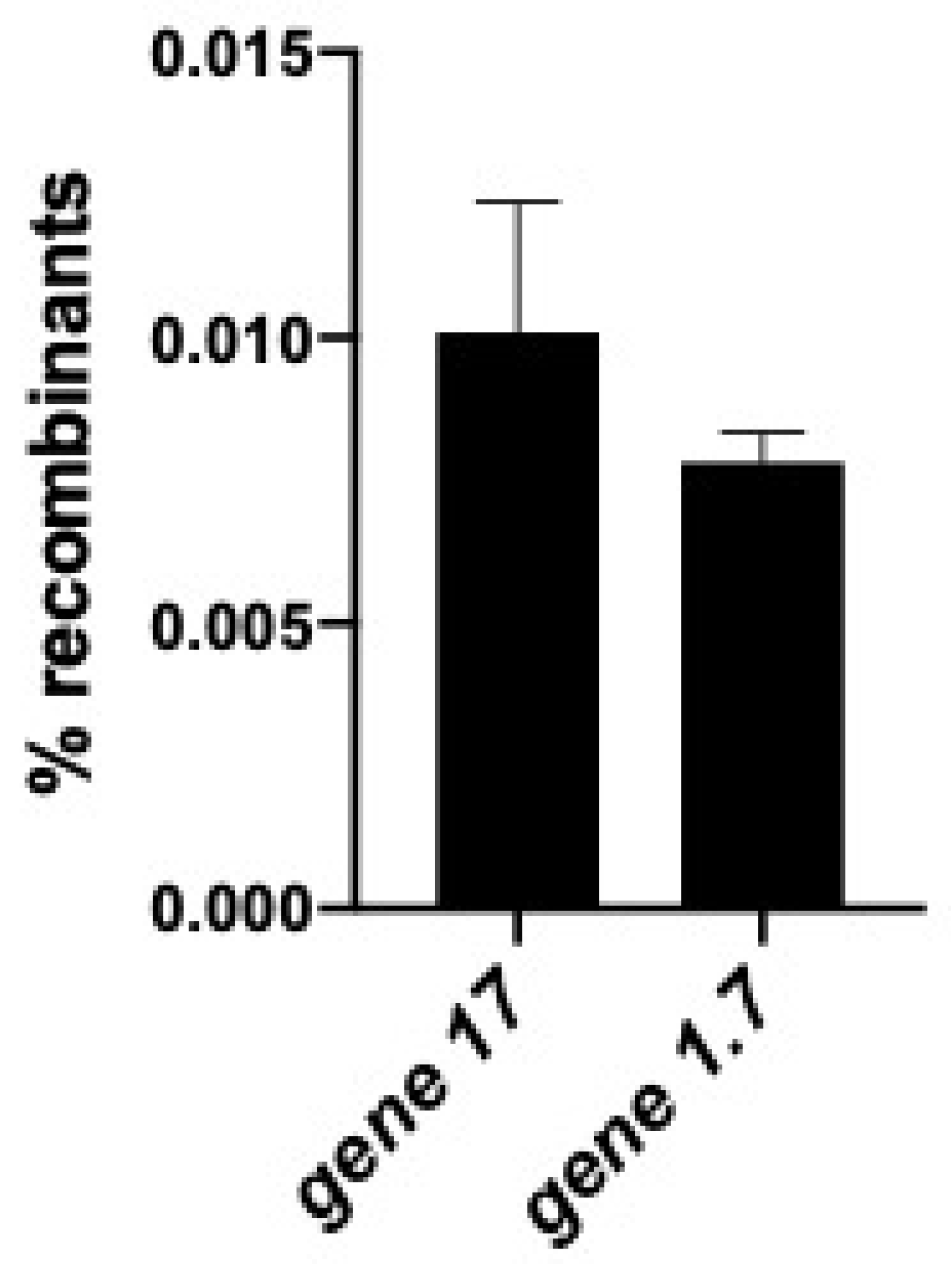
2.4. Gene Replacement by λ Red-Mediated Recombineering
3. Materials and Methods
3.1. Media
3.2. Bacterial Strains
3.3. Bacteriophages
3.4. Preparation of Genomic DNA
3.5. Recombineering Using Transfected Bacteriophage Genomic DNA
3.6. Recombineering Following Infection by Bacteriophage T7
3.7. Quantitation of Phage in Lysates
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chan, L.Y.; Kosuri, S.; Endy, D. Refactoring bacteriophage T7. Mol. Syst. Biol. 2005, 1, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.K.; Koeris, M.S. The next generation of bacteriophage therapy. Curr. Opin. Microbiol. 2011, 14, 524–531. [Google Scholar] [CrossRef] [PubMed]
- Studier, F.W. Bacteriophage T7. Science 1972, 176, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Krüger, D.H.; Schroeder, C. Bacteriophage T3 and bacteriophage T7 virus-host cell interactions. Microbiol. Rev. 1981, 45, 9–51. [Google Scholar] [CrossRef]
- Studier, F. The genetics and physiology of bacteriophage T7. Virology 1969, 39, 562–574. [Google Scholar] [CrossRef]
- Chung, Y.B.; Hinkle, D.C. Bacteriophage T7 DNA packaging. I. Plasmids containing a T7 replication origin and the T7 concatemer junction are packaged into transducing particles during phage infection. J. Mol. Biol. 1990, 216, 911–926. [Google Scholar] [CrossRef]
- Qimron, U.; Marintcheva, B.; Tabor, S.; Richardson, C.C. Genomewide screens for Escherichia coli genes affecting growth of T7 bacteriophage. Proc. Natl. Acad. Sci. USA 2006, 103, 19039–19044. [Google Scholar] [CrossRef] [Green Version]
- Kiro, R.; Shitrit, D.; Qimron, U. Efficient engineering of a bacteriophage genome using the type I-E CRISPR-Cas system. RNA Biol. 2014, 11, 42–44. [Google Scholar] [CrossRef] [Green Version]
- Marinelli, L.J.; Piuri, M.; Swigoňová, Z.; Balachandran, A.; Oldfield, L.M.; Van Kessel, J.C.; Hatfull, G.F. BRED: A Simple and Powerful Tool for Constructing Mutant and Recombinant Bacteriophage Genomes. PLoS ONE 2008, 3, e3957. [Google Scholar] [CrossRef] [Green Version]
- Oppenheim, A.B.; Rattray, A.J.; Bubunenko, M.; Thomason, L.C.; Court, D.L. In vivo recombineering of bacteriophage lambda by PCR fragments and single-strand oligonucleotides. Virology 2004, 319, 185–189. [Google Scholar] [CrossRef] [Green Version]
- Ellis, H.M.; Yu, D.; Ditizio, T.; Court, D.L. High efficiency mutagenesis, repair, and engineering of chromosomal DNA using single-stranded oligonucleotides. Proc. Natl. Acad. Sci. USA 2001, 98, 6742–6746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Datta, S.; Costantino, N.; Zhou, X.; Court, D.L. Identification and analysis of recombineering functions from Gram-negative and Gram-positive bacteria and their phages. Proc. Natl. Acad. Sci. USA 2008, 105, 1626–1631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, K.C. Lambda Gam protein inhibits the helicase and chi-stimulated recombination activities of Escherichia coli RecBCD enzyme. J. Bacteriol. 1991, 173, 5808–5821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ando, H.; Lemire, S.; Pires, D.P.; Lu, T.K. Engineering Modular Viral Scaffolds for Targeted Bacterial Population Editing. Cell Syst. 2015, 1, 187–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molineux, I.J. No syringes please, ejection of phage T7 DNA from the virion is enzyme driven. Mol. Microbiol. 2001, 40, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Molineux, I.J. The T7 group. In The Bacteriophages, 2nd ed.; Calendar, R., Ed.; Oxford University Press: Oxford, UK, 2006; pp. 277–301. [Google Scholar]
- Richardson, C.C. Bacteriophage T7: Minimal requirements for the replication of a duplex DNA molecule. Cell 1983, 33, 315–317. [Google Scholar] [CrossRef]
- Fuller, C.W.; Richardson, C.C. Initiation of DNA replication at the primary origin of bacteriophage T7 by purified proteins. Initiation of bidirectional synthesis. J. Biol. Chem. 1985, 260, 3197–3206. [Google Scholar]
- Scholl, D.; Kieleczawa, J.; Kemp, P.; Rush, J.; Richardson, C.; Merril, C.; Adhya, S.; Molineux, I. Genomic Analysis of Bacteriophages SP6 and K1-5, an Estranged Subgroup of the T7 Supergroup. J. Mol. Biol. 2004, 335, 1151–1171. [Google Scholar] [CrossRef]
- Thomason, L.C.; Costantino, N.; Court, D.L. Examining a DNA Replication Requirement for Bacteriophage λ Red- and Rac Prophage RecET-Promoted Recombination in Escherichia coli. MBio 2016, 7. [Google Scholar] [CrossRef] [Green Version]
- Heumann, H.; Lederer, H.; Kammerer, W.; Palm, P.; Metzger, W.; Baer, G. Large-scale preparation of a DNA fragment containing the strong promoter A1 of the phage T7. Biochim. Biophys. Acta (BBA) Gene Struct. Expr. 1987, 909, 126–132. [Google Scholar] [CrossRef]
- Sawitzke, J.A.; Costantino, N.; Li, X.-T.; Thomason, L.C.; Bubunenko, M.; Court, C.; Court, D.L. Probing Cellular Processes with Oligo-Mediated Recombination and Using the Knowledge Gained to Optimize Recombineering. J. Mol. Biol. 2011, 407, 45–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Springman, R.; Keller, T.; Molineux, I.J.; Bull, J. Evolution at a High Imposed Mutation Rate: Adaptation Obscures the Load in Phage T7. Genetics 2009, 184, 221–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drake, J.W.; Charlesworth, B.; Charlesworth, D.; Crow, J.F. Rates of spontaneous mutation. Genetics 1998, 148, 1667–1686. [Google Scholar] [PubMed]
- Reutimann, H.; Sjoeberg, B.-M.; Holmgren, A. Bacteriophage T7 DNA polymerase: Cloning and high-level expression. Proc. Natl. Acad. Sci. USA 1985, 82, 6783–6787. [Google Scholar] [CrossRef] [Green Version]
- Yosef, I.; Manor, M.; Kiro, R.; Qimron, U. Temperate and lytic bacteriophages programmed to sensitize and kill antibiotic-resistant bacteria. Proc. Natl. Acad. Sci. USA 2015, 112, 7267–7272. [Google Scholar] [CrossRef] [Green Version]
- Yehl, K.; Lemire, S.; Yang, A.C.; Ando, H.; Mimee, M.; Torres, M.D.T.; De La Fuente-Nunez, C.; Lu, T.K. Engineering Phage Host-Range and Suppressing Bacterial Resistance through Phage Tail Fiber Mutagenesis. Cell 2019, 179, 459–469.e9. [Google Scholar] [CrossRef]
- Thomason, L.C.; Sawitzke, J.A.; Li, X.; Costantino, N.; Court, D.L. Recombineering: Genetic Engineering in Bacteria Using Homologous Recombination. Curr. Protoc. Mol. Biol. 2014, 106, 1–16. [Google Scholar] [CrossRef]
- Li, X.-T.; Thomason, L.C.; Sawitzke, J.A.; Costantino, N.; Court, D.L. Positive and negative selection using the tetA-sacB cassette: Recombineering and P1 transduction in Escherichia coli. Nucleic Acids Res. 2013, 41, e204. [Google Scholar] [CrossRef]
- Thomason, L.C.; Costantino, N.; Court, D.L. E. coli Genome Manipulation by P1 Transduction. Curr. Protoc. Mol. Biol. 2007, 79, 1–17. [Google Scholar]
- Thomason, L.C.; Oppenheim, A.B.; Court, D.L. Modifying Bacteriophage λ with Recombineering. Adv. Struct. Saf. Stud. 2009, 501, 239–251. [Google Scholar] [CrossRef]
- Da Silva, J.L.; Piuri, M.; Broussard, G.; Marinelli, L.J.; Bastos, G.M.; Hirata, R.D.; Hatfull, G.F.; Hirata, M. Application of BRED technology to construct recombinant D29 reporter phage expressing EGFP. FEMS Microbiol. Lett. 2013, 344, 166–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheng, Y.; Mancino, V.; Birren, B. Transformation of Escherichia coli with large DNA molecules by electroporation. Nucleic Acids Res. 1995, 23, 1990–1996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, D.; Sun, J.; Shi, J.; Liu, P.; Hao, J. New strategy to improve efficiency for gene replacement in Klebsiella pneumoniae. J. Ind. Microbiol. Biotechnol. 2013, 40, 523–527. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bacterial Strains | Relevant Genotype | Source, Reference |
|---|---|---|
| E. coli strains | ||
| E. coli B | Wild-type | Ian Molineux collection |
| E. coli B40 | E. coli B supD | Ian Molineux collection |
| E. coli B3783 | E. coli B ΔtrxA<>*tetA | This work; recombineering pSIM18; gene replacement hsdR<>bla |
| E. coli K1 | Wild-type | Sankar Adhya collection |
| E. coli K12 strains: | ||
| W3110 | rph-1 inv(rrnD-rrnE) | Laboratory stock strain |
| DY330 | W3110 [λ cI857 gam+bet+exo+ Δ(cro-bioA)] | {Thomason et al., 2014} |
| T-SACK | W3110 araD<>tetA-sacB-amp fliC<>cat argG:kan | {Li et al., 2013} |
| LT976 | W3110 hsdR<>bla λ cI857 gam+bet+exo+ Δ(cro-bioA) | Lynn Thomason collection; recombineering; DY330, gene replacement hsdR<>bla |
| ARP3607 | W3110 hsdR<>bla λ cI857 pL g17 Δ(cro-bioA) | This work; recombineering; gene replacement LT976, (cIII-galK)<>gp17 |
| ARP3750 | W3110 hsdR<>bla ΔtrxA<>tetA λcI857pL g17 Δ(cro-bioA) | This work |
| ARP3620 | LT976 ΔtrxA<>tetA | This work |
| ARP3771 | LT976 supF:Tn10 | This work; transduction LT976 X (P1) |
| ARP3816 | W3110 hsdR<>bla | This work; transduction W3110 X (P1) LT976 |
| DH5α | fhuA2 lac(del)U169 phoA glnV44 Φ80’ lacZ(del)M15 gyrA96 recA1 relA1 endA1 thi-1 hsdR17 | Laboratory stock strain |
| Klebsiella pneumoniae Kpn1760 | Cured of resistance plasmid pKpQIL at 42 °C | Karen Frank collection |
| Bacteriophages and Plasmids | ||
| T7 | Wild-type | Sankar Adhya collection |
| T7 (amber267) | T7 g17Q170UAG | Ian Molineux collection |
| K1-5 | Wild-type | Sankar Adhya collection |
| Pharr | Isolated from sewage | Jason Gill collection |
| Poteet | Isolated from sewage | Jason Gill collection |
| pSIM18 HygroR | pSC101 PLgam+bet+exo+ | {Thomason et al., 2014} |
| pSIM28 HygroR | pSC101, PL-gam+ | (http://redrecombineering.ncifcrf.gov/) |
| Name | Nucleotide Sequence b | Notes |
|---|---|---|
| ARP261 | TAACGCTTCACTCGAGGCGTTTTTCGTTATGTATAAAAAGGAGCACACC/ATGGCTAACGTAATTAAAACCGTTTTGACTTACCAG | λ pL g17 forward oligo |
| ARP262 | GCTTCCCAGCCAGCGTTGCGAGTGCAGTACTCATTCGTTTTATACCTCTG/ATTACTCGTTCTCCACCATGATTGCATTAGG | λ pL g17 reverse oligo |
| ARP292 | ACCAGAACTCATGGCAAGCACGTAATGAAGCCTTA CAGTTCCGTAATGAGGCTGAGACTTTCAGAAACCAA | T7 g17Q170UAG-CAG leading strand oligo |
| ARP293 | TTGGTTTCTGAAAGTCTCAGCCTCATTACGGAA CTGTAAGGCTTCATTACGTGCTTGCCATGAGTTCTGGT | T7 g17Q170UAG-CAG lagging strand oligo |
| ARP301 | GGCAGTGACCCGCTTCCCGTTCGTCCGTCTGTTACTCAAACGAATCAAGGAGGTGTTCTG/ATGAGCGATAAAATTATTCACCTGACTGAC | T7 g1.7<>trxA forward |
| ARP302 | CACTCTGAGCAAGATGTGAAGTCATCAGATAGGCTGTCGGCAGGTGGGGTTGACTTGAAG/TTACGCCAGGTTAGCGTCGAGGA | T7 g1.7<>trxA reverse |
| ARP309 | ACCAACACGCCAGGCTTATTCCTGTGGAGTTATAT/TCCTAATTTTTGTTGACACTCTATC | trxA<>tetA forward |
| ARP310 | TTTTTAGCGACGGGGCACCCGAACATGAAATTCCC/ATCAAAGGGAAAACTGTCCATATGC | trxA<>tetA reverse |
| ARP321 | ACCAGAACTCATGGCAAGCACGTAATGAAGC | g17am267 70 bp dsDNA forward |
| ARP322 | CGCTTGGTTTCTGAAAGTCTCAGCCTCATTACGG | g17am267 70 bp dsDNA forward |
| ARP356 | CGAAATAATCTTCTCCCTGTAGTCTCTTAGATTTACTTTAAGGAGGTCAA/ATGAGCGATAAAATTATTCACCTGACTGAC | g17<>trxA forward |
| ARP357 | AGGTACAGTCATTGTTGTTATCTGACCCTCTACCAATGTACCAGTTATTC/TTACGCCAGGTTAGCGTCGAGGA | g17<>trxA reverse |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jensen, J.D.; Parks, A.R.; Adhya, S.; Rattray, A.J.; Court, D.L. λ Recombineering Used to Engineer the Genome of Phage T7. Antibiotics 2020, 9, 805. https://doi.org/10.3390/antibiotics9110805
Jensen JD, Parks AR, Adhya S, Rattray AJ, Court DL. λ Recombineering Used to Engineer the Genome of Phage T7. Antibiotics. 2020; 9(11):805. https://doi.org/10.3390/antibiotics9110805
Chicago/Turabian StyleJensen, Jordan D., Adam R. Parks, Sankar Adhya, Alison J. Rattray, and Donald L. Court. 2020. "λ Recombineering Used to Engineer the Genome of Phage T7" Antibiotics 9, no. 11: 805. https://doi.org/10.3390/antibiotics9110805
APA StyleJensen, J. D., Parks, A. R., Adhya, S., Rattray, A. J., & Court, D. L. (2020). λ Recombineering Used to Engineer the Genome of Phage T7. Antibiotics, 9(11), 805. https://doi.org/10.3390/antibiotics9110805