Proposal of Potent Inhibitors for a Bacterial Cell Division Protein FtsZ: Molecular Simulations Based on Molecular Docking and ab Initio Molecular Orbital Calculations

 ,
,
Abstract
:1. Introduction
2. Details of Molecular Simulations
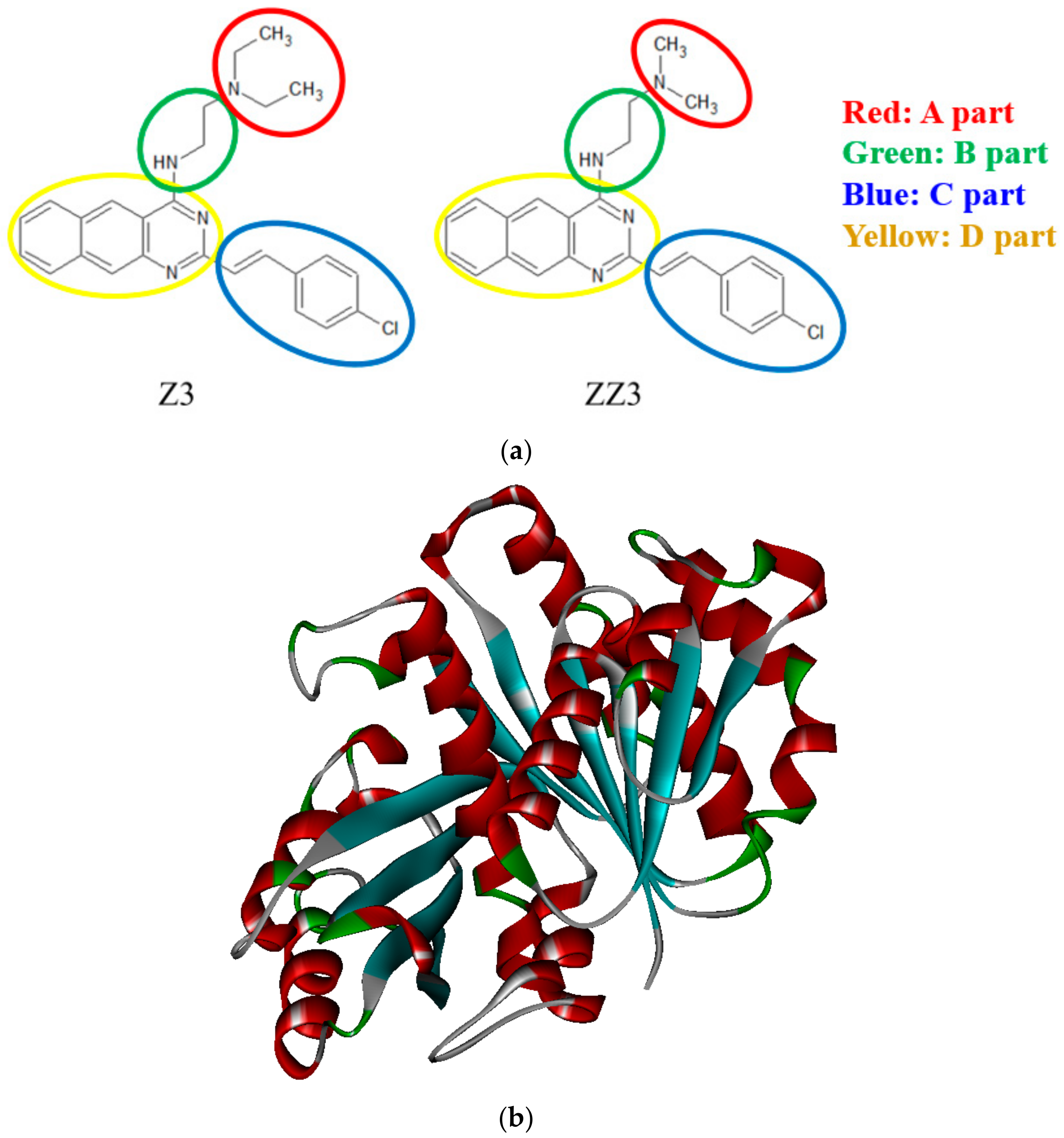
2.1. Proposal of Novel ZZ3 Derivatives as Potent Inhibitors of FtsZ
2.2. Constructions and Optimizations of the FtsZ + Derivative Complexes
2.3. FMO Calculations for the FtsZ + Derivative Complexes
3. Results and Discussion
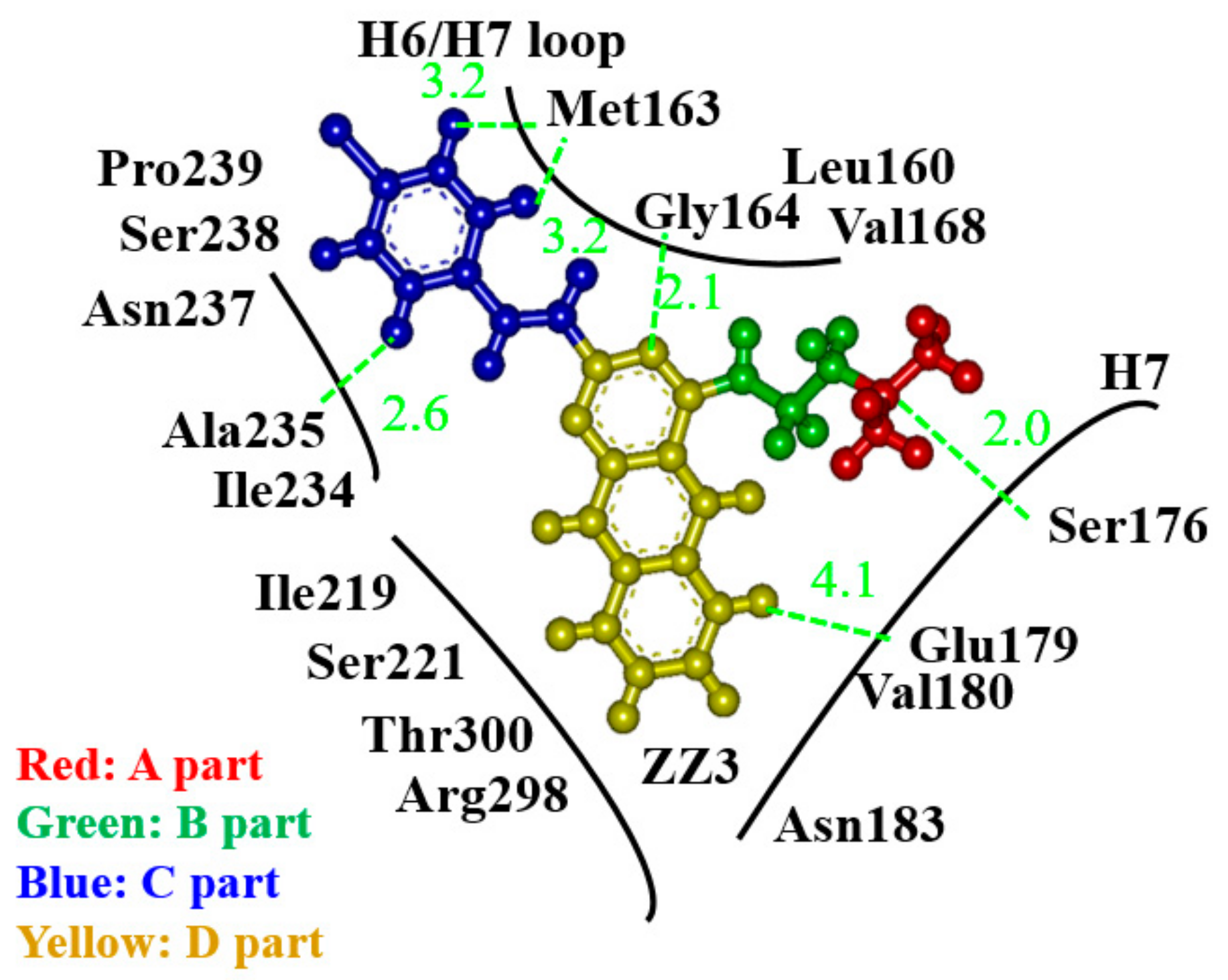
3.1. Binding Properties between FtsZ and the ZZ3 Derivatives by Replacing A-Part
3.2. Binding Properties between FtsZ and the ZZ3 Derivatives by Replacing the B- or D-Part
4. Conclusions
- (1)
- The derivative, ZZ3_X, in which an OH group was introduced in the D-part of ZZ3, possessed the largest BE to FtsZ due to the strong H-bond between the OH group and Asp165 side chain.
- (2)
- Since Asp165 was included in the H6/H7 loop, which was beneficial for the aggregation of FtsZ, ZZ3_X was expected to change the conformation of the loop to inhibit the aggregations.
- (3)
- The replacement of the A- and B-parts of ZZ3 did not exert any positive effect on the enhancement of the interactions between ZZ3 and FtsZ.
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Global Tuberculosis Report 2017; World Health Organization: Geneva, Switzerland, 2017. [Google Scholar]
- World Health Organization Media Centre. WHO. Fact Sheet: Tuberculosis; WHO: Geneva, Switzerland, 2017. [Google Scholar]
- Nagai, H. Present status of multidrug resistant tuberculosis. Iryo 2004, 58, 595–598. (In Japanese) [Google Scholar]
- Kobayashi, H.; Koyanagi, K.; Kato, O.; Oe, T. Outbreak of extensively drug-resistant pulmonary tuberculosis in a hemodialysis facility. Kekkaku 2013, 88, 477–484. (In Japanese) [Google Scholar] [PubMed]
- Karpov, P.A.; Demchuk, O.M.; Britsun, V.M.; Lytvyn, D.I.; Pydiura, N.O.; Rayevsky, A.V.; Samofalova, D.A.; Spivak, S.I.; Volochnyuk, D.M.; Yemets, A.I.; et al. New imidazole inhibitors of mycobacterial FtsZ: The way from high-throughput molecular screening in grid to in vitro verification. Sci. Innov. 2016, 12, 43–55. [Google Scholar] [CrossRef] [Green Version]
- Tomioka, H. Prospects for the development of new antituberculous drugs putting our hopes on new drug targets. Kekkaku 2010, 85, 815–822. (In Japanese) [Google Scholar] [PubMed]
- Margalit, D.N.; Romberg, L.; Mets, R.B.; Hebert, A.M.; Mitchison, T.J.; Kirschner, M.W.; Raychaudhuri, D. Targeting cell division: Small-molecule inhibitors of FtsZ GTPase perturb cytokinetic ring assembly and induce bacterial lethality. Proc. Natl. Acad. Sci. USA 2004, 101, 11821–11826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osawa, M.; Anderson, D.E.; Erickson, H.P. Reconstitution of Contractile FtsZ Rings in Liposomes. Science 2008, 320, 792–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haydon, D.J.; Stokes, N.R.; Ure, R.; Galbraith, G.; Bennett, J.M.; Brown, D.R.; Baker, P.J.; Barynin, V.V.; Rice, D.W.; Sedelnikova, S.E.; et al. An inhibitor of FtsZ with potent and selective anti-staphyloccocal aureus activity. Science 2008, 321, 1673–1675. [Google Scholar] [CrossRef] [PubMed]
- Fujimori, M.; Sogawa, H.; Ota, S.; Karpov, P.; Shulga, S.; Blume, Y.; Kurita, N. Specific interactions between mycobacterial FtsZ protein and curcumin derivatives: Molecular docking and ab initio molecular simulations. Chem. Phys. Lett. 2018, 692, 166–173. [Google Scholar] [CrossRef]
- Sogawa, H.; Sato, R.; Suzuki, K.; Tomioka, S.; Shinzato, T.; Karpov, P.; Shulga, S.; Blume, Y.; Kurita, N. Binding sites of Zantrin inhibitors to the bacterial cell division protein FtsZ: Molecular docking and ab initio molecular orbital calculations. Chem. Phys. 2020, 530, 110603. [Google Scholar] [CrossRef]
- Aggarwal, B.B.; Ichikawa, H.; Garodia, P.; Weerasinghe, P.; Sethi, G.; Bhatt, I.D.; Pandey, M.K.; Shishodia, S.; Nair, M.G. From traditional Ayurvedic medicine to modern medicine: Identification of therapeutic targets for suppression of inflammation and cancer. Expert Opin. Ther. Targets 2006, 10, 87–118. [Google Scholar] [CrossRef] [PubMed]
- Ray, B.; Bisht, S.; Maitra, A.; Maitra, A.; Lahiri, D.K. Neuroprotective and Neurorescue Effects of a Novel Polymeric Nanoparticle Formulation of Curcumin (NanoCurc™) in the Neuronal Cell Culture and Animal Model: Implications for Alzheimer’s disease. J. Alzheimer’s Dis. 2011, 23, 61–77. [Google Scholar] [CrossRef] [PubMed]
- Rai, D.; Singh, J.K.; Roy, N.; Panda, D. Curcumin inhibits FtsZ assembly: An attractive mechanism for its antibacterial activity. Biochem. J. 2008, 410, 147–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nepomuceno, G.M.; Chan, K.M.; Huynh, V.; Martin, K.S.; Moore, J.T.; O’Brien, T.E.; Pollo, L.A.E.; Sarabia, F.J.; Tadeus, C.; Yao, Z.; et al. Synthesis and evaluation of quinazolines as inhibitors of the bacterial cell division protein FtsZ. ACS Med. Chem. Lett. 2015, 6, 308–312. [Google Scholar] [CrossRef] [PubMed]
- Wirth, M.; Zoete, V.; Michielin, O.; Sauer, W.H.B. SwissBioisostere: A database of molecular replacements for ligand design. Nucleic Acids Res. 2013, 41, D1137–D1143. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Shah, S.; Patel, B.; Savjani, J.K. Pharmacophore mapping based virtual screening, molecular docking and ADMET studies of ROCK II inhibitors. Mult. Scler. Relat. Disord. 2018, 21, 35–41. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliver. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Leung, A.K.; White, E.L.; Ross, L.J.; Reynolds, R.C.; DeVito, J.A.; Borhani, D.W. Structure of Mycobacterium tuberculosis FtsZ Reveals Unexpected, G Protein-like Conformational Switches. J. Mol. Biol. 2004, 342, 953–970. [Google Scholar] [CrossRef]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef] [Green Version]
- Roy, A.; Yang, J.; Zhang, Y. COFACTOR: An accurate comparative algorithm for structure-based protein function annotation. Nucleic Acids Res. 2012, 40, W471–W477. [Google Scholar] [CrossRef] [Green Version]
- Søndergaard, C.R.; Olsson, M.H.M.; Rostkowski, M.; Jensen, J.H. Improved Treatment of Ligands and Coupling Effects in Empirical Calculation and Rationalization of pKa Values. J. Chem. Theory Comput. 2011, 7, 2284–2295. [Google Scholar] [CrossRef] [PubMed]
- Olsson, M.H.M.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Calmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Bayly, C.I.; Cieplak, P.; Cornell, W.; Kollman, P.A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: The RESP model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Morris, G.M.; Ruth, H.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Case, D.A.; Darden, T.A.; Cheatham, T.E.; Simmerling, C.L., III; Wang, J.; Duke, R.E.; Luo, R.; Walker, R.C.; Zhang, W.; Merz, K.M.; et al. AMBER 12; University of California: San Francisco, CA, USA, 2012. [Google Scholar]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins Struct. Funct. Bioinform. 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.; Klein, M.L. Compatison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Kitaura, K.; Ikeo, E.; Asada, T.; Nakano, T.; Uebayasi, M. Fragment molecular orbital method: An approximate computational method for large molecules. Chem. Phys. Lett. 1999, 313, 701–706. [Google Scholar] [CrossRef]
- Mochizuki, Y.; Nakano, T.; Koikegami, S.; Tanimori, S.; Abe, Y.; Nagashima, U.; Kitaura, K. A parallelized integral-direct second-order Mφller? Plesset perturbation theory method with a fragment molecular orbital scheme. Theor. Chem. Acc. 2004, 112, 442–452. [Google Scholar] [CrossRef]
- Mochizuki, Y.; Koikegami, S.; Nakano, T.; Amari, S.; Kitaura, K. Large scale MP2 calculations with fragment molecular orbital scheme. Chem. Phys. Lett. 2004, 396, 473–479. [Google Scholar] [CrossRef]
- Mochizuki, Y.; Yamashita, K.; Nakano, T.; Okiyama, Y.; Fukuzawa, K.; Taguchi, N.; Tanaka, S. Higher-order correlated calculations based on fragment molecular orbital scheme. Theor. Chem. Acc. 2011, 130, 515–530. [Google Scholar] [CrossRef]
- Fukuzawa, K.; Komeiji, Y.; Mochizuki, Y.; Kato, A.; Nakano, T.; Tanaka, S. Intra- and intermolecular interactions between cyclic-AMP receptor protein and DNA:Ab initio fragment molecular orbital study. J. Comput. Chem. 2006, 27, 948–960. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | MW | RB | HBA | HBD | LogP | PSA |
|---|---|---|---|---|---|---|
| Z3 | 431.0 | 8 | 3 | 1 | 4.65 | 3.33 |
| ZZ3 | 402.9 | 6 | 3 | 1 | 4.24 | 3.12 |
| ZZ3_II | 402.9 | 7 | 3 | 2 | 4.24 | 3.11 |
| ZZ3_III | 417.0 | 7 | 3 | 1 | 4.45 | 3.22 |
| ZZ3_IV | 388.9 | 6 | 3 | 2 | 4.04 | 3.01 |
| ZZ3_V | 374.9 | 5 | 3 | 2 | 3.83 | 2.87 |
| ZZ3_VI | 403.9 | 7 | 3 | 1 | 4.24 | 3.09 |
| ZZ3_VII | 389.9 | 6 | 3 | 1 | 4.04 | 2.96 |
| ZZ3_VIII | 375.9 | 5 | 3 | 2 | 3.83 | 2.82 |
| ZZ3_IX | 417.0 | 7 | 3 | 1 | 4.45 | 3.21 |
| ZZ3_X | 418.9 | 6 | 4 | 2 | 3.42 | 3.11 |
| ZZ3_XI | 418.9 | 6 | 4 | 2 | 3.42 | 3.09 |
| ZZ3_XII | 418.9 | 6 | 4 | 2 | 3.42 | 3.17 |
| ZZ3_XIII | 417.0 | 6 | 3 | 1 | 4.45 | 3.25 |
| ZZ3_XIV | 417.0 | 6 | 3 | 1 | 4.45 | 3.24 |
| ZZ3_XV | 417.0 | 6 | 3 | 1 | 4.45 | 3.27 |
| Ligand | BBB | Caco2 | HIA | PPB | Mouse | Rat | hERG |
|---|---|---|---|---|---|---|---|
| Z3 | 3.5 | 55.6 | 97.1 | 86.7 | positive | negative | medium |
| ZZ3 | 1.5 | 54.3 | 97.1 | 84.4 | positive | negative | medium |
| ZZ3_II | 4.2 | 48.2 | 95.9 | 91.9 | positive | negative | medium |
| ZZ3_III | 2.4 | 55.0 | 97.1 | 85.1 | positive | negative | medium |
| ZZ3_IV | 3.1 | 45.8 | 95.9 | 86.6 | positive | negative | medium |
| ZZ3_V | 0.5 | 25.8 | 96.3 | 95.8 | positive | negative | medium |
| ZZ3_VI | 1.0 | 53.5 | 97.1 | 91.8 | positive | negative | medium |
| ZZ3_VII | 0.6 | 52.0 | 97.1 | 91.1 | positive | negative | medium |
| ZZ3_VIII | 2.4 | 29.8 | 95.9 | 92.3 | positive | negative | medium |
| ZZ3_IX | 1.7 | 54.9 | 97.1 | 83.3 | positive | negative | medium |
| ZZ3_X | 2.8 | 37.6 | 96.1 | 84.4 | negative | negative | medium |
| ZZ3_XI | 2.8 | 37.6 | 96.1 | 85.3 | positive | negative | medium |
| ZZ3_XII | 2.8 | 37.6 | 96.1 | 85.0 | positive | negative | medium |
| ZZ3_XIII | 2.8 | 54.4 | 97.1 | 84.5 | positive | negative | medium |
| ZZ3_XIV | 3.1 | 54.4 | 97.1 | 84.2 | positive | negative | medium |
| ZZ3_XV | 2.7 | 54.4 | 97.1 | 84.1 | positive | negative | medium |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamamoto, S.; Saito, R.; Nakamura, S.; Sogawa, H.; Karpov, P.; Shulga, S.; Blume, Y.; Kurita, N. Proposal of Potent Inhibitors for a Bacterial Cell Division Protein FtsZ: Molecular Simulations Based on Molecular Docking and ab Initio Molecular Orbital Calculations. Antibiotics 2020, 9, 846. https://doi.org/10.3390/antibiotics9120846
Yamamoto S, Saito R, Nakamura S, Sogawa H, Karpov P, Shulga S, Blume Y, Kurita N. Proposal of Potent Inhibitors for a Bacterial Cell Division Protein FtsZ: Molecular Simulations Based on Molecular Docking and ab Initio Molecular Orbital Calculations. Antibiotics. 2020; 9(12):846. https://doi.org/10.3390/antibiotics9120846
Chicago/Turabian StyleYamamoto, Shohei, Ryosuke Saito, Shunya Nakamura, Haruki Sogawa, Pavel Karpov, Sergey Shulga, Yaroslav Blume, and Noriyuki Kurita. 2020. "Proposal of Potent Inhibitors for a Bacterial Cell Division Protein FtsZ: Molecular Simulations Based on Molecular Docking and ab Initio Molecular Orbital Calculations" Antibiotics 9, no. 12: 846. https://doi.org/10.3390/antibiotics9120846
APA StyleYamamoto, S., Saito, R., Nakamura, S., Sogawa, H., Karpov, P., Shulga, S., Blume, Y., & Kurita, N. (2020). Proposal of Potent Inhibitors for a Bacterial Cell Division Protein FtsZ: Molecular Simulations Based on Molecular Docking and ab Initio Molecular Orbital Calculations. Antibiotics, 9(12), 846. https://doi.org/10.3390/antibiotics9120846