Nonocarbolines A–E, β-Carboline Antibiotics Produced by the Rare Actinobacterium Nonomuraea sp. from Indonesia

 ,
,  and
and 
Abstract
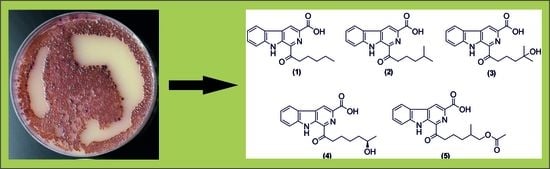
:
1. Introduction
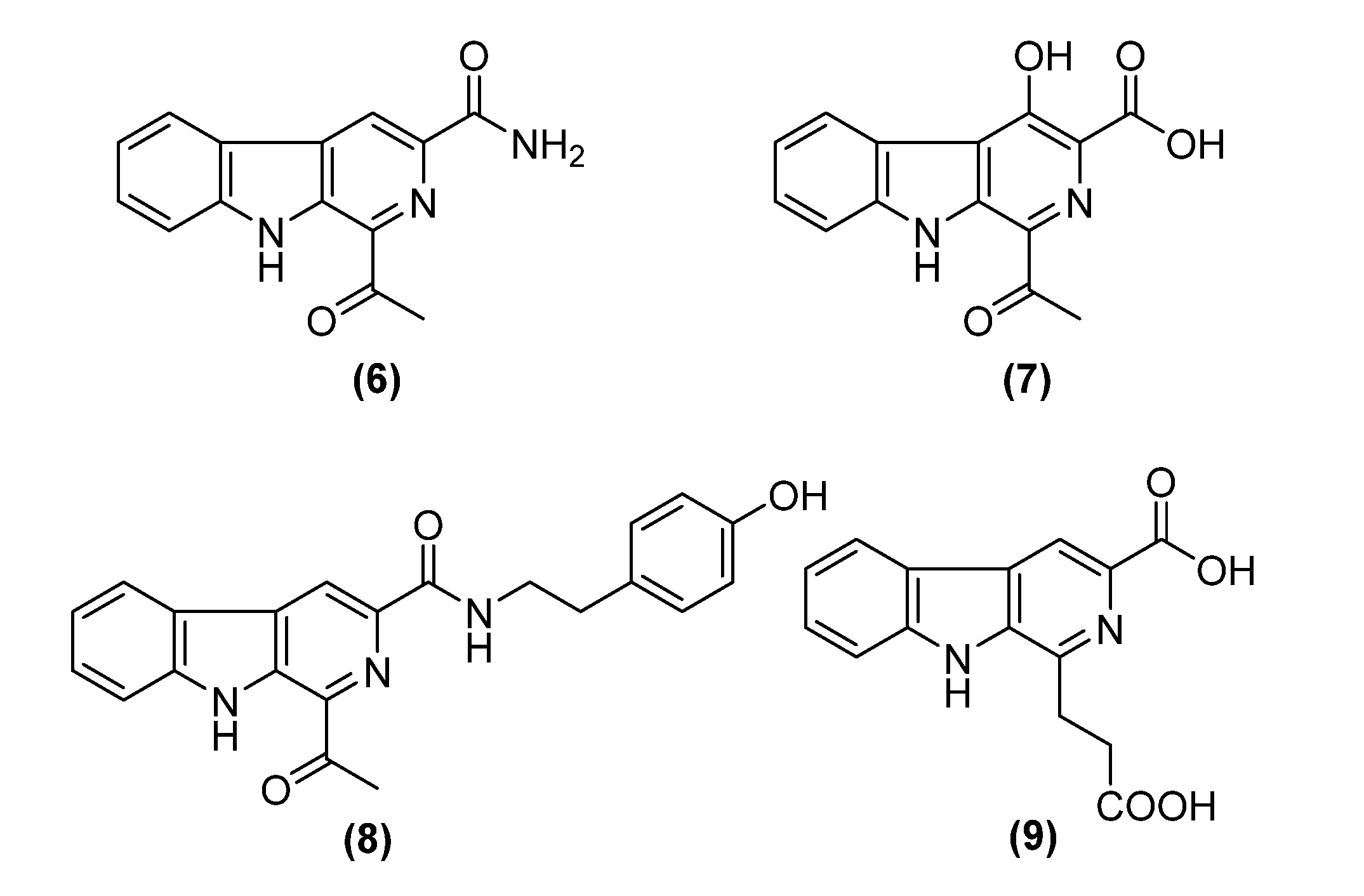
2. Results and Discussion
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Strain Origin and Identification
3.2.1. Sampling and Isolation of the Organism
3.2.2. Analysis of 16S rRNA Sequences
3.3. Scale-up Fermentation, Extraction and Isolation
3.4. Preparation of (R)-and (S)-MTPA Ester Derivatives of 4
3.5. Antimicrobial Assay
3.6. Cytotoxicity Activity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Stadler, M.; Dersch, P. (Eds.) How to overcome the antibiotic crisis–Facts, challenges, technologies & future perspectives. Curr. Top. Microbiol. Immunol. 2016, 398, 496. [Google Scholar]
- Barka, E.A.; Vatsa, P.; Sanchez, L.; Gaveau-Vaillant, N.; Jacquard, C.; Meier-Kolthoff, J.P.; Klenk, H.-P.; Clément, C.; Ouhdouch, Y.; van Wezel, G.P. Taxonomy, Physiology, and Natural Products of Actinobacteria. Microbiol. Mol. Biol. Rev. 2016, 80, 1–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiwari, K.; Gupta, R.K. Rare actinomycetes: A potential storehouse for novel antibiotics. Crit. Rev. Biotechnol. 2012, 32, 108–132. [Google Scholar] [CrossRef] [PubMed]
- Ding, T.; Yang, L.-J.; Zhang, W.-D.; Shen, Y.-H. The secondary metabolites of rare actinomycetes: Chemistry and bioactivity. RSC Adv. 2019, 9, 21964–21988. [Google Scholar] [CrossRef] [Green Version]
- Solecka, J.; Zajko, J.; Postek, M.; Rajnisz, A. Biologically active secondary metabolites from Actinomycetes. Cent. Eur. J. Biol. 2012, 7, 373–390. [Google Scholar] [CrossRef]
- Takahashi, Y. Genus Kitasatospora, taxonomic features and diversity of secondary metabolites. J. Antibiot. 2017, 70, 506–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sungthong, R.; Nakaew, N. The genus Nonomuraea: A review of a rare actinomycete taxon for novel metabolites. J. Basic Microbiol. 2015, 55, 554–565. [Google Scholar] [CrossRef]
- Derewacz, D.K.; McNees, C.R.; Scalmani, G.; Covington, C.L.; Shanmugam, G.; Marnett, L.J.; Polavarapu, P.L.; Bachmann, B.O. Structure and stereochemical determination of hypogeamicins from a cave-derived actinomycete. J. Nat. Prod. 2014, 77, 1759–1763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Supong, K.; Sripreechasak, P.; Phongsopitanun, W.; Tanasupawat, S.; Danwisetkanjana, K.; Bunbamrung, N.; Pittayakhajonwut, P. Antimicrobial substances from the rare actinomycete Nonomuraea rhodomycinica NR4-ASC07T. Nat. Prod. Res. 2019, 33, 2285–2291. [Google Scholar] [CrossRef]
- Shaaban, K.A.; Shaaban, M.; Rahman, H.; Grün-Wollny, I.; Kämpfer, P.; Kelter, G.; Fiebig, H.H.; Laatsch, H. Karamomycins A-C: 2-Naphthalen-2-yl-thiazoles from Nonomuraea endophytica. J. Nat. Prod. 2019, 82, 870–877. [Google Scholar] [CrossRef]
- Sun, B.; Morikawa, T.; Matsuda, H.; Tewtrakul, S.; Wu, L.J.; Harima, S.; Yoshikawa, M. Structures of new β-carboline-type alkaloids with antiallergic-effects from Stellaria dichotoma. J. Nat. Prod. 2004, 67, 1464–1469. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Nam, S.-J.; Lee, B.-C.; Kang, H. β-Carboline alkaloids from a Korean tunicate Eudistoma sp. J. Nat. Prod. 2008, 71, 163–166. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Yao, Y.; He, Z.; Yang, T.; Ma, J.; Tian, X.; Li, Y.; Huang, C.; Chen, X.; Li, W.; et al. Antimalarial β-carboline and indolactam alkaloids from Marinactinospora thermotolerans, a deep sea isolate. J. Nat. Prod. 2011, 74, 2122–2127. [Google Scholar] [CrossRef] [PubMed]
- Sandargo, B.; Michehl, M.; Praditya, D.; Steinmann, E.; Stadler, M.; Surup, F. Antiviral meroterpenoid rhodatin and sesquiterpenoids rhodocoranes A–E from the Wrinkled Peach Mushroom, Rhodotus palmatus. Org. Lett. 2019, 21, 3286–3289. [Google Scholar] [CrossRef]
- Hoye, T.R.; Jeffrey, C.S.; Shao, F. Mosher ester analysis for the determination of absolute configuration of stereogenic (chiral) carbinol carbons. Nat. Protoc. 2007, 2, 2451–2458. [Google Scholar] [CrossRef]
- Laine, A.E.; Lood, C.; Koskinen, A.M.P. Pharmacological importance of optically active tetrahydro-β-carbolines and synthetic approaches to create the C1 stereocenter. Molecules 2014, 19, 1544–1567. [Google Scholar] [CrossRef] [Green Version]
- Kuephadungphan, W.; Macabeo, A.P.G.; Luangsa-ard, J.J.; Tasanathai, K.; Thanakitpipattana, D.; Phongpaichit, S.; Yuyama, K.; Stadler, M. Studies on the biologically active secondary metabolites of the new spider parasitic fungus Gibellula gamsii. Mycol. Prog. 2019, 18, 135–146. [Google Scholar] [CrossRef] [Green Version]
- Chalotra, N.; Ahmed, A.; Rizvi, M.A.; Hussain, Z.; Ahmed, Q.N.; Shah, B.A. Photoredox generated vinyl radicals: Synthesis of bisindoles and β-carbolines. J. Org. Chem. 2018, 83, 14443–14456. [Google Scholar] [CrossRef]
- Kobayashi, J.; Tsuda, M.; Kawasaki, N.; Sasaki, T.; Mikami, Y. 6-Hydroxymanzamine A and 3,4-dihydromanzamine A, new alkaloids from the okinawan marine sponge Aamphimedon sp. J. Nat. Prod. 1994, 57, 1737–1740. [Google Scholar] [CrossRef]
- Schupp, P.; Poehner, T.; Edrada, R.A.; Ebel, R.; Berg, A.; Wray, V.; Proksch, P. Eudistomins W and X, two new β-carbolines from the micronesian tunicate Eudistoma sp. J. Nat. Prod. 2003, 66, 272–275. [Google Scholar] [CrossRef]
- Prinsep, M.R.; Blunt, J.W.; Munro, M.H.G. New cytotoxic β-carboline alkaloids from the marine bryozoan, Cribricellina cribraria. J. Nat. Prod. 1991, 54, 1068–1076. [Google Scholar] [CrossRef] [PubMed]
- Ichiba, T.; Corgiat, J.M.; Scheuer, P.J.; Kelly-Borges, M. 8-Hydroxymanzamine a, a β-carboline alkaloid from a sponge, Pachypellina sp. J. Nat. Prod. 1994, 57, 168–170. [Google Scholar] [CrossRef]
- Rao, K.V.; Santarsiero, B.D.; Mesecar, A.D.; Schinazi, R.F.; Tekwani, B.L.; Hamann, M.T. New manzamine alkaloids with activity against infectious and tropical parasitic diseases from an Indonesian sponge. J. Nat. Prod. 2003, 66, 823–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Giorgio, C.; Delmas, F.; Ollivier, E.; Elias, R.; Balansard, G.; Timon-David, P. In vitro activity of the β-carboline alkaloids harmane, harmine, and harmaline toward parasites of the species Leishmania infantum. Exp. Parasitol. 2004, 106, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Chang, F.R.; Wang, H.K.; Kashiwada, Y.; McPhail, A.T.; Bastow, K.F.; Tachibana, Y.; Cosentino, M.; Lee, K.H. Anti-HIV agents 451 and antitumor agents 205.2 Two new sesquiterpenes, leitneridanins A and B, and the cytotoxic and anti-HIV principles from Leitneria floridana. J. Nat. Prod. 2000, 63, 1712–1715. [Google Scholar] [CrossRef]
- Chen, Y.X.; Xu, M.Y.; Li, H.J.; Zeng, K.J.; Ma, W.Z.; Tian, G.B.; Xu, J.; Yang, D.P.; Lan, W.J. Diverse secondary metabolites from the marine-derived fungus Dichotomomyces cejpii F31-1. Mar. Drugs 2017, 15, 3–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helaly, S.E.; Kuephadungphan, W.; Phainuphong, P.; Ibrahim, M.A.A.; Tasanathai, K.; Mongkolsamrit, S.; Luangsa-ard, J.J.; Phongpaichit, S.; Rukachaisirikul, V.; Stadler, M. Pigmentosins from Gibellula sp. as antibiofilm agents and a new glycosylated asperfuran from Cordyceps javanica. Beilstein J. Org. Chem. 2019, 15, 2968–2981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aroonsri, A.; Kitani, S.; Hashimoto, J.; Kosone, I.; Izumikawa, M.; Komatsu, M.; Fujita, N.; Takahashi, Y.; Shin-ya, K.; Ikeda, H.; et al. Pleiotropic control of secondary metabolism and morphological development by KsbC, a butyrolactone autoregulator receptor homologue in Kitasatospora setae. Appl. Environ. Microbiol. 2012, 78, 8015–8024. [Google Scholar] [CrossRef] [Green Version]
- Mori, T.; Hoshino, S.; Sahashi, S.; Wakimoto, T.; Matsui, T.; Morita, H.; Abe, I. Structural basis for β-carboline alkaloid production by the microbial homodimeric enzyme McbB. Chem. Biol. 2015, 22, 898–906. [Google Scholar] [CrossRef] [Green Version]
- Hayakawa, M.; Otoguro, M.; Takeuchi, T.; Yamazaki, T.; Iimura, Y. Application of a method incorporating differential centrifugation for selective isolation of motile actinomycetes in soil and plant litter. Antonie van Leeuwenhoek 2000, 78, 171–185. [Google Scholar] [CrossRef]
- Mohr, K.I.; Stechling, M.; Wink, J.; Wilharm, E.; Stadler, M. Comparison of myxobacterial diversity and evaluation of isolation success in two niches: Kiritimati Island and German compost. Microbiol. Open 2016, 5, 268–278. [Google Scholar] [CrossRef] [PubMed]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl. Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Yoon, S.H.; Ha, S.M.; Kwon, S.; Lim, J.; Kim, Y.; Seo, H.; Chun, J. Introducing EzBioCloud: A taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int. J. Syst. Evol. Microbiol. 2017, 67, 1613–1617. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar]
- Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef]
- Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef]
- Babadi, Z.K.; Sudarman, E.; Ebrahimipour, G.H.; Primahana, G.; Stadler, M.; Wink, J. Structurally diverse metabolites from the rare actinobacterium Saccharothrix xinjiangensis. J. Antibiot. 2019, 73, 48–55. [Google Scholar] [CrossRef]
- Phukhamsakda, C.; Macabeo, A.P.G.; Huch, V.; Cheng, T.; Hyde, K.D.; Stadler, M. Sparticolins A–G, biologically active oxidized spirodioxynaphthalene derivatives from the ascomycete Sparticola junci. J. Nat. Prod. 2019, 82, 2878–2885. [Google Scholar] [CrossRef]




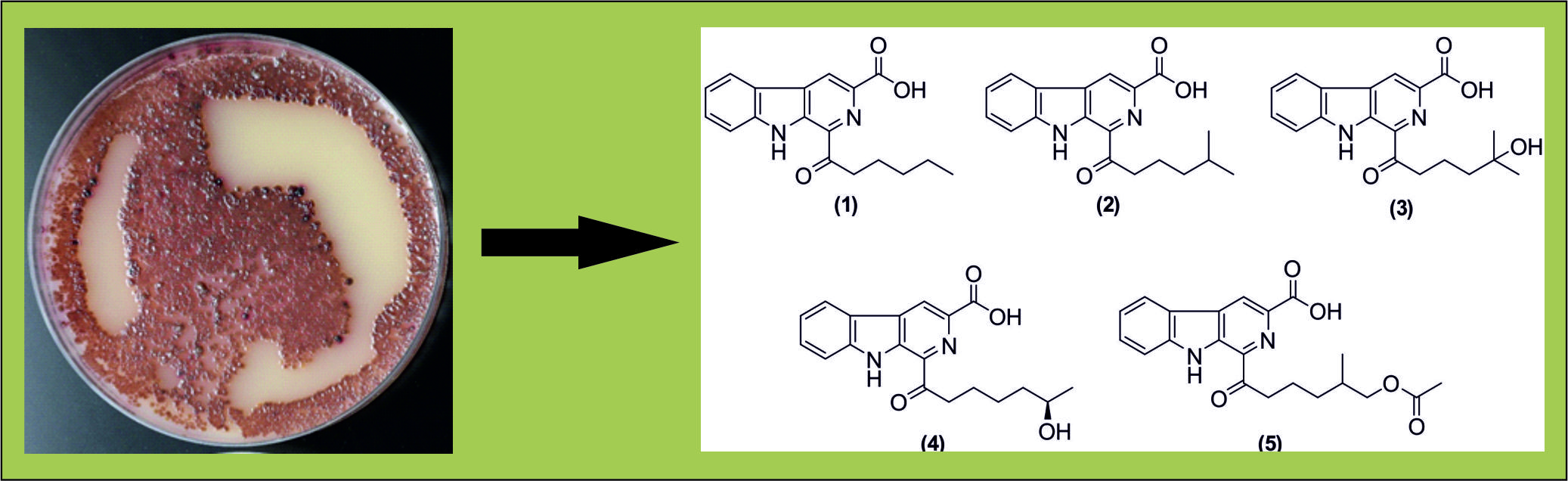{kind=link}
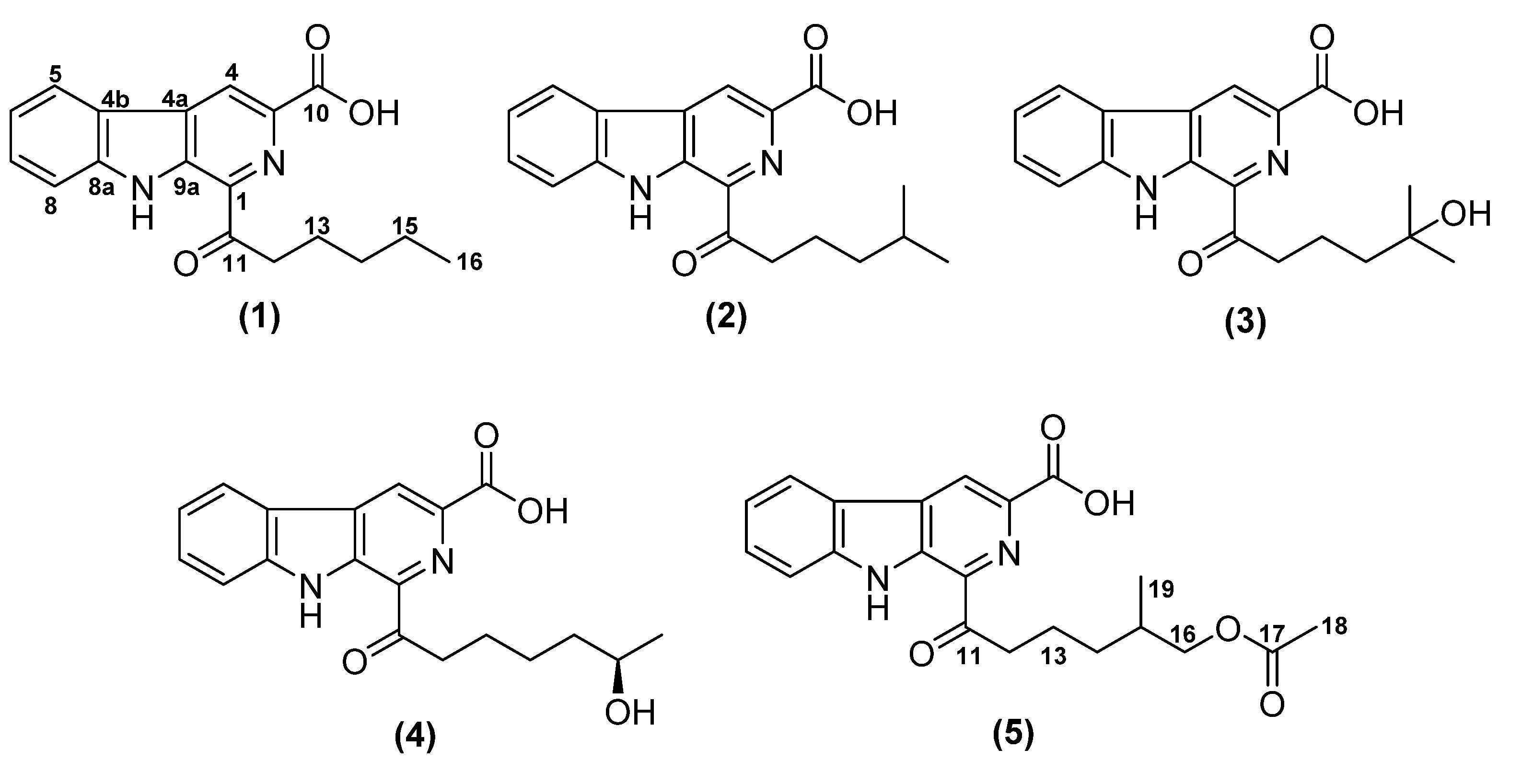{kind=link}
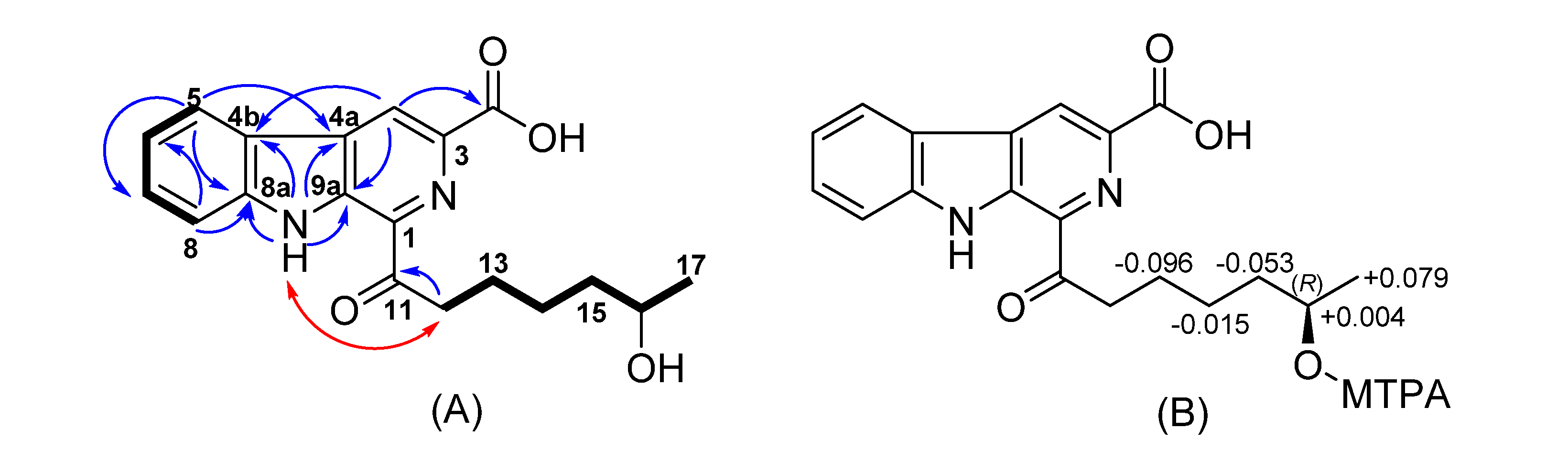{kind=link}
{kind=link}
| Pos. | 1 | 2 | 3 | 4 | 5 | Mar. B [13] | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| δH, m (J in Hz) | δC, Type | δH, m (J in Hz) | δC, Type | δH, m (J in Hz) | δC,, Type | δH, m (J in Hz) | δC,, Type | δH, m (J in Hz) | δC,, Type | δC,, Type | |
| 1 | - | 134.8, C | - | 134.8, C | - | 134.6, C | - | 134.6, C | - | 134.3, C | 133.9, C |
| 2 | - | - | - | - | - | - | - | - | - | - | - |
| 3 | - | 136.5, C | - | 136.4, C | - | 138.0, C | - | 138.8, C | - | 134.5, C | 138.8, C |
| 4 | 9.14, s | 120.8, CH | 9.14, s | 120.8, CH | 9.05, s | 120.2, CH | 9.10, s | 120.5, CH | 9.04, s | 120.2, CH | 117.8, CH |
| 4a | - | 131.4, C | - | 131.4, C | - | 131.2, C | - | 131.3, C | - | 131.2, C | 131.8, C |
| 4b | - | 120.2, C | - | 120.2, C | - | 120.5, C | - | 120.3, C | - | 120.5, C | 120.2, C |
| 5 | 8.44, d (7.5) | 122.1, CH | 8.44, d (7.8) | 122.1, CH | 8.38, d (7.7) | 120.3, CH | 8.41, d (7.8) | 122.0, CH | 8.37, d (7.5) | 121.9, CH | 122.2, CH |
| 6 | 7.35, t (7.5) | 120.9, CH | 7.35, t (7.8) | 120.9, CH | 7.32, t (7.7) | 121.9, CH | 7.32, t (7.8) | 120.6, CH | 7.31, t (7.5) | 120.4, CH | 120.7, CH |
| 7 | 7.63, t (7.5) | 129.2, CH | 7.63, t (7.8) | 129.2, CH | 7.60, t (7.7) | 128.9, CH | 7.61, t (7.8) | 129.0, CH | 7.59, t (7.5) | 128.8, CH | 129.2, CH |
| 8 | 7.83, d (7.5) | 113.3, CH | 7.83, d (7.8) | 113.3, CH | 7.81, d (7.7) | 113.1, CH | 7.81, d (7.8) | 113.2, CH | 7.81, d (7.5) | 113.1, CH | 113.2, CH |
| 8a | - | 142.2, C | - | 142.2, C | - | 142.1, C | - | 142.2, C | - | 142.1, C | 142.3, C |
| 9 | 12.27, s | - | 12.28, s | - | 12.08, s | - | 12.17, s | - | 12.06, s | - | - |
| 9a | - | 135.0, C | - | 135.0, C | - | 134.4, C | - | 134.8, C | - | 134.6, C | 134.7, C |
| 10 | - | 166.3, C | - | 166.3, C | - | 167.1, C | - | 166.8, C | - | 167.1, C | 163.7, C |
| 11 | - | 203.1, C | - | 203.0, C | - | 203.4, C | - | 203.2, C | - | 203.2, C | 202.2, C |
| 12 | 3.41, t (7.4) | 36.7, CH2 | 3.40, t (7.2) | 36.9, CH2 | 3.39, t (7.0) ov | 37.3, CH2 | 3.41, t (7.2) | 36.8, CH2 | 3.40, t (7.0) | 36.9, CH2 | - |
| 13 | 1.75, m | 23.2, CH2 | 1.76, m | 21.4, CH2 | 1.79, m | 18.8, CH2 | 1.74, m | 23.8, CH2 | 1.83; 1.73, m | 20.9, CH2 | - |
| 14 | 1.39, m ov | 30.9, CH2 | 1.30, m | 38.1, CH2 | 1.48, m | 43.2, CH2 | 1.46, m | 25.1, CH2 | 1.48; 1.27, m | 32.5, CH2 | - |
| 15 | 1.37, m ov | 22.0, CH | 1.61, m | 27.3,CH | - | 68.8, C | 1.38, m | 39.1, CH2 | 1.81, m | 32.0, CH | - |
| 16 | 0.90, t (7.0) | 13.6, CH3 | 0.90, d (6.5) | 22.4, CH3 | 1.10, s | 29.3, CH3 | 3.59, m | 65.7, CH | 3.92; 3.84, dd (10.5; 6.0) | 68.5, CH2 | - |
| 17 | - | - | 0.90, d (6.5) | 22.4, CH3 | 1.10, s | 29.3, CH3 | 1.04, d (6.0) | 23.6, CH3 | - | 170.5, C | - |
| 18 | - | - | 2.00, s | 20.6, CH3 | - | ||||||
| 19 | - | - | 0.92, d (6.6) | 16.6, CH3 | - | ||||||
| Microorganism | [1] | [2] | [3] | [4] | [5] | Reference [µg/mL] |
|---|---|---|---|---|---|---|
| MIC [µg/mL] | ||||||
| Escherichia coli acrB JW0451-2 | - | 66.7 | - | - | - | 8.33 a |
| Bacillus subtilis DSM 10 | 66.7 | 33.4 | 66.7 | 16.7 | - | 0.52 b |
| Staphylococcus aureus Newman | - | 66.7 | - | 66.7 | - | 0.52 a |
| Mycobacterium smegmatis ATCC 700084 | - | 66.7 | - | - | - | 0.52 c |
| Mucor hiemalis DSM 2656 | 33.4 | 8.3 | - | - | - | 4.20 d |
| Cell line | IC50 [µM] | Ref e [µM] | ||||
| mouse fibroblast L-929 | 35.4 | 89.5 | - | - | - | 1.1 × 10−3 |
| HeLa cells KB-3.1 | - | 51.6 | 77.4 | 2.3 | - | 5.9 × 10−5 |
| human breast adenocarcinoma MCF-7 | - | - | - | 77.4 | - | - |
| human lung carcinoma A-549 | - | - | - | 1.7 | - | - |
| human prostate cancC-3 | - | - | - | 10.6 | - | - |
| ovarian carcinoma SKOV-3 | - | - | - | 17.1 | - | 2.9 × 10−4 |
| squamous cell carcinoma A-431 | - | - | - | 58.1 | - | 6.7 × 10−5 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Primahana, G.; Risdian, C.; Mozef, T.; Sudarman, E.; Köck, M.; Wink, J.; Stadler, M. Nonocarbolines A–E, β-Carboline Antibiotics Produced by the Rare Actinobacterium Nonomuraea sp. from Indonesia. Antibiotics 2020, 9, 126. https://doi.org/10.3390/antibiotics9030126
Primahana G, Risdian C, Mozef T, Sudarman E, Köck M, Wink J, Stadler M. Nonocarbolines A–E, β-Carboline Antibiotics Produced by the Rare Actinobacterium Nonomuraea sp. from Indonesia. Antibiotics. 2020; 9(3):126. https://doi.org/10.3390/antibiotics9030126
Chicago/Turabian StylePrimahana, Gian, Chandra Risdian, Tjandrawati Mozef, Enge Sudarman, Matthias Köck, Joachim Wink, and Marc Stadler. 2020. "Nonocarbolines A–E, β-Carboline Antibiotics Produced by the Rare Actinobacterium Nonomuraea sp. from Indonesia" Antibiotics 9, no. 3: 126. https://doi.org/10.3390/antibiotics9030126
APA StylePrimahana, G., Risdian, C., Mozef, T., Sudarman, E., Köck, M., Wink, J., & Stadler, M. (2020). Nonocarbolines A–E, β-Carboline Antibiotics Produced by the Rare Actinobacterium Nonomuraea sp. from Indonesia. Antibiotics, 9(3), 126. https://doi.org/10.3390/antibiotics9030126