Benzamide Derivatives Targeting the Cell Division Protein FtsZ: Modifications of the Linker and the Benzodioxane Scaffold and Their Effects on Antimicrobial Activity

 , , ,
, , , 
Abstract
:1. Introduction
2. Results and Discussion
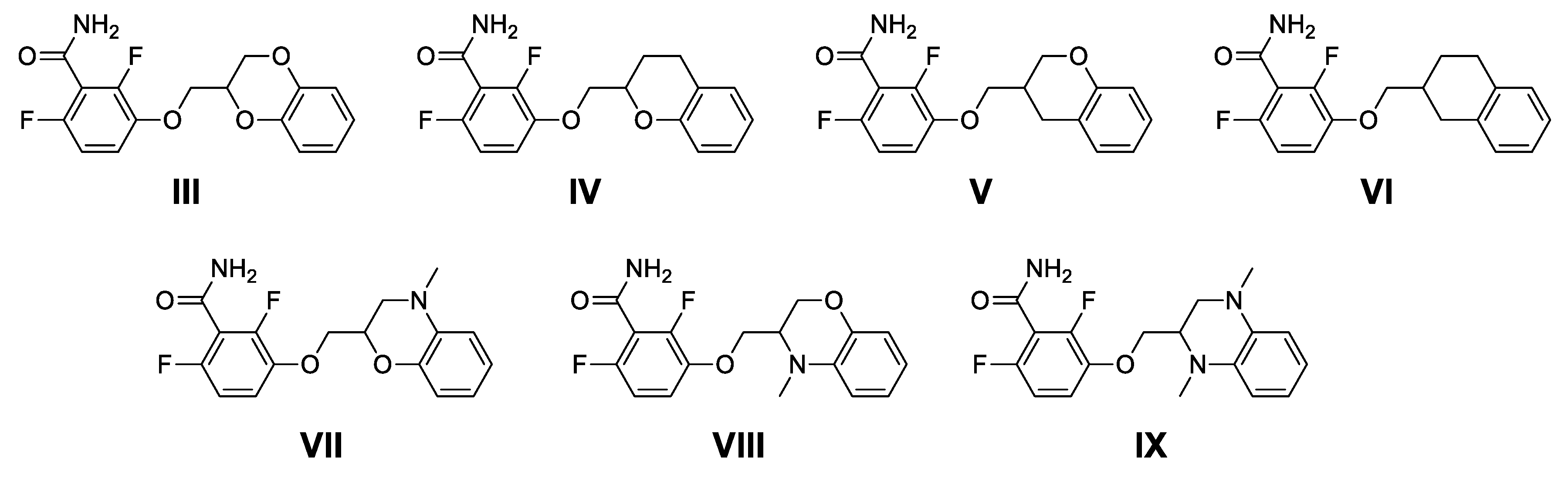
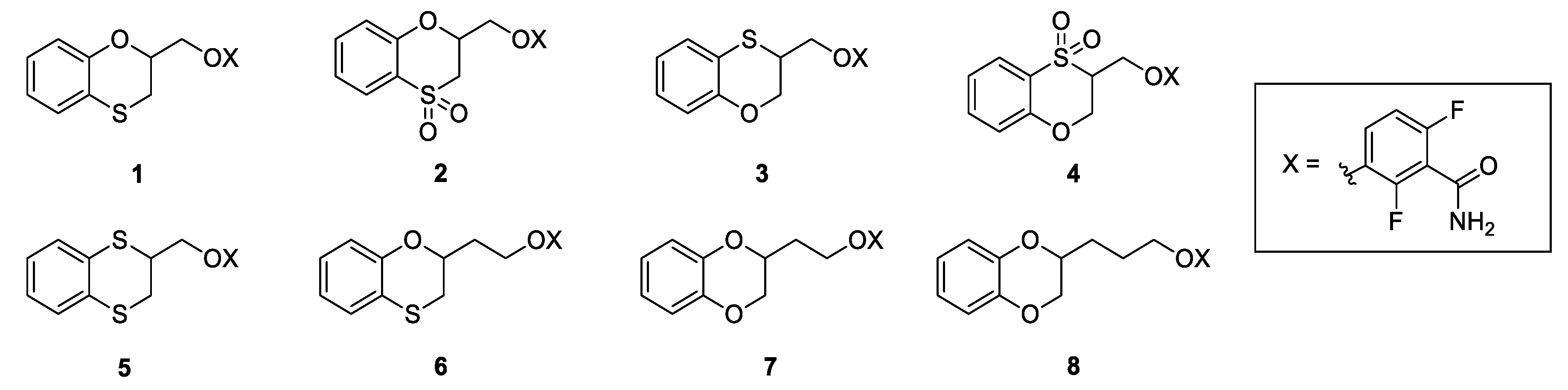
2.1. Design and Physico-chemical Profile Calculations
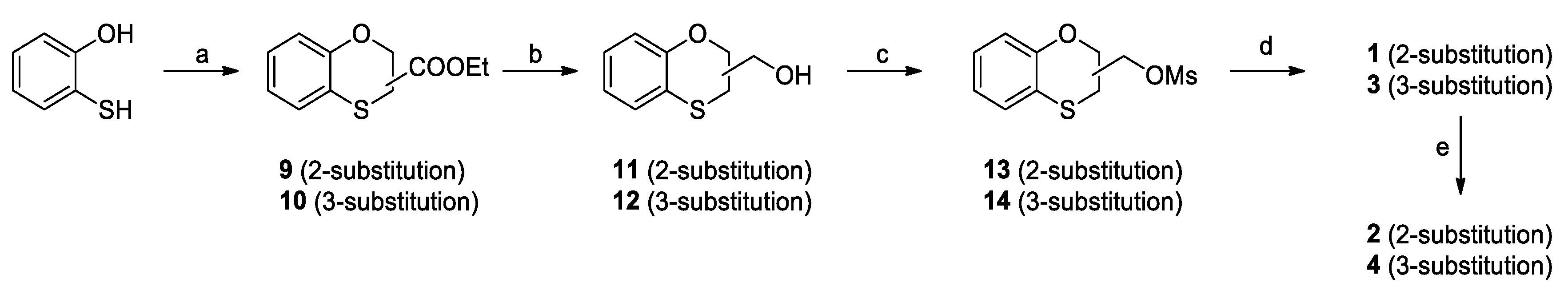
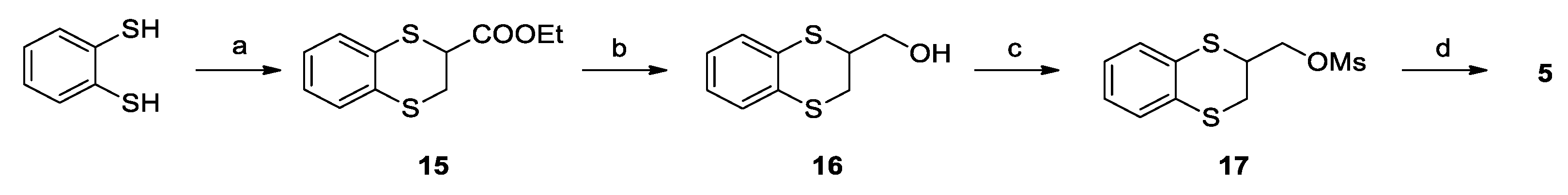
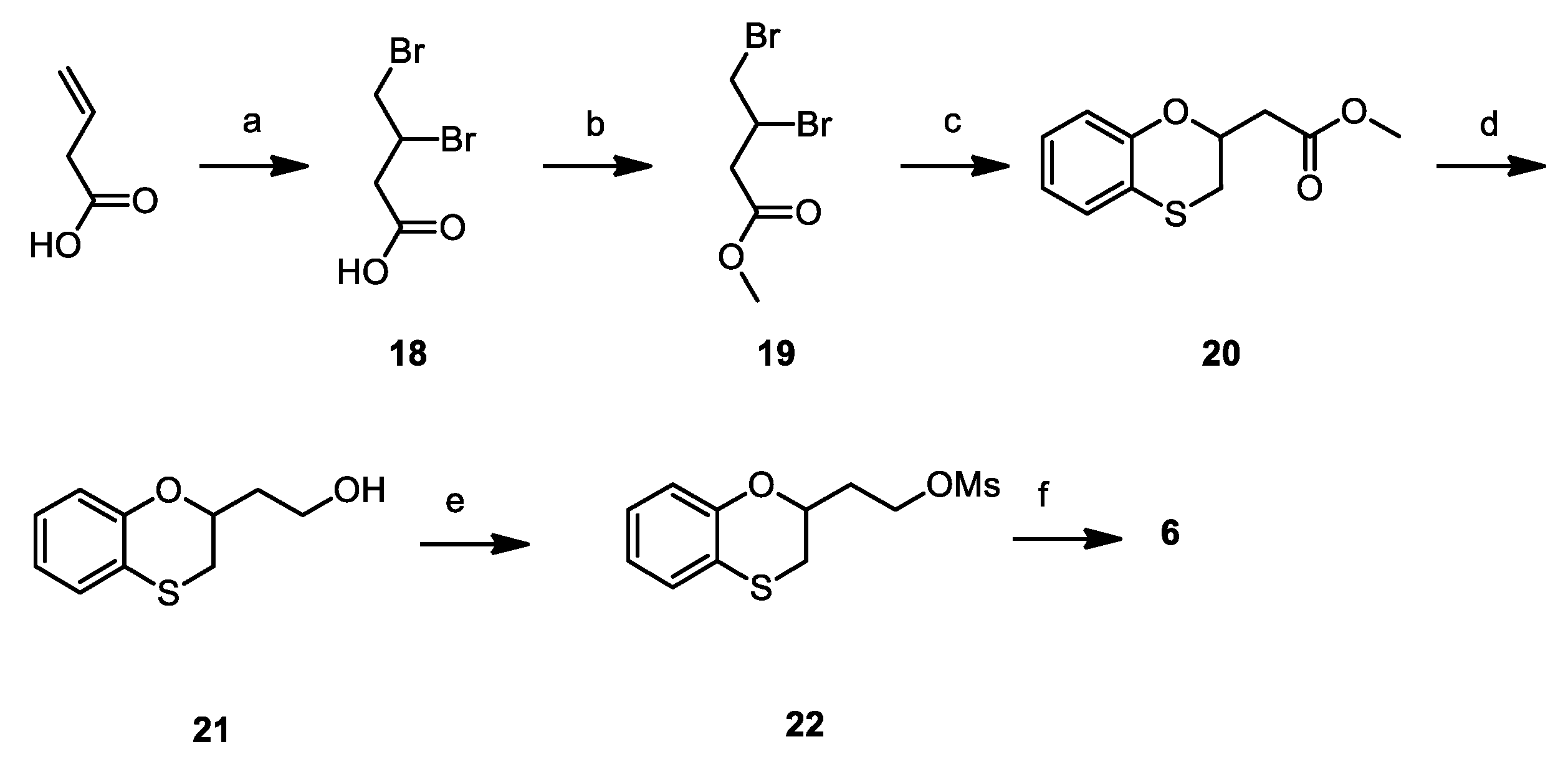
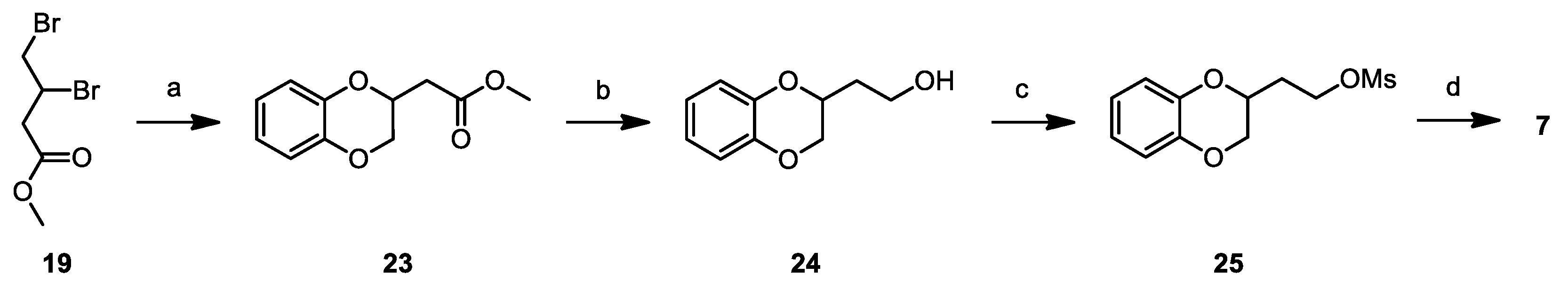
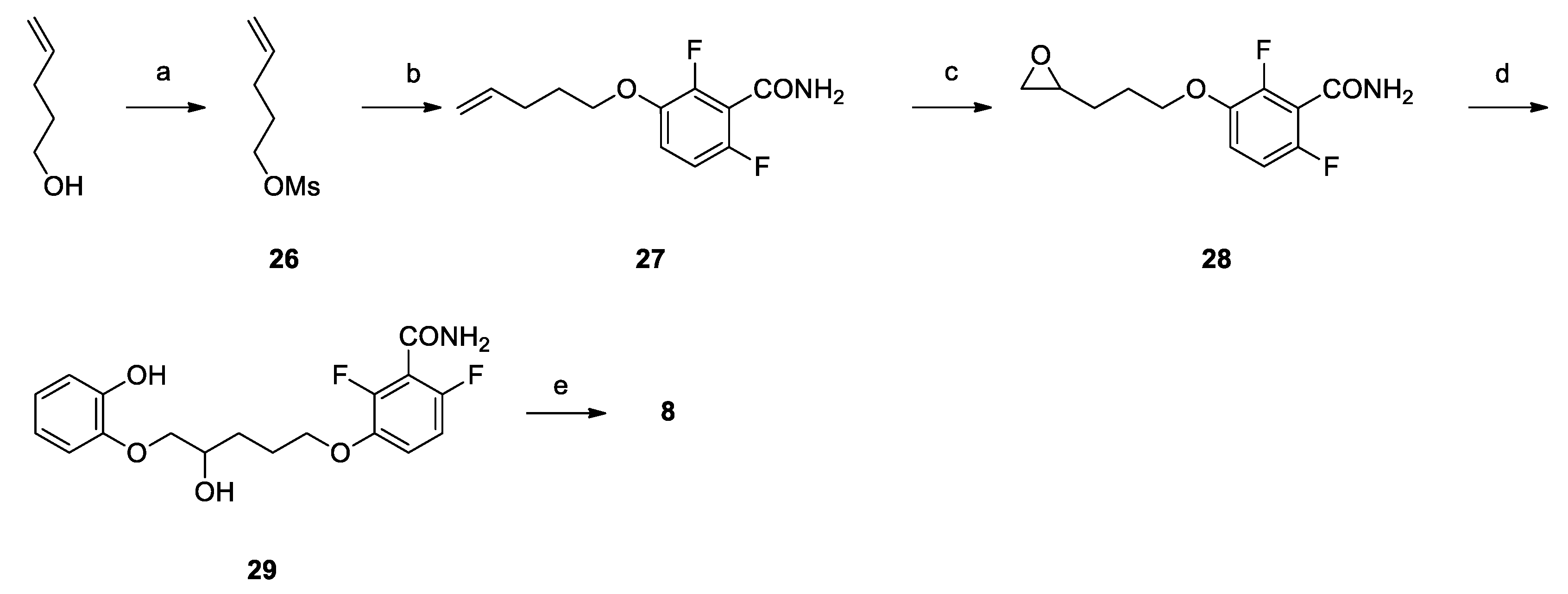
2.2. Chemistry
2.3. Antimicrobial Activity
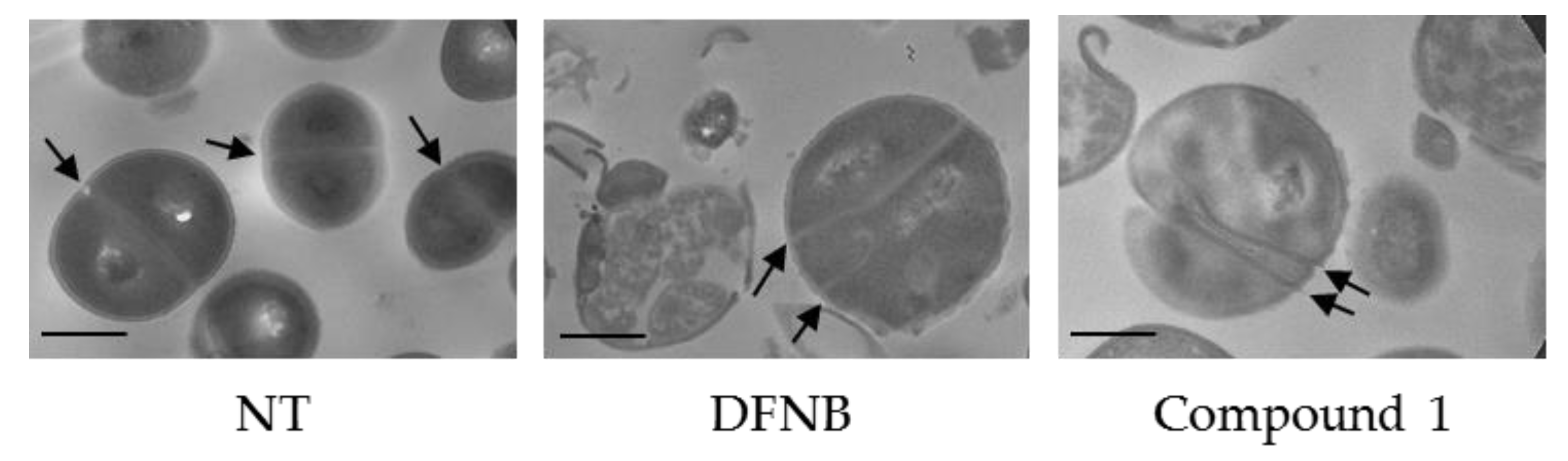
2.4. Microscopy Evaluation
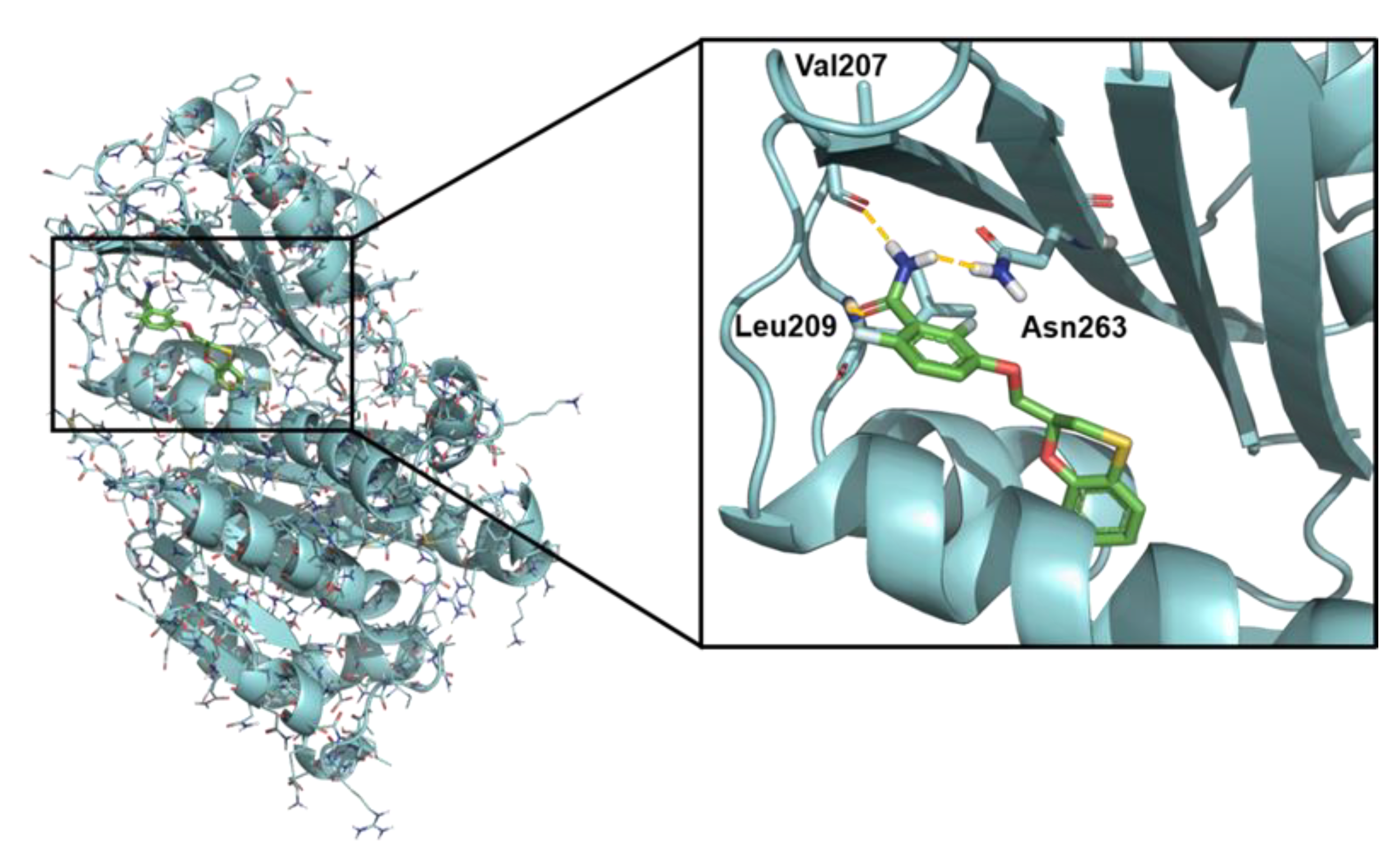
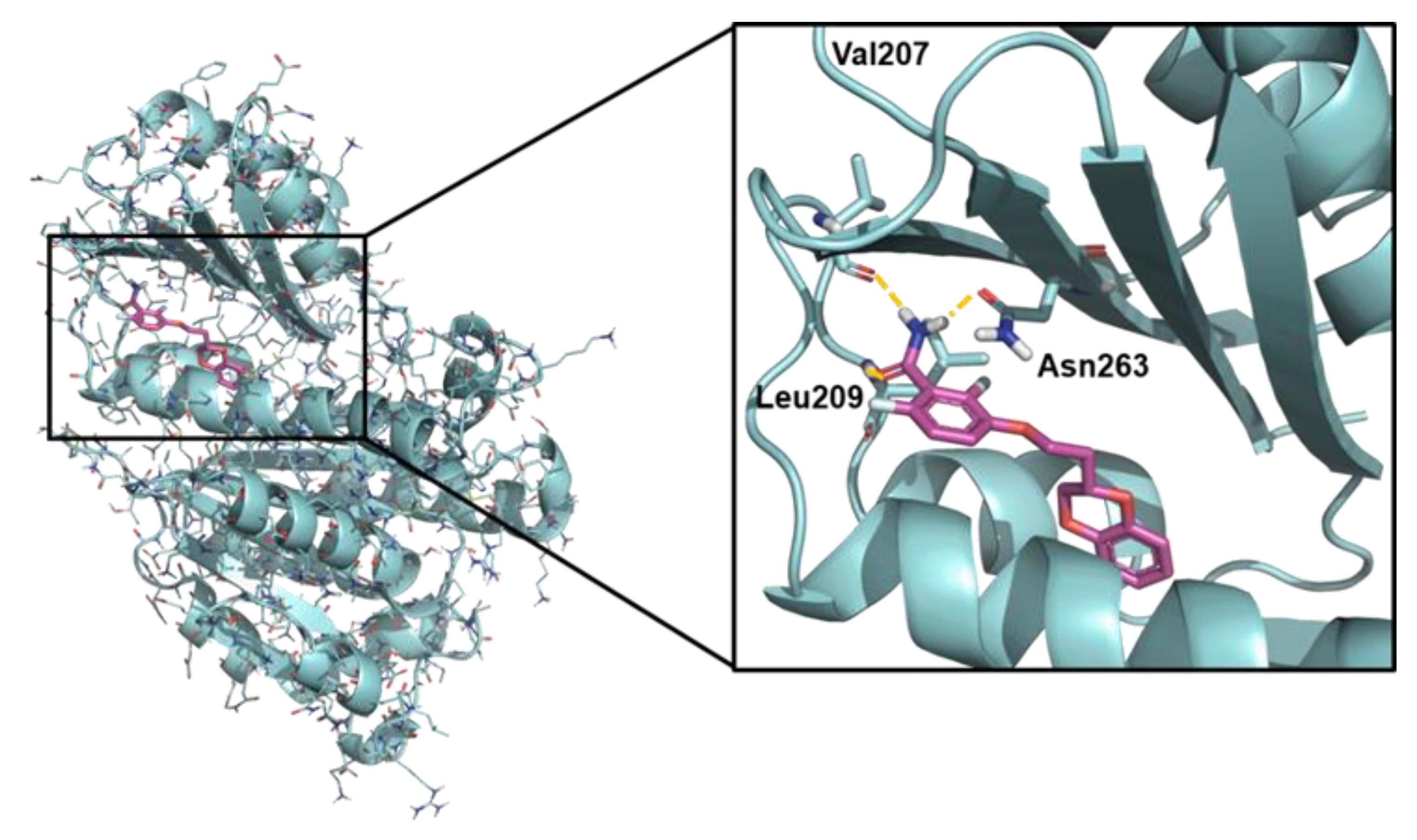
2.5. Computational Studies
3. Materials and Methods
3.1. Chemistry
Synthesis
3.2. Cells
3.3. Antibacterial Activity
3.3.1. MSSA, MRSA and ESBL Protocols
3.3.2. MDRSA, E. coli N43, E. coli D22 Protocols
3.4. Thiazolyl Blue Tetrazolium Bromide (MTT) Cytotoxicity Assay
3.5. Transmission Electron Microscopy
3.6. Computational Studies
Supplementary Files
Supplementary File 1Author Contributions
Funding
Conflicts of Interest
References
- CDC. Antibiotic Resistance Threats in the United States; CDC: Atlanta, GA, USA, 2019. [Google Scholar]
- Luepke, K.H.; Suda, K.J.; Boucher, H.; Russo, R.L.; Bonney, M.W.; Hunt, T.D.; Mohr, J.F. Past, present, and future of antibacterial economics: Increasing bacterial resistance, limited antibiotic pipeline, and societal implications. Pharmacotherapy 2017, 37, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Tacconelli, E.; Carrara, E.; Savoldi, A.; Harbarth, S.; Mendelson, M.; Monnet, D.L.; Pulcini, C.; Kahlmeter, G.; Kluytmans, J.; Carmeli, Y.; et al. Discovery, research, and development of new antibiotics: The WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis. 2018, 18, 318–327. [Google Scholar] [CrossRef]
- Rice, L.B. Federal funding for the study of antimicrobial resistance in nosocomial pathogens: No ESKAPE. J. Infect. Dis. 2008, 197, 1079–1081. [Google Scholar] [CrossRef] [PubMed]
- Munita, J.M.; Arias, C.A. Mechanisms of antibiotic resistance. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laws, M.; Shaaban, A.; Rahman, K.M. Antibiotic resistance breakers: Current approaches and future directions. FEMS Microbiol. Rev. 2019, 43, 490–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.; Deng, Z.; Yan, A. Bacterial multidrug efflux pumps: Mechanisms, physiology and pharmacological exploitations. Biochem. Biophys. Res. Commun. 2014, 453, 254–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piddock, L.J.V. Clinically relevant chromosomally encoded multidrug resistance efflux pumps in bacteria. Clin. Microbiol. Rev. 2006, 19, 382–402. [Google Scholar] [CrossRef] [Green Version]
- Nikaido, H. Structure and mechanism of RND-type multidrug efflux pumps. Adv. Enzymol. Relat. Areas Mol. Biol. 2011, 77, 1–60. [Google Scholar] [CrossRef] [Green Version]
- Margalit, D.N.; Romberg, L.; Mets, R.B.; Hebert, A.M.; Mitchison, T.J.; Kirschner, M.W.; RayChaudhuri, D. Targeting cell division: Small-molecule inhibitors of FtsZ GTPase perturb cytokinetic ring assembly and induce bacterial lethality. Proc. Natl. Acad. Sci. USA. 2004, 101, 11821–11826. [Google Scholar] [CrossRef] [Green Version]
- Kaul, M.; Zhang, Y.; Parhi, A.K.; LaVoie, E.J.; Pilch, D.S. Inhibition of RND-type efflux pumps confers the FtsZ-directed prodrug TXY436 with activity against Gram-negative bacteria. Biochem. Pharmacol. 2014, 89, 321–328. [Google Scholar] [CrossRef]
- Straniero, V.; Sebastián Pérez, V.; Hrast, M.; Zanotto, C.; Casiraghi, A.; Suigo, L.; Zdovc, I.; Radaelli, A.; de Giuli Morghen, C.; Valoti, E.; et al. Benzodioxane-benzamides as antibacterial agents: Computational and SAR studies to evaluate the influence of the 7-substitution in FtsZ interaction. ChemMedChem 2020, 2, 195–209. [Google Scholar] [CrossRef] [PubMed]
- Erickson, H.P. FtsZ, a tubulin homologue in prokaryote cell division. Trends Cell Biol. 1997, 7, 362–367. [Google Scholar] [CrossRef]
- Vaughan, S.; Wickstead, B.; Gull, K.; Addinall, S.G. Molecular evolution of FtsZ protein sequences encoded within the genomes of archaea, bacteria, and eukaryota. J. Mol. Evol. 2004, 58, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Casiraghi, A.; Suigo, L.; Valoti, E.; Straniero, V. Targeting bacterial cell division: A binding site-centered approach to the most promising inhibitors of the essential protein FtsZ. Antibiotics 2020, 9, 69. [Google Scholar] [CrossRef] [Green Version]
- Haeusser, D.P.; Margolin, W. Splitsville: Structural and functional insights into the dynamic bacterial Z ring. Nat. Rev. Microbiol. 2016, 14, 305–319. [Google Scholar] [CrossRef] [Green Version]
- Margolin, W. FtsZ and the division of prokaryotic cells and organelles. Nat. Rev. Mol. Cell Biol. 2005, 6, 862–871. [Google Scholar] [CrossRef] [Green Version]
- Lomovskaya, O.; Lee, A.; Hoshino, K.; Ishida, H.; Mistry, A.; Warren, M.S.; Boyer, E.; Chamberland, S.; Lee, V.J. Use of a genetic approach to evaluate the consequences of inhibition of efflux pumps in pseudomonas aeruginosa. Antimicrob. Agents Chemother. 1999, 43, 1340–1346. [Google Scholar] [CrossRef] [Green Version]
- Sulavik, M.C.; Houseweart, C.; Cramer, C.; Jiwani, N.; Murgolo, N.; Greene, J.; DiDomenico, B.; Shaw, K.J.; Miller, G.H.; Hare, R.; et al. Antibiotic susceptibility profiles of Escherichia coli strains lacking multidrug efflux pump genes. Antimicrob. Agents Chemother. 2001, 45, 1126–1136. [Google Scholar] [CrossRef] [Green Version]
- Poole, K. Efflux-mediated multiresistance in Gram-negative bacteria. Clin. Microbiol. Infect. 2004, 10, 12–26. [Google Scholar] [CrossRef] [Green Version]
- Chiodini, G.; Pallavicini, M.; Zanotto, C.; Bissa, M.; Radaelli, A.; Straniero, V.; Bolchi, C.; Fumagalli, L.; Ruggeri, P.; de Giuli Morghen, C.; et al. Benzodioxane-benzamides as new bacterial cell division inhibitors. Eur. J. Med. Chem. 2015, 89, 252–265. [Google Scholar] [CrossRef]
- Straniero, V.; Pallavicini, M.; Chiodini, G.; Zanotto, C.; Volontè, L.; Radaelli, A.; Bolchi, C.; Fumagalli, L.; Sanguinetti, M.; Menchinelli, G.; et al. 3-(Benzodioxan-2-ylmethoxy)-2,6-difluorobenzamides bearing hydrophobic substituents at the 7-position of the benzodioxane nucleus potently inhibit methicillin-resistant Sa and Mtb cell division. Eur. J. Med. Chem. 2016, 120, 227–243. [Google Scholar] [CrossRef] [PubMed]
- Straniero, V.; Zanotto, C.; Straniero, L.; Casiraghi, A.; Duga, S.; Radaelli, A.; de Giuli Morghen, C.; Valoti, E. 2,6-Difluorobenzamide inhibitors of bacterial cell division protein FtsZ: Design, synthesis, and structure-activity relationships. ChemMedChem 2017, 12, 1303–1318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casiraghi, A.; Valoti, E.; Suigo, L.; Artasensi, A.; Sorvillo, E.; Straniero, V. How reaction conditions may influence the regioselectivity in the synthesis of 2,3-Dihydro-1,4-benzoxathiine Derivatives. J. Org. Chem. 2018, 83, 13217–13227. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.D.; Kanwar, S.; Rajpoot, S. Aziridines as templates: A general strategy for the stereospecific synthesis of 2-azetidinones. J. Heterocycl. Chem. 2006, 43, 11–19. [Google Scholar] [CrossRef]
- Fischer, D.F.; Sarpong, R. Total synthesis of (+)-complanadine A using an iridium-catalyzed pyridine C-H functionalization. J. Am. Chem. Soc. 2010, 132, 5926–5927. [Google Scholar] [CrossRef] [Green Version]
- Czaplewski, L.G.; Collins, I.; Boyd, E.A.; Brown, D.; East, S.P.; Gardiner, M.; Fletcher, R.; Haydon, D.J.; Henstock, V.; Ingram, P.; et al. Antibacterial alkoxybenzamide inhibitors of the essential bacterial cell division protein FtsZ. Bioorg. Med. Chem. Lett. 2009, 19, 524–527. [Google Scholar] [CrossRef]
- Bissa, M.; Zanotto, C.; Pacchioni, S.; Volonté, L.; Venuti, A.; Lembo, D.; de Giuli Morghen, C.; Radaelli, A. The L1 protein of human papilloma virus 16 expressed by a fowlpox virus recombinant can assemble into virus-like particles in mammalian cell lines but elicits a non-neutralising humoral response. Antivir. Res. 2015, 116, 67–75. [Google Scholar] [CrossRef]
- Schrödinger Release 2015-4: LigPrep; Schrödinger LLC: New York, NY, USA, 2015.
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Banks, J.L.; Beard, H.S.; Cao, Y.; Cho, A.E.; Damm, W.; Farid, R.; Felts, A.K.; Halgren, T.A.; Mainz, D.T.; Maple, J.R.; et al. Integrated modeling program, applied chemical theory (IMPACT). J. Comput. Chem. 2005, 26, 1752–1780. [Google Scholar] [CrossRef] [Green Version]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed] [Green Version]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Structure | MIC (μG/ML) | FtsZ Binding Site | Reference | ||
|---|---|---|---|---|---|---|
| Methicillin Sensitive S. aureus | E. coli WildType | E. coli AcrAB– | ||||
| Z2 |  | 0.6 | 20 | 2.5 | GTP-binding site | [10] |
| Z3 |  | 2.1 | >40 | 4 | GTP-binding site | [10] |
| TXY436 |  | 1.0 | >40 | 8 | Interdomain site | [11] |
| Compound I |  | 0.6 | >40 | 16 | Interdomain site | [12] |
| Compound II |  | 0.6 | >40 | 16 | Interdomain site | [12] |
| Dipole | QlogPo/w | PSA | QPPCaco | QPPMDCK | |
|---|---|---|---|---|---|
| III | 7.215 | 2.920 | 75.393 | 1.036.486 | 1.249.904 |
| 1 | 6.647 | 3.417 | 66.311 | 1.032.637 | 2.032.874 |
| 2 | 8.801 | 1.756 | 105.241 | 105.625 | 103.165 |
| 3 | 7.504 | 3.301 | 66.536 | 1.033.096 | 1.677.064 |
| 4 | 7.720 | 1.674 | 104.281 | 316.001 | 306.684 |
| 5 | 6.968 | 3.739 | 58.274 | 1.032.481 | 2.579.522 |
| 6 | 6.036 | 3.735 | 64.832 | 1.033.074 | 1.953.979 |
| 7 | 6.615 | 3.294 | 74.783 | 1.032.977 | 1.213.317 |
| 8 | 7.188 | 3.660 | 74.847 | 1.032.656 | 1.212.923 |
| Cpd | MSSA ATCC 29213 | MRSA ATCC 43300 | MDRSA 12.1 | MDRSA 11.7 | MRC-5 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| MIC (µg/mL) | MBC (µg/mL) | TI90 | MIC (µg/mL) | MBC (µg/mL) | TI90 | MIC (µg/mL) | MIC (µg/mL) | TD90 (µg/mL) | |||
| III | 5 | 80 | 16 | - | 5 | 80 | 16 | - | 8 | 16 | - |
| 1 | 1 | 1 | 1 | >800 | 1 | 1 | 1 | >800 | 2 | 2 | >800 |
| 2 | >100 | >100 | - | - | >100 | >100 | - | - | - | - | - |
| 3 | 20 | 20 | 1 | 10 | 20 | 20 | 1 | 10 | 32 | 32 | 200 |
| 4 | >100 | >100 | - | - | >100 | >100 | - | - | - | - | - |
| 5 | 5 | 10 | 2 | 20 | 5 | 10 | 2 | 20 | 8 | 8 | 200 |
| 6 | 2.5 | 5 | 2 | - | 2.5 | 5 | 2 | - | 4 | 8 | - |
| 7 | 5 | 5 | 1 | 76 | 5 | 5 | 1 | 76 | 4 | 4 | 380 |
| 8 | 10 | 100 | 10 | - | 10 | 100 | 10 | - | 16 | 32 | - |
| Cpd | E. coli ATCC 25922 | ESBL E. coli | E. coli N43 |
|---|---|---|---|
| MIC (µg/mL) | MIC (µg/mL) | MIC (µg/mL) | |
| I | >100 | >100 | 16 |
| II | >100 | >100 | 16 |
| III | >100 | >100 | >128 |
| 1 | >100 | >100 | 64 |
| 2 | >100 | >100 | - |
| 3 | >100 | >100 | 128 |
| 4 | >100 | >100 | - |
| 5 | >100 | >100 | >128 |
| 6 | >100 | >100 | >128 |
| 7 | >100 | >100 | 8 |
| 8 | >100 | >100 | >128 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Straniero, V.; Suigo, L.; Casiraghi, A.; Sebastián-Pérez, V.; Hrast, M.; Zanotto, C.; Zdovc, I.; De Giuli Morghen, C.; Radaelli, A.; Valoti, E. Benzamide Derivatives Targeting the Cell Division Protein FtsZ: Modifications of the Linker and the Benzodioxane Scaffold and Their Effects on Antimicrobial Activity. Antibiotics 2020, 9, 160. https://doi.org/10.3390/antibiotics9040160
Straniero V, Suigo L, Casiraghi A, Sebastián-Pérez V, Hrast M, Zanotto C, Zdovc I, De Giuli Morghen C, Radaelli A, Valoti E. Benzamide Derivatives Targeting the Cell Division Protein FtsZ: Modifications of the Linker and the Benzodioxane Scaffold and Their Effects on Antimicrobial Activity. Antibiotics. 2020; 9(4):160. https://doi.org/10.3390/antibiotics9040160
Chicago/Turabian StyleStraniero, Valentina, Lorenzo Suigo, Andrea Casiraghi, Victor Sebastián-Pérez, Martina Hrast, Carlo Zanotto, Irena Zdovc, Carlo De Giuli Morghen, Antonia Radaelli, and Ermanno Valoti. 2020. "Benzamide Derivatives Targeting the Cell Division Protein FtsZ: Modifications of the Linker and the Benzodioxane Scaffold and Their Effects on Antimicrobial Activity" Antibiotics 9, no. 4: 160. https://doi.org/10.3390/antibiotics9040160
APA StyleStraniero, V., Suigo, L., Casiraghi, A., Sebastián-Pérez, V., Hrast, M., Zanotto, C., Zdovc, I., De Giuli Morghen, C., Radaelli, A., & Valoti, E. (2020). Benzamide Derivatives Targeting the Cell Division Protein FtsZ: Modifications of the Linker and the Benzodioxane Scaffold and Their Effects on Antimicrobial Activity. Antibiotics, 9(4), 160. https://doi.org/10.3390/antibiotics9040160