Proteomic Analysis of Biomaterial Surfaces after Contacting with Body Fluids by MALDI-ToF Mass Spectroscopy

 ,
,
Abstract
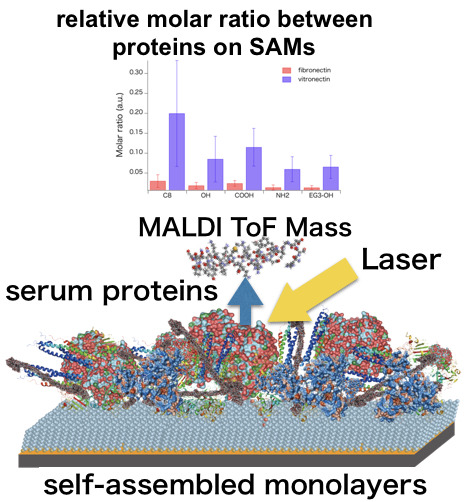
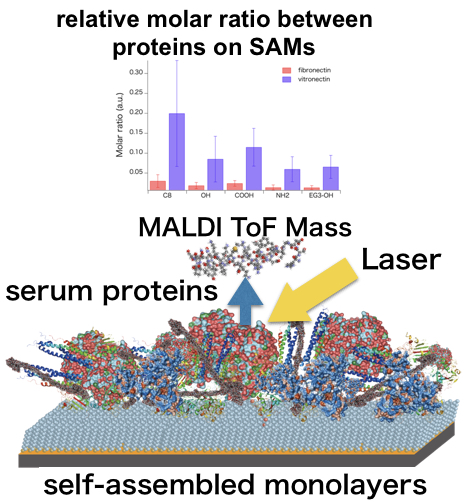
:
1. Introduction
2. Materials and Methods
2.1. Self-Assembled Monolayers and Their Substrates
2.2. QCM Measurements
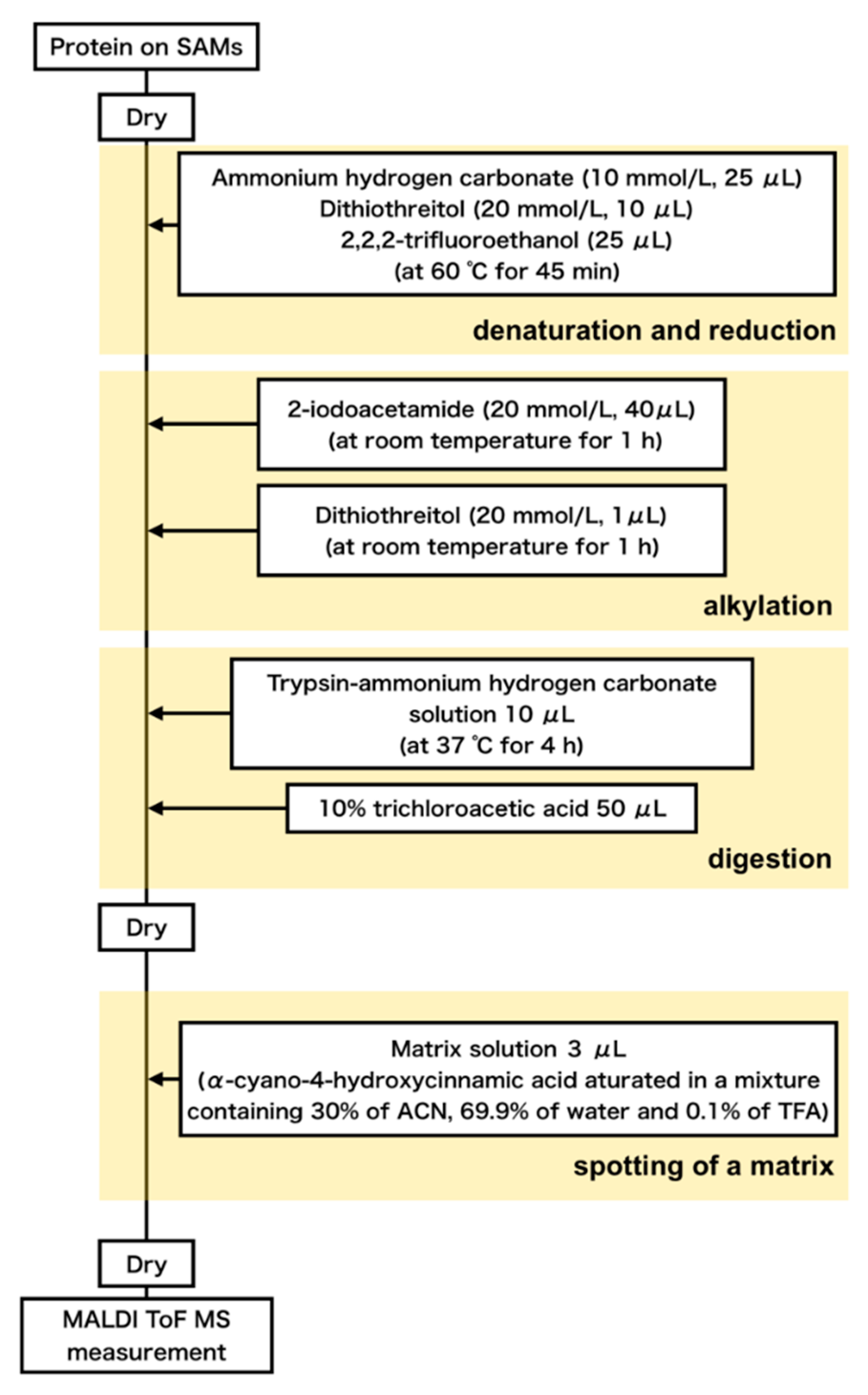
2.3. Formation of Protein Layer on SAMs from FBS for Peptide Mass Fingerprinting
2.4. Digestion of the Serum Proteins Adsorbed on the SAMs
2.5. Standard Samples to Calibrate Peak Intensity
2.6. MALDI-ToF Mass Spectroscopic Measurements
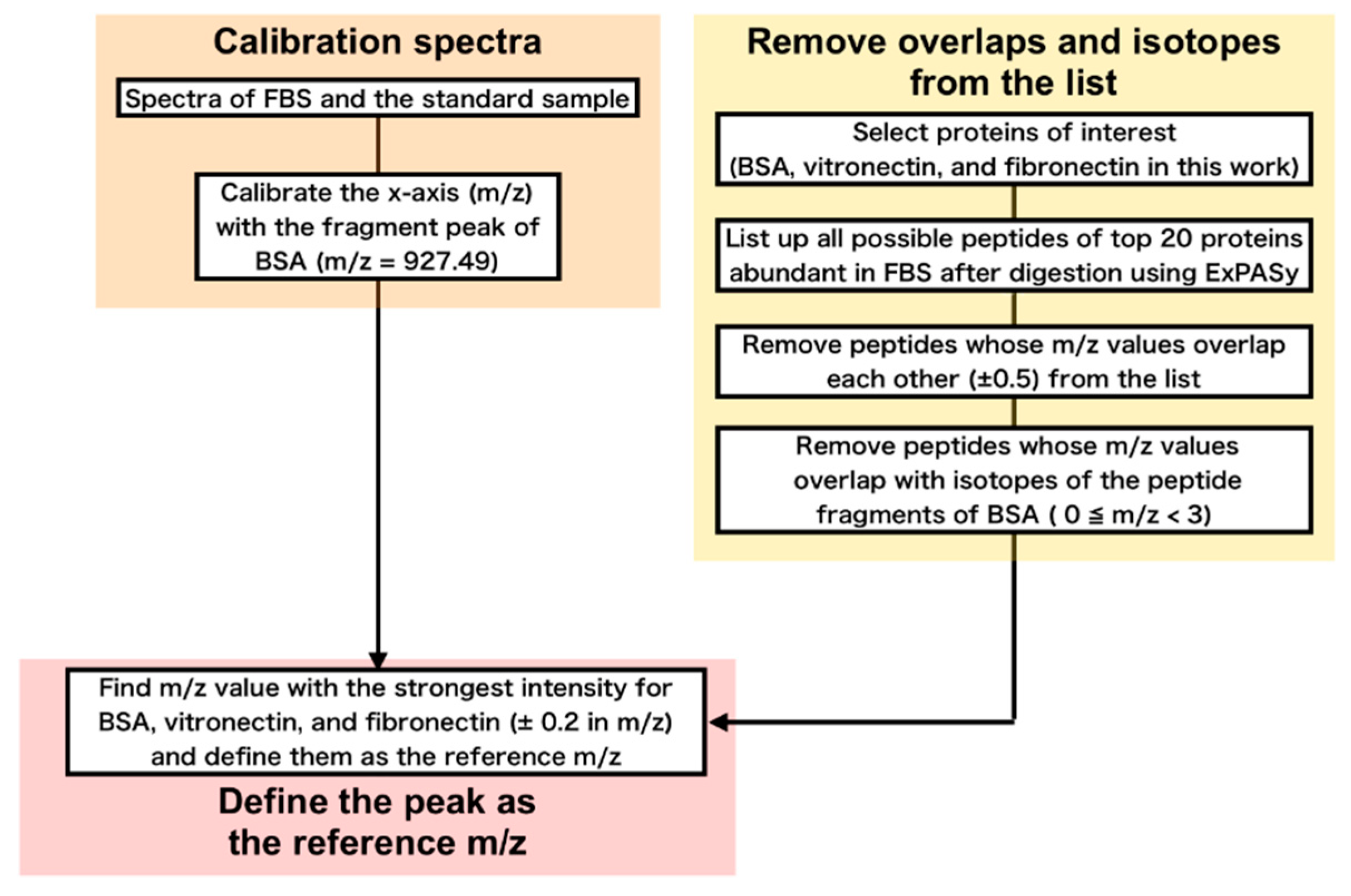
2.7. Analysis of the Obtained Mass Spectra
2.7.1. Finding m/z Values Unique for Proteins of Interest
2.7.2. Conversion from Peak Intensities to Molar Ratio
3. Results and Discussion
4. Summary and Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tanaka, M.; Hayashi, T.; Morita, S. The roles of water molecules at the biointerface of medical polymers. Polym. J. 2013, 45, 701–710. [Google Scholar] [CrossRef]
- Kane, R.S.; Takayama, S.; Ostuni, E.; Ingber, D.E.; Whitesides, G.M. Patterning proteins and cells using soft lithography. Biomaterials 1999, 20, 2363–2376. [Google Scholar] [CrossRef]
- Flaim, C.J.; Chien, S.; Bhatia, S.N. An extracellular matrix microarray for probing cellular differentiation. Nat. Methods 2005, 2, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Watt, F.M.; Huck, W.T. Role of the extracellular matrix in regulating stem cell fate. Nat. Rev. Mol. Cell Biol. 2013, 14, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Silin, V.V.; Weetall, H.; Vanderah, D.J. SPR Studies of the Nonspecific Adsorption Kinetics of Human IgG and BSA on Gold Surfaces Modified by Self-Assembled Monolayers (SAMs). J. Colloid Interface Sci. 1997, 185, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Nakata, S.; Kido, N.; Hayashi, M.; Hara, M.; Sasabe, H.; Sugawara, T.; Matsuda, T. Chemisorption of proteins and their thiol derivatives onto gold surfaces: Characterization based on electrochemical nonlinearity. Biophys. Chem. 1996, 62, 63–72. [Google Scholar] [CrossRef]
- Caruso, F.; Furlong, D.N.; Kingshott, P. Characterization of Ferritin Adsorption onto Gold. J. Colloid Interface Sci. 1997, 186, 129–140. [Google Scholar] [CrossRef]
- Hook, F.; Rodahl, M.; Kasemo, B.; Brzezinski, P. Structural changes in hemoglobin during adsorption to solid surfaces: Effects of pH, ionic strength, and ligand binding. Proc. Natl. Acad. Sci. USA 1998, 95, 12271–12276. [Google Scholar] [CrossRef] [Green Version]
- Renken, J.; Dahint, R.; Grunze, M.; Josse, F. Multifrequency evaluation of different immunosorbents on acoustic plate mode sensors. Anal. Chem. 1996, 68, 176–182. [Google Scholar] [CrossRef]
- Cavic, B.A.; Thompson, M. Adsorptions of plasma proteins and their elutabilities from a polysiloxane surface studied by an on-line acoustic wave sensor. Anal. Chem. 2000, 72, 1523–1531. [Google Scholar] [CrossRef]
- Gizeli, E.; Bender, F.; Rasmusson, A.; Saha, K.; Josse, F.; Cernosek, R. Sensitivity of the acoustic waveguide biosensor to protein binding as a function of the waveguide properties. Biosens. Bioelectron. 2003, 18, 1399–1406. [Google Scholar] [CrossRef]
- Labarre, D.; Vauthier, C.; Chauvierre, C.; Petri, B.; Muller, R.; Chehimi, M.M. Interactions of blood proteins with poly(isobutylcyanoacrylate) nanoparticles decorated with a polysaccharidic brush. Biomaterials 2005, 26, 5075–5084. [Google Scholar] [CrossRef] [PubMed]
- Zenhausern, F.; Adrian, M.; Descouts, P. Solution structure and direct imaging of fibronectin adsorption to solid surfaces by scanning force microscopy and cryo-electron microscopy. J. Electron Microsc. 1993, 42, 378–388. [Google Scholar]
- Lestelius, M.; Liedberg, B.; Lundstrom, I.; Tengvall, P. In vitro plasma protein adsorption and kallikrein formation on 3-mercaptopropionic acid, L-cysteine and glutathione immobilized onto gold. J. Biomed. Mater. Res. 1994, 28, 871–880. [Google Scholar] [CrossRef] [PubMed]
- Caruso, F.; Furlong, D.N.; Ariga, K.; Ichinose, I.; Kunitake, T. Characterization of polyelectrolyte-protein multilayer films by atomic force microscopy, scanning electron microscopy, and Fourier transform infrared reflection-absorption spectroscopy. Langmuir 1998, 14, 4559–4565. [Google Scholar] [CrossRef]
- Schwendel, D.; Dahint, R.; Herrwerth, S.; Schloerholz, M.; Eck, W.; Grunze, M. Temperature dependence of the protein resistance of poly- and oligo(ethylene glycol)-terminated alkanethiolate monolayers. Langmuir 2001, 17, 5717–5720. [Google Scholar] [CrossRef]
- Model, M.A.; Healy, K.E. Quantification of the surface density of a fluorescent label with the optical microscope. J. Biomed. Mater. Res. 2000, 50, 90–96. [Google Scholar] [CrossRef]
- Beverloo, H.B.; Vanschadewijk, A.; Bonnet, J.; Vandergeest, R.; Runia, R.; Verwoerd, N.P.; Vrolijk, J.; Ploem, J.S.; Tanke, H.J. Preparation and microscopic visualization of multicolor luminescent immunophosphors. Cytometry 1992, 13, 561–570. [Google Scholar] [CrossRef]
- Pappin, D.J.; Hojrup, P.; Bleasby, A.J. Rapid identification of proteins by peptide-mass fingerprinting. Curr. Biol. 1993, 3, 327–332. [Google Scholar] [CrossRef]
- Scheler, C.; Lamer, S.; Pan, Z.; Li, X.P.; Salnikow, J.; Jungblut, P. Peptide mass fingerprint sequence coverage from differently stained proteins on two-dimensional electrophoresis patterns by matrix assisted laser desorptiodionization-mass spectrometry (MALDI-MS). Electrophoresis 1998, 19, 918–927. [Google Scholar] [CrossRef]
- Sempf, K.; Arrey, T.; Gelperina, S.; Schorge, T.; Meyer, B.; Karas, M.; Kreuter, J. Adsorption of plasma proteins on uncoated PLGA nanoparticles. Eur. J. Pharm. Biopharm. 2013, 85, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Urbani, A.; Lupisella, S.; Sirolli, V.; Bucci, S.; Amoroso, L.; Pavone, B.; Pieroni, L.; Sacchetta, P.; Bonomini, M. Proteomic analysis of protein adsorption capacity of different haemodialysis membranes. Mol. BioSyst. 2012, 8, 1029–1039. [Google Scholar] [CrossRef] [PubMed]
- Urbani, A.; Sirolli, V.; Lupisella, S.; Levi-Mortera, S.; Pavone, B.; Pieroni, L.; Amoroso, L.; Di Vito, R.; Bucci, S.; Bernardini, S.; et al. Proteomic investigations on the effect of different membrane materials on blood protein adsorption during haemodialysis. Blood Transfus. 2012, 10, s101–s112. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, K.; Henriksen, J.R.; Andresen, T.L. Adsorption of cationic peptides to solid surfaces of glass and plastic. PLoS ONE 2015, 10, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirschhofer, F.; Rieder, A.; Prechtl, C.; Kuhl, B.; Sabljo, K.; Woll, C.; Obst, U.; Brenner-Weiss, G. Quartz crystal microbalance with dissipation coupled to on-chip MALDI-ToF mass spectrometry as a tool for characterising proteinaceous conditioning films on functionalised surfaces. Anal. Chim. Acta 2013, 802, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.Y.; Baker, H.; Hancock, W.S.; Fawaz, F.; McCaman, M.; Pungor, E. Proteomic analysis for the assessment of different lots of fetal bovine serum as a raw material for cell culture. Part IV. Application of proteomics to the manufacture of biological drugs. Biotechnol. Prog. 2006, 22, 1294–1300. [Google Scholar] [CrossRef]
- Shannahan, J.H.; Brown, J.M.; Chen, R.; Ke, P.C.; Lai, X.; Mitra, S.; Witzmann, F.A. Comparison of nanotube-protein corona composition in cell culture media. Small 2013, 9, 2171–2181. [Google Scholar] [CrossRef]
- Sakulkhu, U.; Mahmoudi, M.; Maurizi, L.; Coullerez, G.; Hofmann-Amtenbrink, M.; Vries, M.; Motazacker, M.; Rezaee, F.; Hofmann, H. Significance of surface charge and shell material of superparamagnetic iron oxide nanoparticle (SPION) based core/shell nanoparticles on the composition of the protein corona. Biomater. Sci. 2015, 3, 265–278. [Google Scholar] [CrossRef]
- Strojan, K.; Leonardi, A.; Bregar, V.B.; Krizaj, I.; Svete, J.; Pavlin, M. Dispersion of nanoparticles in different media importantly determines the composition of their protein corona. PLoS ONE 2017, 12, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Artimo, P.; Jonnalagedda, M.; Arnold, K.; Baratin, D.; Csardi, G.; De Castro, E.; Duvaud, S.; Flegel, V.; Fortier, A.; Gasteiger, E.; et al. ExPASy: SIB bioinformatics resource portal. Nucleic Acids Res. 2012, 40, W597–W603. [Google Scholar] [CrossRef]
- Available online: https://www.expasy.org/ (accessed on 22 December 2019).
- Bucknall, M.; Fung, K.Y.C.; Duncan, M.W. Practical quantitative biomedical applications of MALDI-TOF mass spectrometry. J. Am. Soc. Mass Spectrom. 2002, 13, 1015–1027. [Google Scholar] [CrossRef] [Green Version]
- Chang, R.; Asatyas, S. Water near bioinert self-assembled monolayers. Polym. J. 2018, 50, 563–571. [Google Scholar] [CrossRef]
- Hayashi, T.; Tanaka, Y. Mechanism underlying bioinertness of self-assembled monolayers of oligo(ethyleneglycol)-terminated alkanethiols on gold: protein adsorption, platelet adhesion, and surface forces. Phys. Chem. Chem. Phys. 2012, 14, 10196–10206. [Google Scholar] [CrossRef] [PubMed]
- Sekine, T.; Tanaka, Y. Evaluation of Factors To Determine Platelet Compatibility by Using Self-Assembled Monolayers with a Chemical Gradient. Langmuir 2015, 31, 7100–7105. [Google Scholar] [CrossRef]
- Harder, P.; Grunze, M. Molecular conformation in oligo(ethylene glycol)-terminated self-assembled monolayers on gold and silver surfaces determines their ability to resist protein adsorption. J. Phys. Chem. B 1998, 102, 426–436. [Google Scholar] [CrossRef]
- Sekine, T.; Asatyas, S. Surface force and vibrational spectroscopic analyses of interfacial water molecules in the vicinity of methoxy-tri(ethylene glycol)-terminated monolayers: mechanisms underlying the effect of lateral packing density on bioinertness. J Biomater Sci Polym Ed 2017, 28, 1231–1243. [Google Scholar] [CrossRef]
- Hayashi, T.; Hara, M. Nonfouling self-assembled monolayers: mechanisms underlying protein and cell resistance. Curr. Phys. Chem. 2011, 1, 90–98. [Google Scholar] [CrossRef]
- Yang, Z.; Jaeckisch, S.M. Enhanced binding of Aspergillus fumigatus spores to A549 epithelial cells and extracellular matrix proteins by a component from the spore surface and inhibition by rat lung lavage fluid. Thorax 2000, 55, 579–584. [Google Scholar] [CrossRef] [Green Version]
- Cuypers, P.A.; Willems, G.M. Adsorption kinetics of protein mixtures. A tentative explanation of the Vroman effect. Ann. N. Y. Acad. Sci. 1987, 516, 244–252. [Google Scholar] [CrossRef] [Green Version]
- Leonard, E.F.; Vroman, L. Is the Vroman effect of importance in the interaction of blood with artificial materials? J. Biomater. Sci. Polym. Ed 1991, 3, 95–107. [Google Scholar] [CrossRef]
- Yang, D.; Ramkissoon, K. High-accuracy peptide mass fingerprinting using peak intensity data with machine learning. J. Proteome Res. 2008, 7, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Timm, W.; Scherbart, A. Peak intensity prediction in MALDI-TOF mass spectrometry: a machine learning study to support quantitative proteomics. Bmc Bioinformatics 2008, 9, 443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gay, S.; Binz, P.A. Peptide mass fingerprinting peak intensity prediction: extracting knowledge from spectra. Proteomics 2002, 2, 1374–1391. [Google Scholar] [CrossRef]
- Stead, D.A.; Preece, A. Universal metrics for quality assessment of protein identifications by mass spectrometry. Mol. Cell. Proteomics 2006, 5, 1205–1211. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
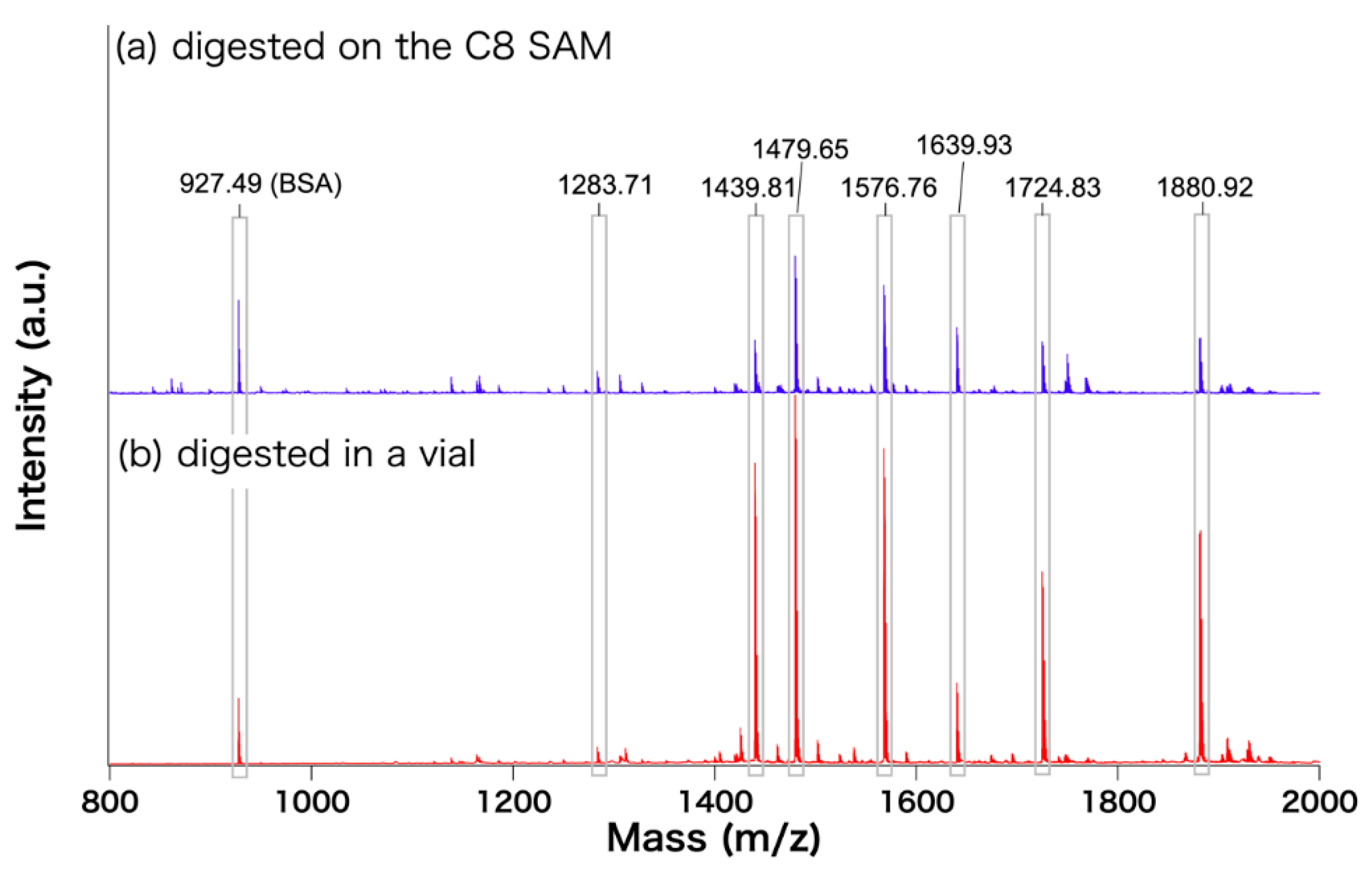{kind=link}
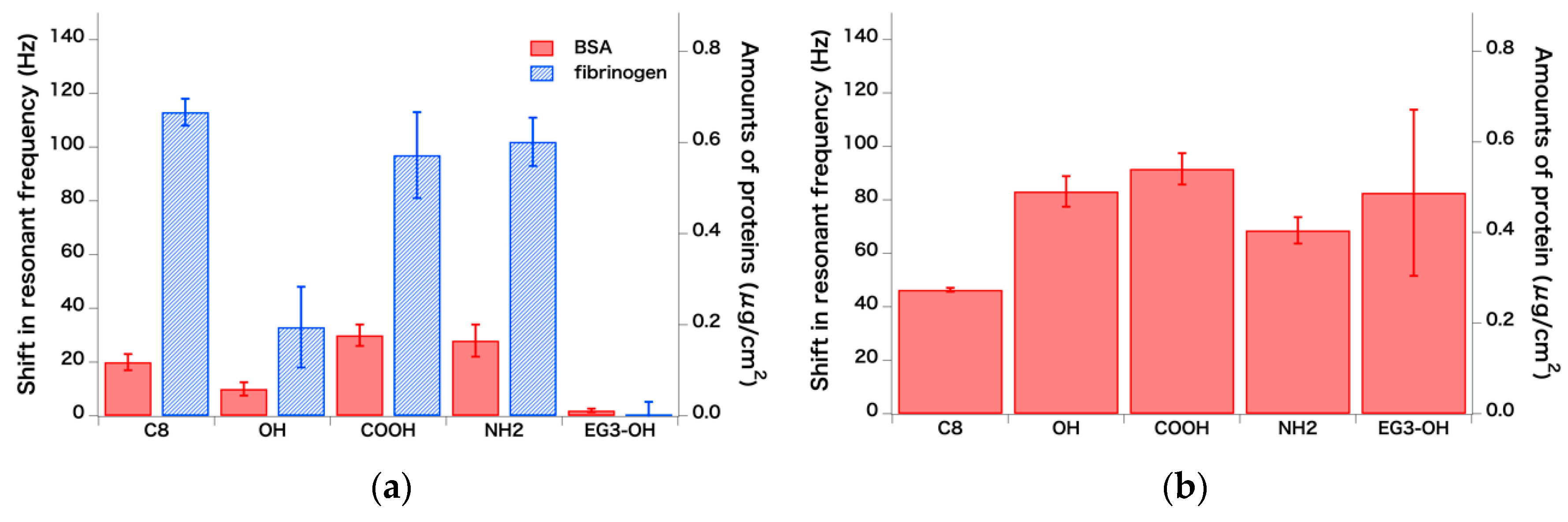{kind=link}
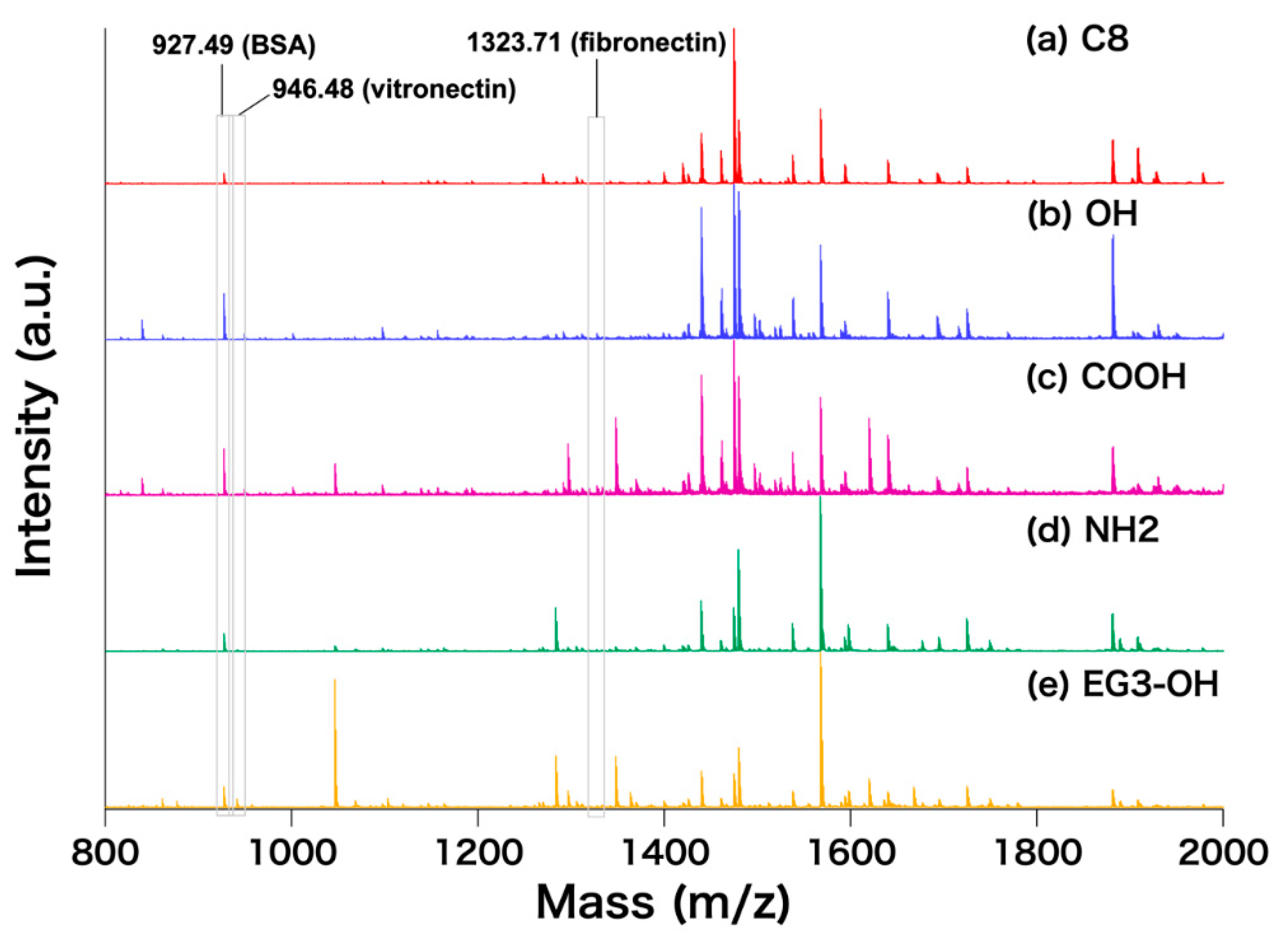{kind=link}
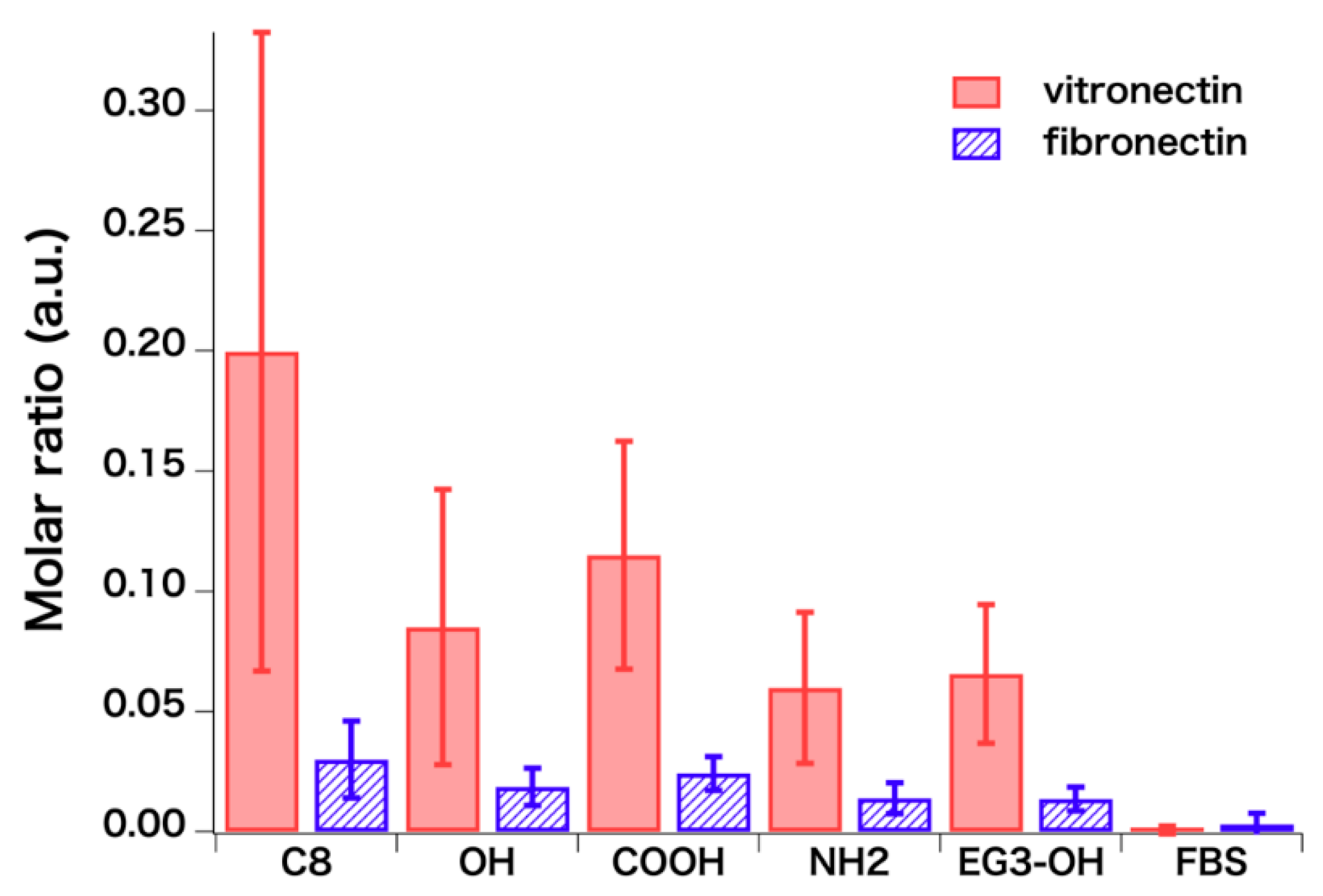{kind=link}
| Abbreviation | Chemical Structure of the Thiol Molecules |
|---|---|
| C8 | HS–(CH2)7–CH3 |
| COOH | HS–(CH2)11–COOH |
| OH | HS–(CH2)11–OH |
| NH2 | HS–(CH2)11–NH2 |
| EG3–OH | HS–(CH2)11–(O–CH2–CH2)3–OH |
| Protein | Molecular Weight | Reference m/z |
|---|---|---|
| Serum albumin | 69,293 | 927.493 |
| Alpha-1-antiproteinase | 46,104 | 873.468 |
| Plasminogen | 91,216 | 1426.640 |
| Cone cGMP-specific 3′,5′-cyclic phosphodiesterase subunit alpha’ | 98,798 | 1476.690 |
| Lactoperoxidase | 80,642 | 1423.830 |
| NADH-ubiquinone oxidoreductase 75 kDa subunit | 79,468 | 2048.070 |
| Alpha-2-HS-glycoprotein | 38,419 | 2058.130 |
| Kininogen-2 | 68,710 | 1998.000 |
| hemiferrin | 24,091 | 1715.900 |
| Integrin beta-1 | 88,094 | 1436.670 |
| Prothrombin | 70,506 | 1716.890 |
| Apolipoprotein A-I | 30,276 | 1772.920 |
| Antithrombin-III | 52,347 | 2062.050 |
| Beta-2-glycoprotein 1 | 38,252 | 1475.770 |
| Alpha-2-antiplasmin | 54,711 | 1728.820 |
| Protein AMBP | 39,235 | 2058.930 |
| Hemoglobin fetal subunit beta | 15,859 | 1434.670 |
| Alpha 1-antichymotrypsin | 28,571 | 2210.180 |
| Apolipoprotein A-II | 11,202 | 657.324 |
| Hemoglobin subunit alpha | 15,184 | 2986.440 |
| fibronectin | 272,154 | 1323.710 |
| vitronectin | 53,615 | 946.489 |
| Protein | Relative Peak Intensity of the Reference m/z for Each Protein with Respect to that for BSA | |||||
|---|---|---|---|---|---|---|
| FBS | C8 | OH | COOH | NH2 | EG3-OH | |
| Serum albumin | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| Alpha-1-antiproteinase | 0.00719 ± 0.00421 | 0.212 ± 0.138 | 0.0843 ± 0.0333 | 0.109 ± 0.076 | 0.172 ± 0.084 | 0.111 ± 0.056 |
| Plasminogen | 0.950 ± 0.526 | 2.59 ± 1.81 | 0.746 ± 0.609 | 1.31 ± 0.50 | 1.54 ± 0.69 | 0.846 ± 0.314 |
| Cone cGMP-specific 3′,5′-cyclic phosphodiesterase subunit alpha’ | 0.0639 ± 0.0442 | 1.24 ± 0.82 | 0.279 ± 0.177 | 0.298 ± 0.186 | 0.972 ± 0.537 | 1.17 ± 0.77 |
| Lactoperoxidase | 0.0132 ± 0.0075 | 0.166 ± 0.171 | 0.0575 ± 0.0308 | 0.0692 ± 0.0269 | 0.311 ± 0.539 | 0.222 ± 0.160 |
| NADH-ubiquinone oxidoreductase 75 kDa subunit | 0.0763 ± 0.0658 | 1.62 ± 2.03 | 0.101 ± 0.063 | 0.231 ± 0.157 | 0.533 ± 0.247 | 0.242 ± 0.062 |
| Alpha-2-HS-glycoprotein | 1.02 ± 0.97 | 0.707 ± 0.775 | 0.0908 ± 0.0489 | 0.152 ± 0.150 | 0.317 ± 0.194 | 0.179 ± 0.029 |
| Kininogen-2 | 0.0118 ± 0.0055 | 0.135 ± 0.112 | 0.0731 ± 0.0489 | 0.0916 ± 0.0345 | 0.137 ± 0.088 | 0.103 ± 0.045 |
| Hemiferrin | 0.138 ± 0.123 | 0.155 ± 0.050 | 0.114 ± 0.057 | 0.142 ± 0.062 | 0.110 ± 0.039 | 0.100 ± 0.020 |
| Integrin beta-1 | 0.0117 ± 0.00609 | 0.123 ± 0.039 | 0.114 ± 0.047 | 0.140 ± 0.052 | 0.0825 ± 0.0347 | 0.0676 ± 0.0361 |
| Prothrombin | 0.125 ± 0.118 | 0.212 ± 0.111 | 0.241 ± 0.075 | 0.234 ± 0.107 | 0.121 ± 0.057 | 0.143 ± 0.081 |
| Apolipoprotein A-I | 0.0242 ± 0.0237 | 0.157 ± 0.079 | 0.0925 ± 0.0457 | 0.107 ± 0.070 | 0.143 ± 0.108 | 0.0798 ± 0.0403 |
| Antithrombin-III | 0.0641 ± 0.0715 | 2.56 ± 2.17 | 0.786 ± 0.296 | 0.757 ± 0.676 | 1.01 ± 0.51 | 0.482 ± 0.122 |
| Beta-2-glycoprotein 1 | 0.582 ± 0.481 | 0.117 ± 0.061 | 0.0784 ± 0.0327 | 0.128 ± 0.064 | 0.0953 ± 0.0403 | 0.0948 ± 0.0556 |
| Alpha-2-antiplasmin | 0.0355 ± 0.0297 | 0.256 ± 0.133 | 0.129 ± 0.038 | 0.153 ± 0.097 | 0.159 ± 0.092 | 0.0913 ± 0.0375 |
| Protein AMBP | 0.0596 ± 0.0383 | 0.204 ± 0.077 | 0.0859 ± 0.0331 | 0.131 ± 0.056 | 0.108 ± 0.038 | 0.0894 ± 0.0375 |
| Hemoglobin fetal subunit beta | 0.0187 ± 0.0064 | 0.154 ± 0.121 | 0.0531 ± 0.0277 | 0.0703 ± 0.031 | 0.151 ± 0.082 | 0.216 ± 0.158 |
| Alpha 1-antichymotrypsin | 0.00869 ± 0.00451 | 0.192 ± 0.062 | 0.475 ± 0.310 | 0.467±0.191 | 0.208 ± 0.068 | 0.130 ± 0.054 |
| Apolipoprotein A-II | 0.00390 ± 0.00420 | 0.227 ± 0.263 | 0.110 ± 0.047 | 0.109±0.072 | 0.133 ± 0.061 | 0.115 ± 0.044 |
| Hemoglobin subunit alpha | 0.00598 ± 0.00580 | 0.166 ± 0.145 | 0.212 ± 0.121 | 0.346 ± 0.334 | 0.136 ± 0.064 | 0.125 ± 0.124 |
| Fibronectin | 0.00317 ± 0.00226 | 0.100 ± 0.051 | 0.0620 ± 0.0259 | 0.0803 ± 0.0365 | 0.0461 ± 0.0241 | 0.0451 ± 0.0176 |
| Vitronectin | 0.00297±0.00160 | 0.132 ± 0.129 | 0.0563 ± 0.0379 | 0.0761 ± 0.0329 | 0.0396 ± 0.0249 | 0.0435 ± 0.0200 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hirohara, M.; Maekawa, T.; Mondarte, E.A.Q.; Nyu, T.; Mizushita, Y.; Hayashi, T. Proteomic Analysis of Biomaterial Surfaces after Contacting with Body Fluids by MALDI-ToF Mass Spectroscopy. Coatings 2020, 10, 12. https://doi.org/10.3390/coatings10010012
Hirohara M, Maekawa T, Mondarte EAQ, Nyu T, Mizushita Y, Hayashi T. Proteomic Analysis of Biomaterial Surfaces after Contacting with Body Fluids by MALDI-ToF Mass Spectroscopy. Coatings. 2020; 10(1):12. https://doi.org/10.3390/coatings10010012
Chicago/Turabian StyleHirohara, Makoto, Tatsuhiro Maekawa, Evan Angelo Quimada Mondarte, Takashi Nyu, Yoshiki Mizushita, and Tomohiro Hayashi. 2020. "Proteomic Analysis of Biomaterial Surfaces after Contacting with Body Fluids by MALDI-ToF Mass Spectroscopy" Coatings 10, no. 1: 12. https://doi.org/10.3390/coatings10010012
APA StyleHirohara, M., Maekawa, T., Mondarte, E. A. Q., Nyu, T., Mizushita, Y., & Hayashi, T. (2020). Proteomic Analysis of Biomaterial Surfaces after Contacting with Body Fluids by MALDI-ToF Mass Spectroscopy. Coatings, 10(1), 12. https://doi.org/10.3390/coatings10010012