A Review on Anodizing of Aerospace Aluminum Alloys for Corrosion Protection

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Anodizing
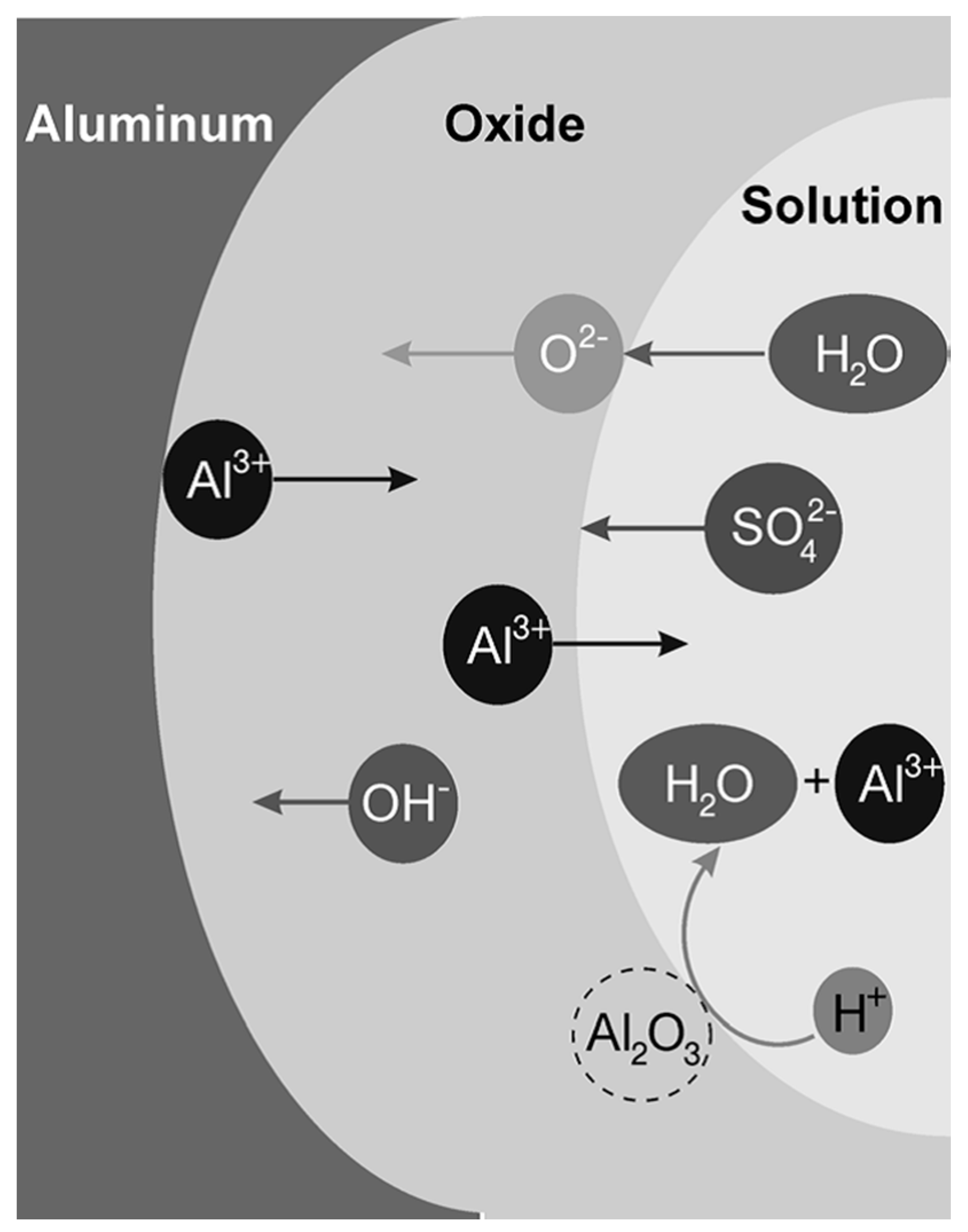
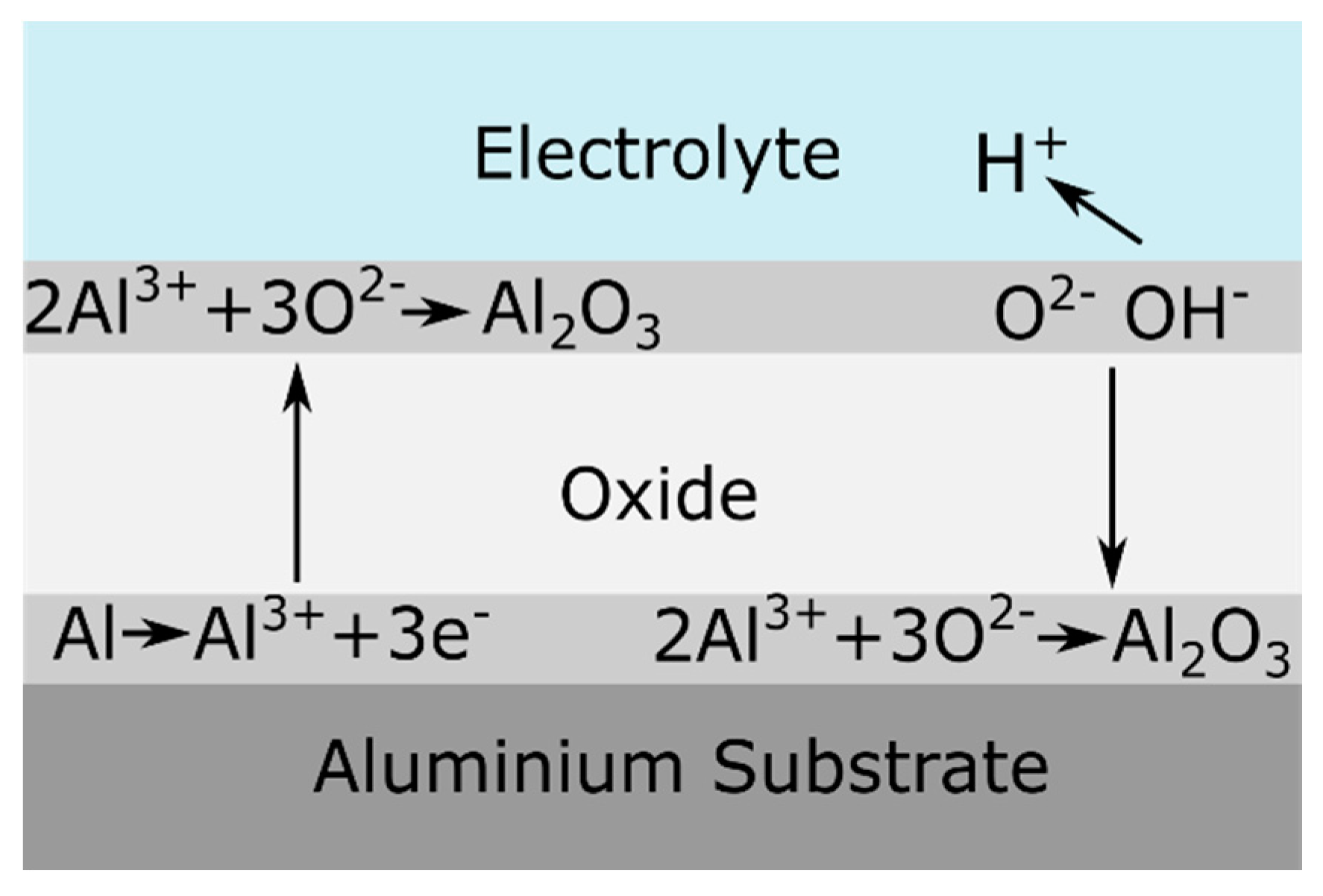
2.1. Working Principles
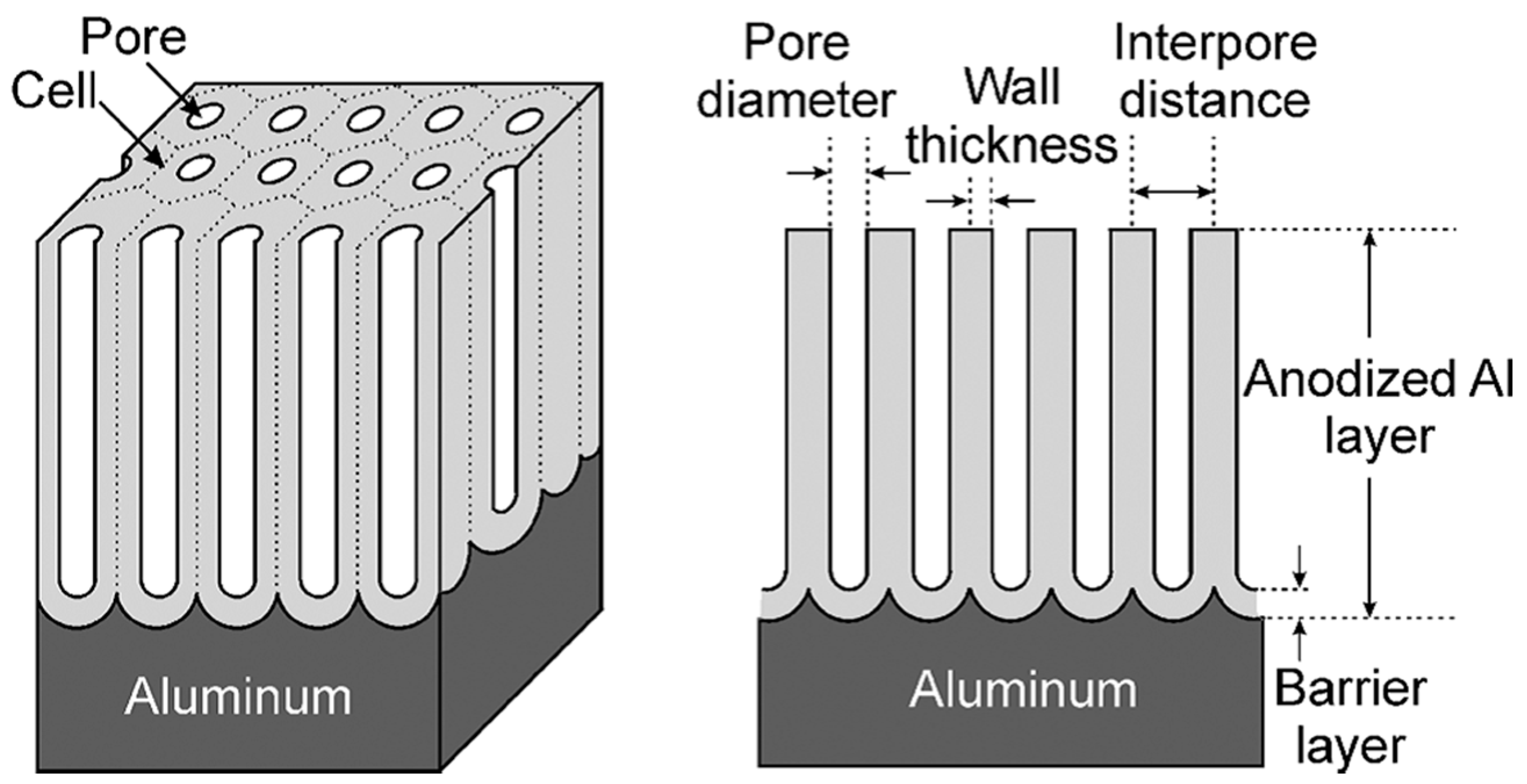
2.2. Barrier Anodic Layers and Porous Anodic Layers
2.3. Effect of the Applied Voltage or the Applied Current
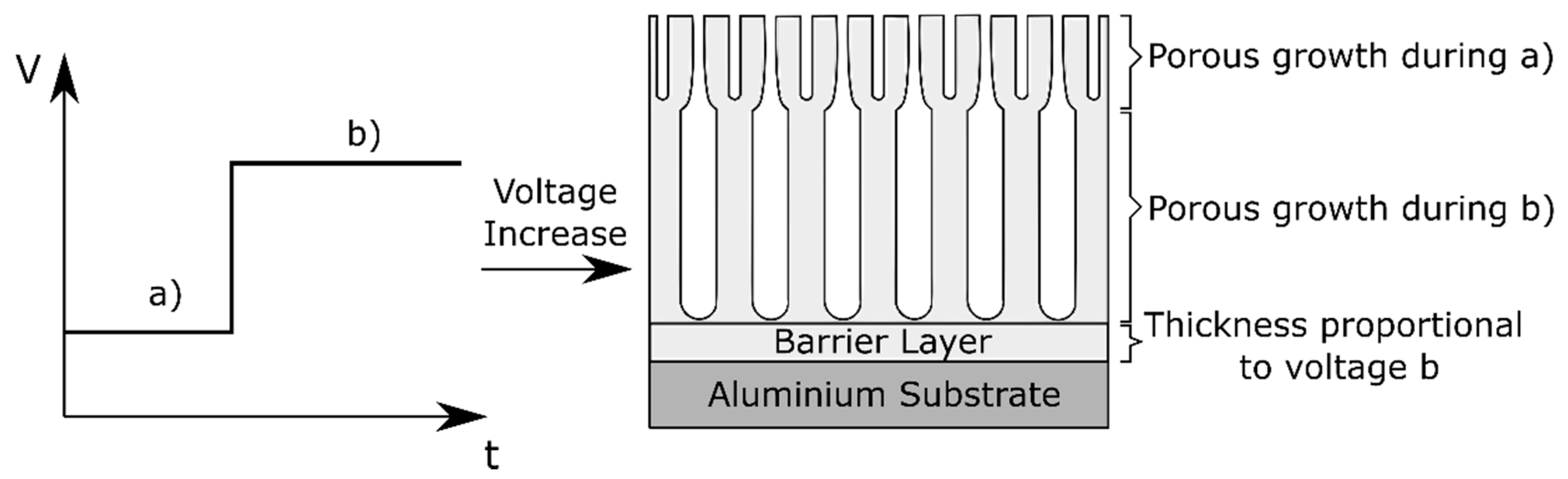
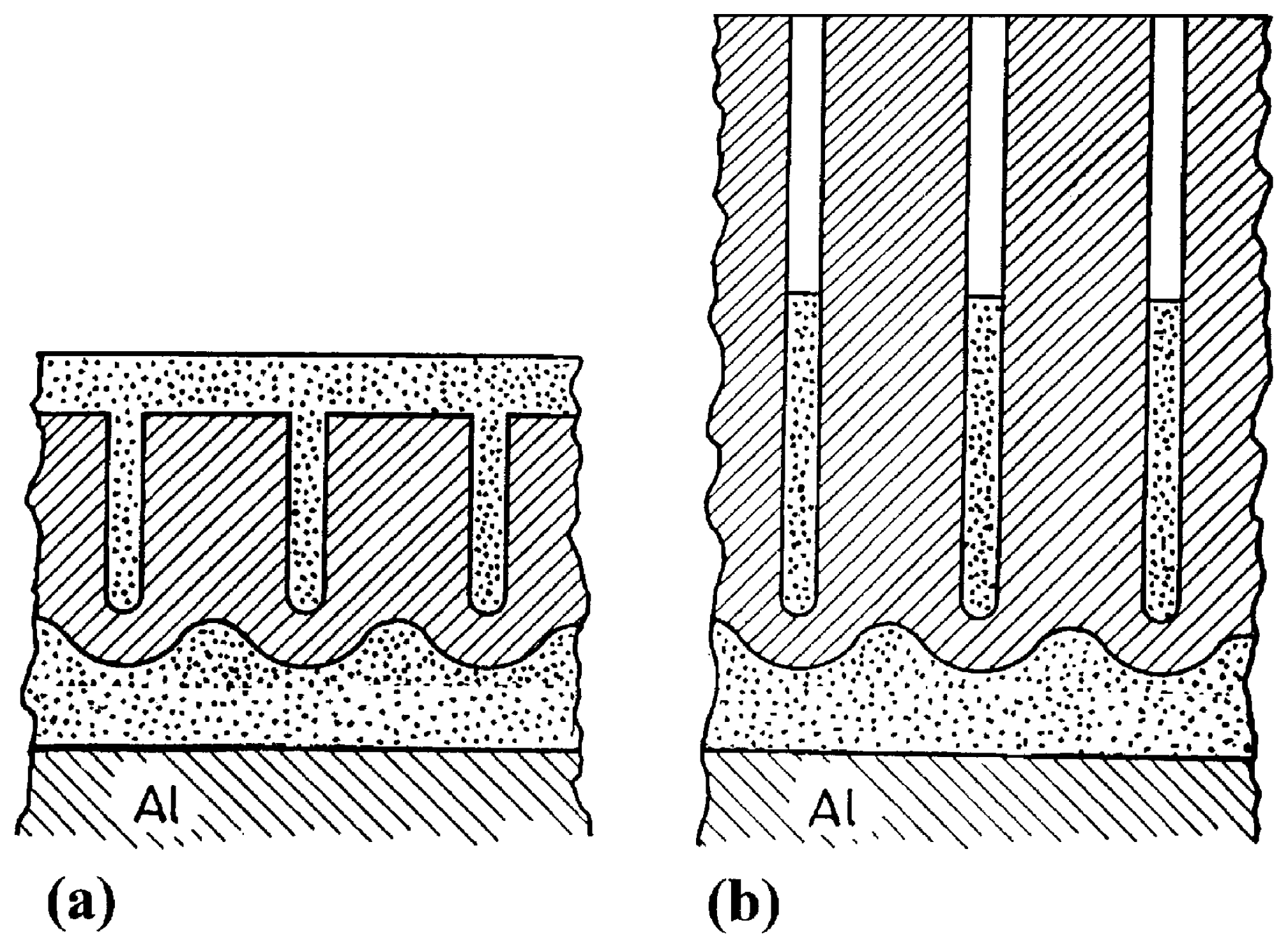
2.4. 2-Step Anodizing: Complex Anodic Layers
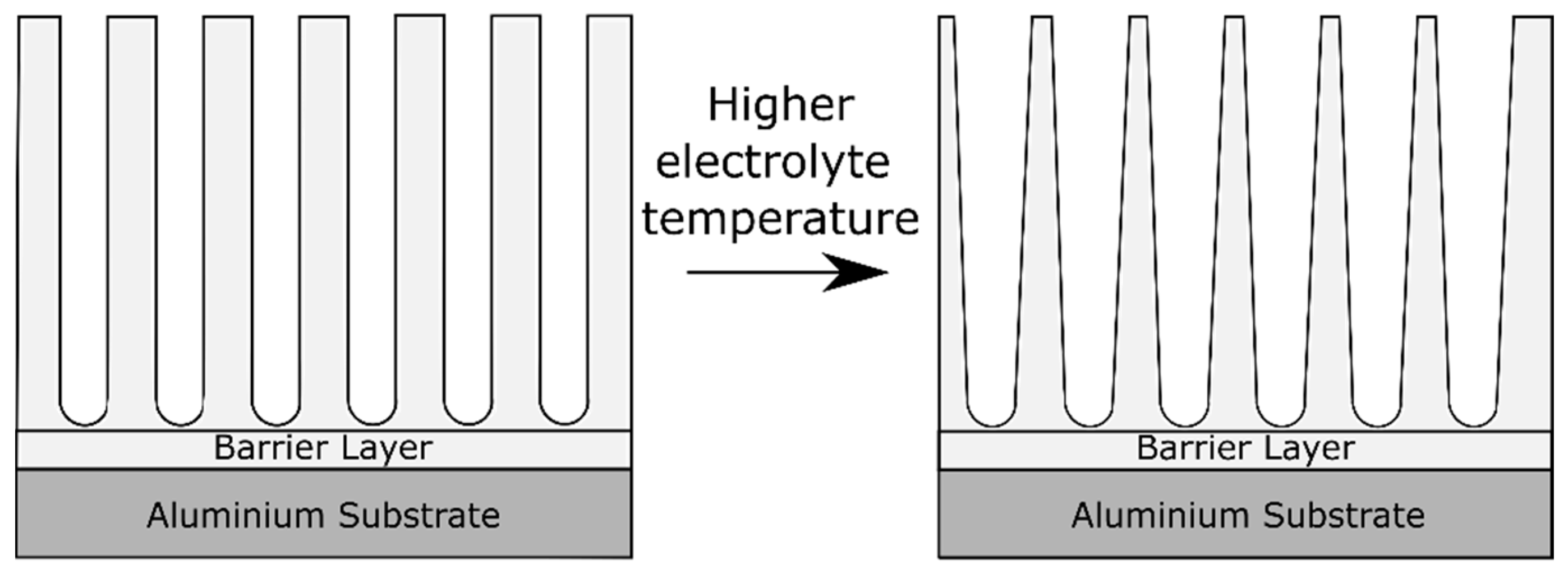
2.5. Effect of the Electrolyte Temperature
3. Anodic Layer Requirements for Aerospace Applications
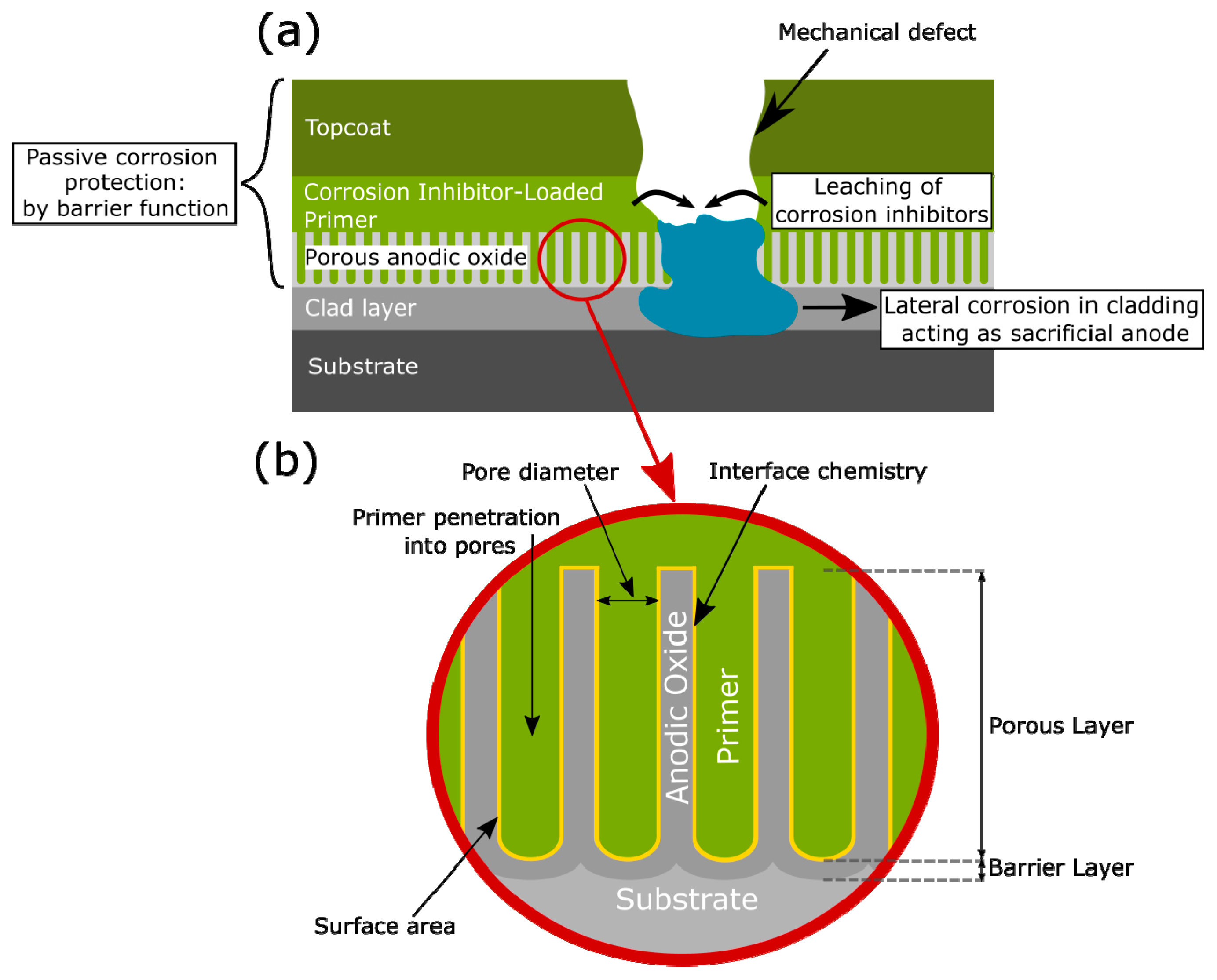
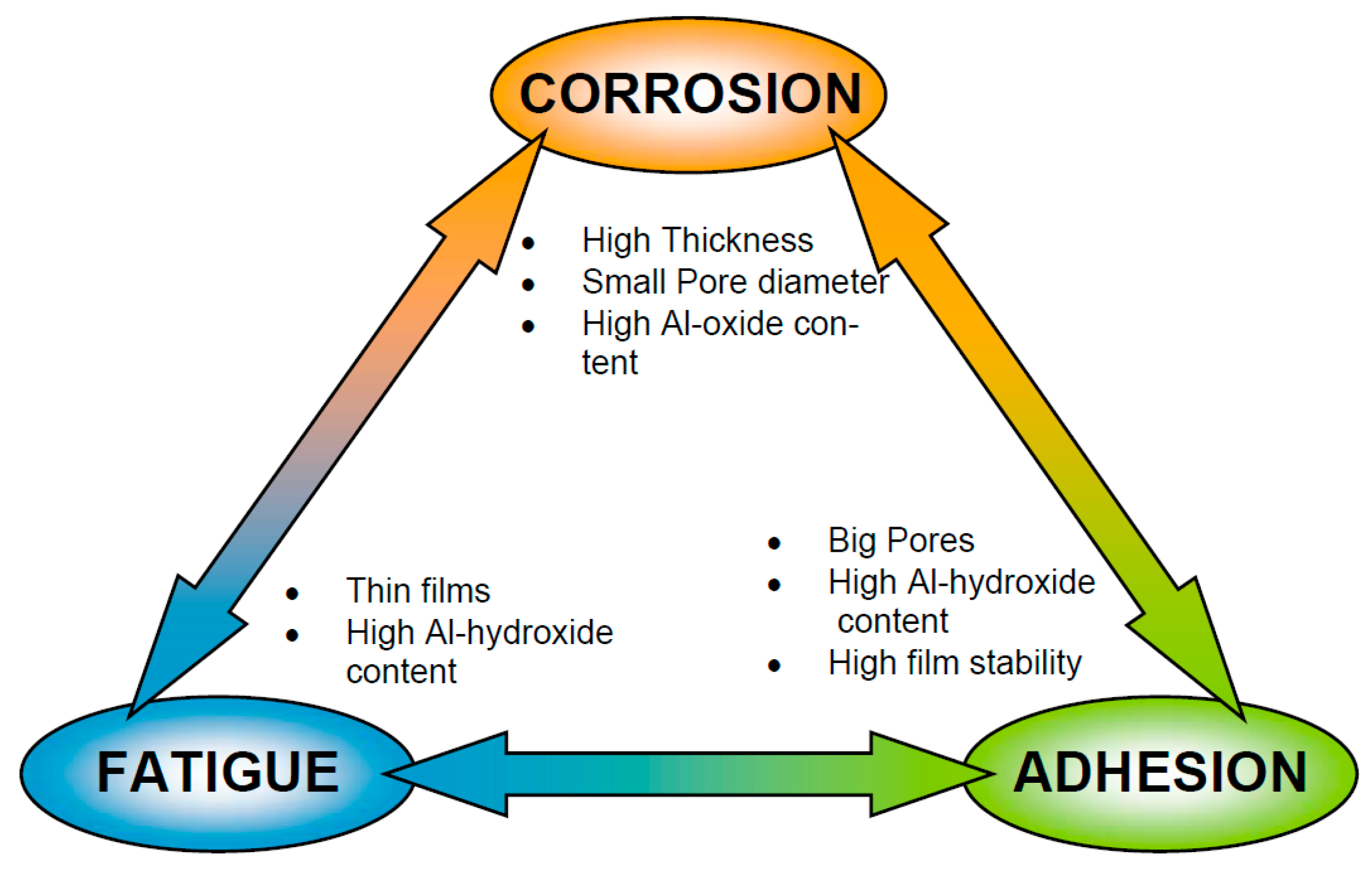
3.1. Corrosion Protection
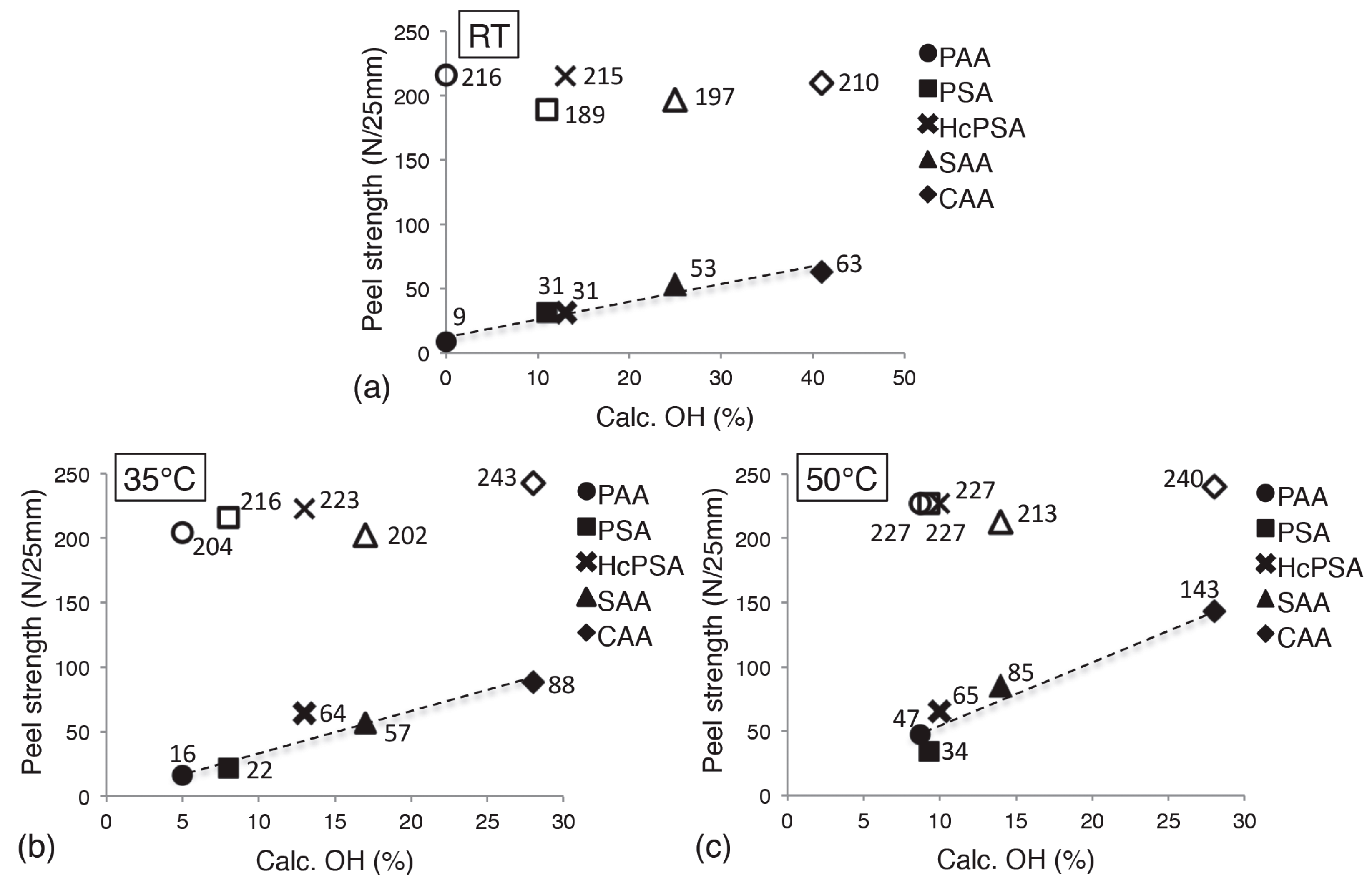
3.2. Adhesion
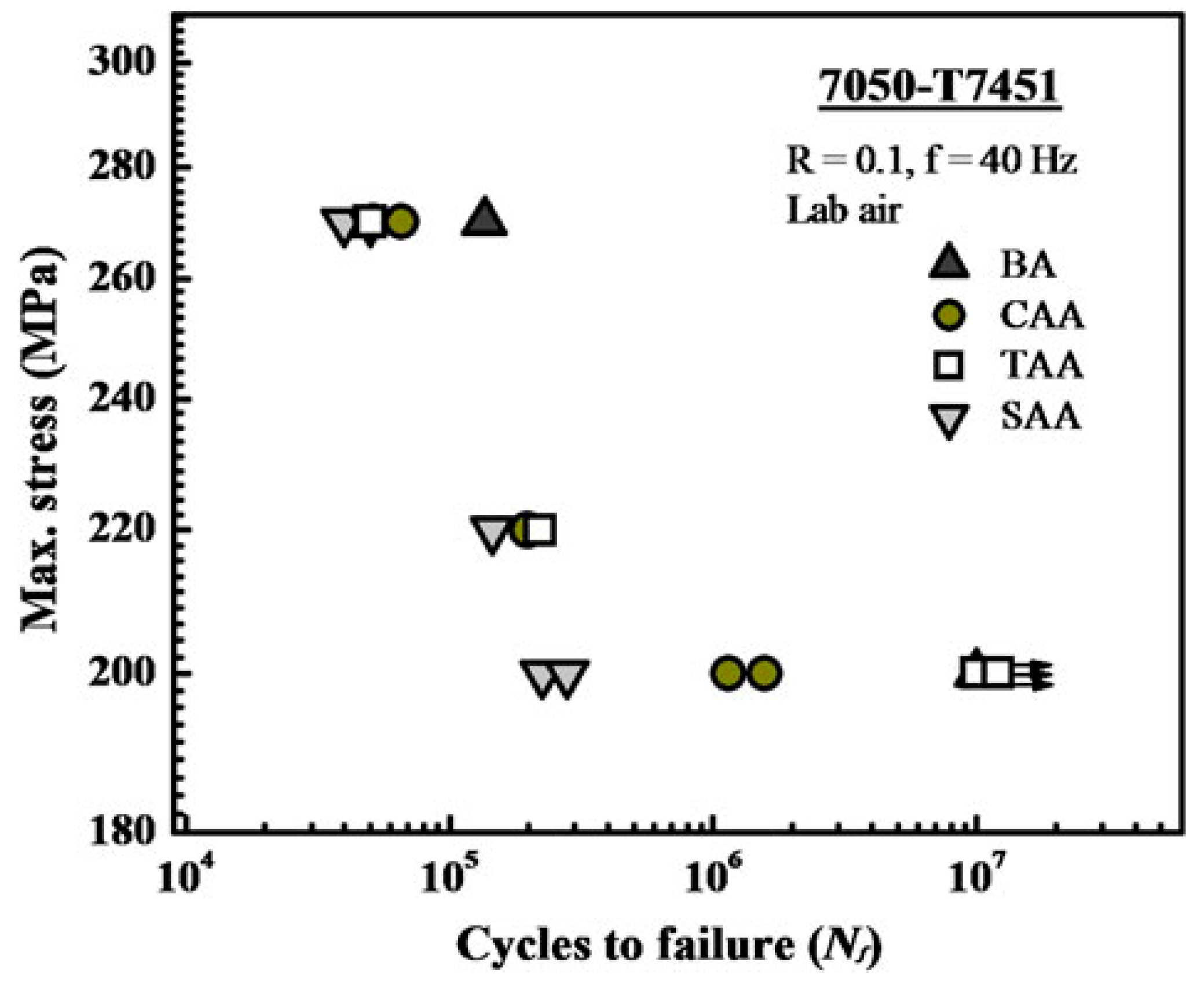
3.3. Fatigue
4. Substitution of the Chromic Acid Anodizing Process
4.1. Tartaric Sulfuric Acid Anodizing (TSA)
5. Anodizing as a Step in a Process
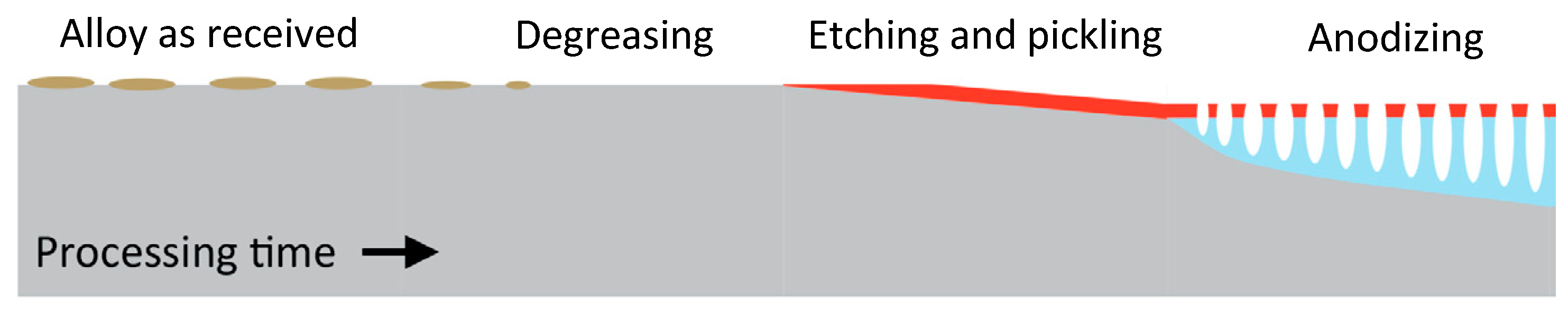
5.1. Pre-Treatment
5.1.1. Degreasing
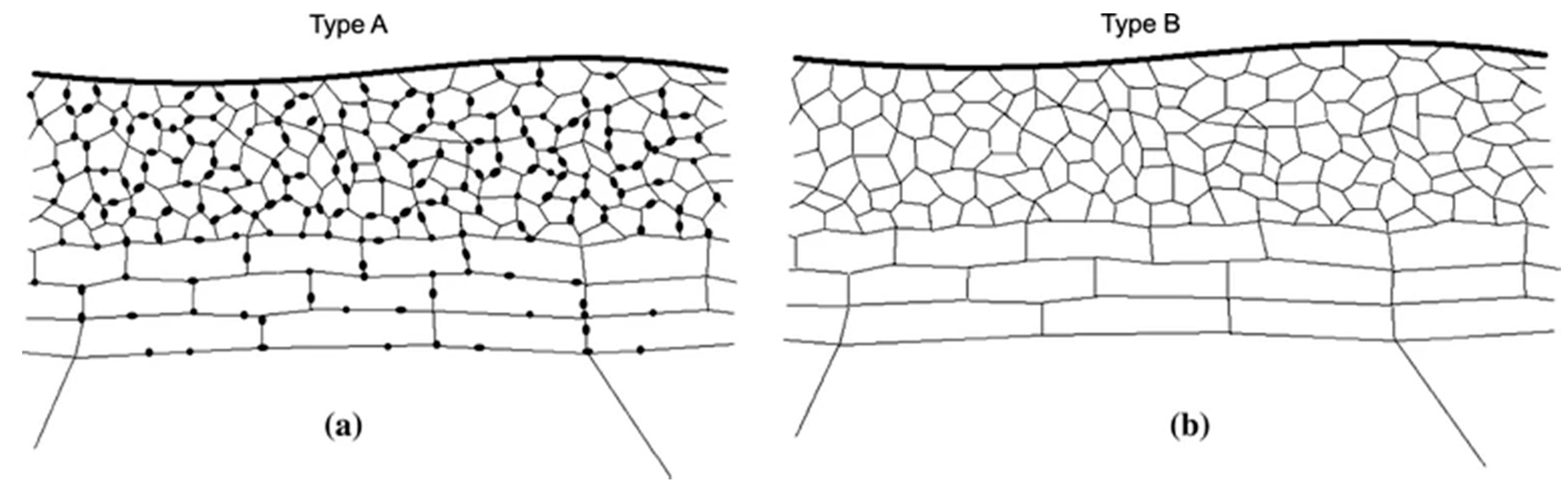
5.1.2. Alkaline Etching
5.1.3. Acidic Pickling
5.2. Post-Treatments
5.2.1. Sealing
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References and Notes
- Budinski, K.G.; Budinski, M.K. Engineering Materials: Properties and Selection; Prentice Hall: Upper Saddle River, NJ, USA, 2010; ISBN 978-0-13-712842-6. [Google Scholar]
- Gialanella, S.; Malandruccolo, A. Aerospace Alloys; Topics in Mining, Metallurgy and Materials Engineering; Springer International Publishing: Basel, Switzerland, 2019; ISBN 978-3-030-24440-8. [Google Scholar]
- Anthony, E. Hughes High Strength Al-Alloys: Microstructure, Corrosion and Principles of Protection. In Recent Trends in Processing and Degradation of Aluminum Alloys; Birbilis, N., Ed.; IntechOpen: Rijeka, Croatia, 2011; p. 223, Chapter 10. [Google Scholar]
- Boag, A.; Hughes, A.E.; Wilson, N.; Torpy, A.; MacRae, C.; Glenn, M.; Muster, T. How complex is the microstructure of AA2024-T3? Corros. Sci. 2009, 51, 1565–1568. [Google Scholar] [CrossRef]
- Becker, M. Chromate-free chemical conversion coatings for aluminum alloys. Corros. Rev. 2019, 37, 321–342. [Google Scholar] [CrossRef]
- Matykina, E.; Arrabal, R.; Mohedano, M.; Mingo, B.; Gonzalez, J.; Pardo, A.; Merino, M.C. Recent advances in energy efficient PEO processing of aluminum alloys. Trans. Nonferrous Met. Soc. China 2017, 27, 1439–1454. [Google Scholar] [CrossRef]
- Del Olmo, R.; Mohedano, M.; Visser, P.; Matykina, E.; Arrabal, R. Flash-PEO coatings loaded with corrosion inhibitors on AA2024. Surf. Coat. Technol. 2020, 126317. [Google Scholar] [CrossRef]
- Hagans, P.L.; Haas, C.M. Chromate Conversion Coatings. In Surface Engineering; Cotell, C.M., Sprague, J.A., Smidt, F.A., Jr., Eds.; ASM International: Cleveland, OH, USA, 1994; Volume 5, pp. 405–411. ISBN 978-1-62708-170-2. [Google Scholar]
- Simchen, F.; Sieber, M.; Kopp, A.; Lampke, T. Introduction to plasma electrolytic oxidation—An overview of the process and applications. Coatings 2020, 10, 628. [Google Scholar]
- McCafferty, E. Introduction to Corrosion Science; Springer: New York, NY, USA, 2010; ISBN 978-1-4419-0455-3. [Google Scholar]
- Benavides, S. Corrosion Control in the Aerospace Industry; Woodhead Publishing Series in Metals and Surface Engineering; Elsevier Science: Amsterdam, The Netherlands, 2009; ISBN 978-1-84569-553-8. [Google Scholar]
- United States Department of Labor OSHA, Toxic and Hazardous Substances in Occupational Exposure to Hexavalent Chromium. Regulatory standard 29 CFR 1910.1026 and 29 CFR 1926.1126 Federal Register Number 71:10099-10385 29 2006.
- Regulation (EC) No 1907/2006 of the European Parliament and of the Council of 18 December 2006 concerning the Registration, Evaluation, Authorisation and Restriction of Chemicals (REACH), establishing a European Chemicals Agency, amending Directive 1999/45/EC and repealing Council Regulation (EEC) No 793/93 and Commission Regulation (EC) No 1488/94 as well as Council Directive 76/769/EEC and Commission Directives 91/155/EEC, 93/67/EEC, 93/105/EC and 2000/21/EC 2006. Official journal L396, pp. 1–849.
- Garcia Rubio, M.; Ocon, P.; Climent-Font, A.; Smith, R.W.; Curioni, M.; Thompson, G.; Skeldon, P.; Lavia, A.; Garcia, I. Influence of molybdate species on the tartaric acid/sulfuric acid anodic films grown on AA2024 T3 aerospace alloy. Corros. Sci. 2009, 51, 2034–2042. [Google Scholar] [CrossRef]
- Curioni, M.; Skeldon, P.; Koroleva, E.; Thompson, G.E.; Ferguson, J. Role of Tartaric Acid on the Anodizing and Corrosion Behavior of AA 2024 T3 Aluminum Alloy. J. Electrochem. Soc. 2009, 156, C147–C153. [Google Scholar] [CrossRef]
- Abrahami, S.T.; de Kok, J.M.M.; Terryn, H.; Mol, J.M.C. Towards Cr(VI)-free anodization of aluminum alloys for aerospace adhesive bonding applications: A review. Front. Chem. Sci. Eng. 2017, 11, 465–482. [Google Scholar] [CrossRef]
- Visser, P.; Gonzalez-Garcia, Y.; Mol, J.M.C.; Terryn, H. Mechanism of Passive Layer Formation on AA2024-T3 from Alkaline Lithium Carbonate Solutions in the Presence of Sodium Chloride. J. Electrochem. Soc. 2018, 165, C60–C70. [Google Scholar] [CrossRef] [Green Version]
- Moutarlier, V.; Gigandet, M.P.; Normand, B.; Pagetti, J. EIS characterisation of anodic films formed on 2024 aluminum alloy, in sulfuric acid containing molybdate or permanganate species. Corros. Sci. 2005, 47, 937–951. [Google Scholar] [CrossRef]
- Hu, T.; Shi, H.; Wei, T.; Liu, F.; Fan, S.; Han, E.-H. Cerium tartrate as a corrosion inhibitor for AA 2024-T3. Corros. Sci. 2015, 95, 152–161. [Google Scholar] [CrossRef]
- Zubillaga, O.; Cano, F.J.; Azkarate, I.; Molchan, I.S.; Thompson, G.E.; Skeldon, P. Anodic films containing polyaniline and nanoparticles for corrosion protection of AA2024T3 aluminum alloy. Surf. Coat. Technol. 2009, 203, 1494–1501. [Google Scholar] [CrossRef]
- Tavandashti, N.P.; Ghorbani, M.; Shojaei, A.; Gonzalez-Garcia, Y.; Terryn, H.; Mol, J.M.C. pH responsive Ce(III) loaded polyaniline nanofibers for self-healing corrosion protection of AA2024-T3. Prog. Org. Coat. 2016, 99, 197–209. [Google Scholar] [CrossRef]
- Yoganandan, G.; Balaraju, J.N. Synergistic effect of V and Mn oxyanions for the corrosion protection of anodized aerospace aluminum alloy. Surf. Coat. Technol. 2014, 252, 35–47. [Google Scholar] [CrossRef] [Green Version]
- Yabuki, A.; Nagayama, Y.; Fathona, I.W. Porous anodic oxide film with self-healing ability for corrosion protection of aluminum. Electrochim. Acta 2019, 296, 662–668. [Google Scholar] [CrossRef]
- Whelan, M.; Barton, K.; Cassidy, J.; Colreavy, J.; Duffy, B. Corrosion inhibitors for anodised aluminum. Surf. Coat. Technol. 2013, 227, 75–83. [Google Scholar] [CrossRef]
- Anthes, A.; Weidmann, S.K.; Fürbeth, W.; Kremmer, K.; Schneider, M. Nanoparticle-based impregnation of chromate-free anodizing layers for corrosion protection and adhesive bonding. Surf. Coat. Technol. 2018, 348, 121–129. [Google Scholar] [CrossRef]
- Costenaro, H.; Lanzutti, A.; Paint, Y.; Fedrizzi, L.; Terada, M.; de Melo, H.G.; Olivier, M.-G. Corrosion resistance of 2524 Al alloy anodized in tartaric-sulfuric acid at different voltages and protected with a TEOS-GPTMS hybrid sol-gel coating. Surf. Coat. Technol. 2017, 324, 438–450. [Google Scholar] [CrossRef]
- Renaud, A.; Poorteman, M.; Escobar, J.; Dumas, L.; Paint, Y.; Bonnaud, L.; Dubois, P.; Olivier, M.-G. A new corrosion protection approach for aeronautical applications combining a Phenol-paraPhenyleneDiAmine benzoxazine resin applied on sulfo-tartaric anodized aluminum. Prog. Org. Coat. 2017, 112, 278–287. [Google Scholar] [CrossRef]
- Wang, P.; Dong, X.; Schaefer, D.W. Structure and water-barrier properties of vanadate-based corrosion inhibitor films. Corros. Sci. 2010, 52, 943–949. [Google Scholar] [CrossRef]
- Mata, D.; Serdechnova, M.; Mohedano, M.; Mendis, C.L.; Lamaka, S.V.; Tedim, J.; Hack, T.; Nixon, S.; Zheludkevich, M.L. Hierarchically organized Li–Al-LDH nano-flakes: A low-temperature approach to seal porous anodic oxide on aluminum alloys. RSC Adv. 2017, 7, 35357–35367. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.; Dong, H.; Shi, H.; Xiong, L.; He, C.; Liu, J.; Li, S. Effects of graphene oxide-filled sol-gel sealing on the corrosion resistance and paint adhesion of anodized aluminum. Appl. Surf. Sci. 2019, 479, 105–113. [Google Scholar] [CrossRef]
- Suzuki, Y.; Kawahara, K.; Kikuchi, T.; Suzuki, R.O.; Natsui, S. Corrosion-Resistant Porous Alumina Formed via Anodizing Aluminum in Etidronic Acid and Its Pore-Sealing Behavior in Boiling Water. J. Electrochem. Soc. 2019, 166, C261–C269. [Google Scholar] [CrossRef]
- Marzocchi, V.; Iglesias-Rubianes, L.; Thompson, G.; Bellucci, F. The influence of tartaric acid additions on the anodizing behavior of AA2024-T3 alloy in sulfuric acid. Corros. Rev. 2007, 25. [Google Scholar] [CrossRef]
- Boisier, G.; Lamure, A.; Pébère, N.; Portail, N.; Villatte, M. Corrosion protection of AA2024 sealed anodic layers using the hydrophobic properties of carboxylic acids. Surf. Coat. Technol. 2009, 203, 3420–3426. [Google Scholar] [CrossRef] [Green Version]
- Vignoli Machado, T.; Atz Dick, P.; Knörnschild, G.H.; Dick, L.F.P. The effect of different carboxylic acids on the sulfuric acid anodizing of AA2024. Surf. Coat. Technol. 2020, 383, 125283. [Google Scholar] [CrossRef]
- Terada, M.; Queiroz, F.M.; Aguiar, D.B.S.; Ayusso, V.H.; Costenaro, H.; Olivier, M.-G.; de Melo, H.G.; Costa, I. Corrosion resistance of tartaric-sulfuric acid anodized AA2024-T3 sealed with Ce and protected with hybrid sol–gel coating. Surf. Coat. Technol. 2019, 372, 422–426. [Google Scholar] [CrossRef]
- Kosari, A.; Visser, P.; Tichelaar, F.; Eswara, S.; Audinot, J.-N.; Wirtz, T.; Zandbergen, H.; Terryn, H.; Mol, J.M.C. Cross-sectional characterization of the conversion layer formed on AA2024-T3 by a lithium-leaching coating. Appl. Surf. Sci. 2020, 512, 145665. [Google Scholar] [CrossRef]
- Marcoen, K.; Visser, P.; Trindade, G.F.; Abel, M.-L.; Watts, J.F.; Mol, J.M.C.; Terryn, H.; Hauffman, T. Compositional study of a corrosion protective layer formed by leachable lithium salts in a coating defect on AA2024-T3 aluminum alloys. Prog. Org. Coat. 2018, 119, 65–75. [Google Scholar] [CrossRef] [Green Version]
- Doublet, A.; Kjellberg, M.; Jousselme, B.; Pinault, M.; Deniau, G.; Cornut, R.; Charrier, G. Bifunctional coatings: Coupling an organic adhesion promoter with an anticorrosion inorganic layer. RSC Adv. 2019, 9, 24043–24049. [Google Scholar] [CrossRef] [Green Version]
- Brace, A.W.; Sheasby, P.G. The Technology of Anodizing Aluminum; Technicopy Limited: Gloucestershire, UK, 1979; ISBN 0-905228-08-1. [Google Scholar]
- Aerts, T. Study of the Influence of Temperature and Heat Transfer during Anodic Oxide Growth on Aluminum. Ph.D. Thesis, Vrije Universiteit Brussel, Ixelles, Belgium, 2009. [Google Scholar]
- Thompson, G.E. Porous anodic alumina: Fabrication, characterization and applications. Thin Solid Film. 1997, 297, 192–201. [Google Scholar] [CrossRef]
- Abrahami, S. Cr(VI)-Free Pre-Treatments for Adhesive Bonding of Aerospace Aluminum Alloys. Ph.D. Thesis, Delft University of Technology, Delft, The Netherlands, 2016. [Google Scholar]
- Sulka, G.D. Highly Ordered Anodic Porous Alumina Formation by Self-Organized Anodizing. In Nanostructured Materials in Electrochemistry; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2008; pp. 1–116. ISBN 978-3-527-62150-7. [Google Scholar]
- Xu, Y.; Thompson, G.E.; Wood, G.C.; Bethune, B. Anion incorporation and migration during barrier film formation on aluminum. Corros. Sci. 1987, 27, 83–102. [Google Scholar] [CrossRef]
- Garcia Rubio, M. Optimisation of a Non-Chromium Containing Tartaric Acid/Sulfuric Acid Anodising Bath for Aluminum Alloy for Aerospace Industry Application. Ph.D. Thesis, Universidad Auntonoma de Madrid, Madrid, Spain, 2009. [Google Scholar]
- De Miera, M.S.; Curioni, M.; Skeldon, P.; Thompson, G.E. The behavior of second phase particles during anodizing of aluminum alloys. Corros. Sci. 2010, 52, 2489–2497. [Google Scholar] [CrossRef]
- De Miera, M.S.; Curioni, M.; Skeldon, P.; Thompson, G.E. Modelling the anodizing behavior of aluminum alloys in sulfuric acid through alloy analogues. Corros. Sci. 2008, 50, 3410–3415. [Google Scholar] [CrossRef]
- Curioni, M.; Saenz de Miera, M.; Skeldon, P.; Thompson, G.E.; Ferguson, J. Macroscopic and Local Filming Behavior of AA2024 T3 Aluminum Alloy during Anodizing in Sulfuric Acid Electrolyte. J. Electrochem. Soc. 2008, 155, C387–C395. [Google Scholar] [CrossRef]
- Torrescano-Alvarez, J.M.; Curioni, M.; Skeldon, P. Effects of oxygen evolution on the voltage and film morphology during galvanostatic anodizing of AA 2024-T3 aluminum alloy in sulfuric acid at −2 and 24 °C. Electrochim. Acta 2018, 275, 172–181. [Google Scholar] [CrossRef]
- Thompson, G.E.; Wood, G.C. 5-Anodic Films on Aluminum. In Treatise on Materials Science and Technology; Scully, J.C., Ed.; Elsevier: Amsterdam, The Netherlands, 1983; Volume 23, pp. 205–329. ISBN 0161-9160. [Google Scholar]
- Ono, S.; Ichinose, H.; Kawaguchi, T.; Masuko, N. The observation of anodic oxide films on aluminum by high resolution electron microscopy. Corros. Sci. 1990, 31, 249–254. [Google Scholar] [CrossRef]
- Palibroda, E.; Marginean, P. Considerations on the adsorbed water concentration of sulfuric porous aluminum oxide. Thin Solid Film. 1994, 240, 73–75. [Google Scholar] [CrossRef]
- Wernick, S.; Pinner, R. Surface Treatment and Finishing of Aluminum and Its Alloys, 4th ed.; Robert Draper Ltd.: Sevenoaks, UK, 1972; Volume I. [Google Scholar]
- Voon, C.H.; Derman, M.N.; Hashim, U.; Ahmad, K.R.; Foo, K.L. Effect of Temperature of Oxalic Acid on the Fabrication of Porous Anodic Alumina from Al-Mn Alloys. J. Nanomater. 2013, 2013, 167047. [Google Scholar] [CrossRef]
- Lee, W.; Park, S.-J. Porous Anodic Aluminum Oxide: Anodization and Templated Synthesis of Functional Nanostructures. Chem. Rev. 2014, 114, 7487–7556. [Google Scholar] [CrossRef]
- Takahashi, H.; Nagayama, M. The determination of the porosity of anodic oxide films on aluminum by the pore-filling method. Corros. Sci. 1978, 18, 911–925. [Google Scholar] [CrossRef]
- O’Sullivan, J.P.; Wood, G.C. The morphology and mechanism of formation of porous anodic films on aluminum. Proc. R. Soc. London. A. Math. Phys. Sci. 1970, 317, 511–543. [Google Scholar] [CrossRef]
- Oh, J.; Thompson, C.V. The role of electric field in pore formation during aluminum anodization. Electrochim. Acta 2011, 56, 4044–4051. [Google Scholar] [CrossRef]
- Garcia-Vergara, S.J.; Skeldon, P.; Thompson, G.E.; Hashimoto, T.; Habazaki, H. Compositional Evidence for Flow in Anodic Films on Aluminum under High Electric Fields. J. Electrochem. Soc. 2007, 154, C540–C545. [Google Scholar] [CrossRef]
- Garcia-Vergara, S.J.; Skeldon, P.; Thompson, G.E.; Habazaki, H. Stress generated porosity in anodic alumina formed in sulfuric acid electrolyte. Corros. Sci. 2007, 49, 3772–3782. [Google Scholar] [CrossRef]
- Skeldon, P.; Thompson, G.E.; Garcia-Vergara, S.J.; Iglesias-Rubianes, L.; Blanco-Pinzon, C.E. A Tracer Study of Porous Anodic Alumina. Electrochem. Solid-State Lett. 2006, 9, B47–B51. [Google Scholar] [CrossRef]
- Sato, N. A theory for breakdown of anodic oxide films on metals. Electrochim. Acta 1971, 16, 1683–1692. [Google Scholar]
- Nelson, J.; Oriani, R. Stress generation during anodic oxidation of titanium and aluminum. Corros. Sci. 1993, 34, 307–326. [Google Scholar]
- Bradhurst, D.H.; Llewelyn Leach, J.S. The Mechanical Properties of Thin Anodic Films on Aluminum. J. Electrochem. Soc. 1966, 113, 1245. [Google Scholar] [CrossRef]
- Wüthrich, N. Intrinsic stresses in anodic films on aluminum. Electrochim. Acta 1981, 26, 1617–1623. [Google Scholar] [CrossRef]
- Moon, S.-M.; Pyun, S.-I. The mechanism of stress generation during the growth of anodic oxide films on pure aluminum in acidic solutions. Electrochim. Acta 1998, 43, 3117–3126. [Google Scholar] [CrossRef]
- Sacco, L.; Florea, I.; Cojocaru, C.-S. Fabrication of porous anodic alumina (PAA) templates with straight pores and with hierarchical structures through exponential voltage decrease technique. Surf. Coat. Technol. 2019, 364, 248–255. [Google Scholar] [CrossRef] [Green Version]
- Curioni, M.; Skeldon, P.; Thompson, G.E.; Ferguson, J. Graded Anodic Film Morphologies for Sustainable Exploitation of Aluminum Alloys in Aerospace. Adv. Mater. Res. 2008, 38, 48–55. [Google Scholar] [CrossRef]
- Van Put, M.A.; Abrahami, S.T.; Elisseeva, O.; de Kok, J.M.M.; Mol, J.M.C.; Terryn, H. Potentiodynamic anodizing of aluminum alloys in Cr(VI)-free electrolytes. Surf. Interface Anal. 2016, 48, 946–952. [Google Scholar] [CrossRef]
- Iglesias-Rubianes, L.; Garcia-Vergara, S.J.; Skeldon, P.; Thompson, G.E.; Ferguson, J.; Beneke, M. Cyclic oxidation processes during anodizing of Al–Cu alloys. Electrochim. Acta 2007, 52, 7148–7157. [Google Scholar] [CrossRef]
- Ono, S.; Saito, M.; Asoh, H. Self-ordering of anodic porous alumina induced by local current concentration: Burning. Electrochem. Solid-State Lett. 2004, 7, B21–B24. [Google Scholar] [CrossRef]
- De Graeve, I.; Terryn, H.; Thompson, G.E. Influence of local heat development on film thickness for anodizing aluminum in sulfuric acid. J. Electrochem. Soc. 2003, 150, B158. [Google Scholar] [CrossRef]
- Tu, G.; Chen, I.; Shimizu, K. The temperature rise and burning for high rate anodizing of aluminum in oxalic acid. J. Jpn. Inst. Light Met. 1990, 40, 382–389. [Google Scholar] [CrossRef] [Green Version]
- Chung, C.K.; Liao, M.W.; Chang, H.C.; Lee, C.T. Effects of temperature and voltage mode on nanoporous anodic aluminum oxide films by one-step anodization. Thin Solid Film. 2011, 520, 1554–1558. [Google Scholar] [CrossRef]
- Graeve, I.D.; Terryn, H.; Thompson, G.E. AC-anodising of aluminum: Contribution to electrical and efficiency study. Electrochim. Acta 2006, 52, 1127–1134. [Google Scholar] [CrossRef]
- Ashcroft, I.; Cartwright, T.; Bahrani, D.; Critchlow, G. Anodising aluminum alloy. U.S. Patent 7922889B2, 12 April 2011. [Google Scholar]
- Girginov, A.A.; Zahariev, A.S.; Machkova, M.S. Kinetics of formation of complex anodic oxide films on aluminum. Mater. Chem. Phys. 2002, 76, 274–278. [Google Scholar] [CrossRef]
- Li, D.; Jiang, C.; Ren, X.; Long, M.; Jiang, J. Fabrication of porous anodic alumina membranes with ultrathick barrier layer. Mater. Lett.-Mater Lett 2008, 62, 3228–3231. [Google Scholar] [CrossRef]
- Aerts, T.; Jorcin, J.-B.; De Graeve, I.; Terryn, H. Comparison between the influence of applied electrode and electrolyte temperatures on porous anodizing of aluminum. Electrochim. Acta 2010, 55, 3957–3965. [Google Scholar] [CrossRef]
- Abd-Elnaiem, A.M.; Gaber, A. Parametric study on the anodization of pure aluminum thin film used in fabricating nano-pores template. Int. J. Electrochem. Sci 2013, 8, 9741–9751. [Google Scholar]
- Schnaut, U.; Gammel, F.; Fangmeier, A.; Kock, E. Surface Treatment of Aluminum in Aerospace: Today and Tomorrow. In Proceedings of the 3rd Aluminium Surface Science & Technology Symposium, Bonn, Germany, 18–22 May 2003; Available online: http://www.asst2018.com/home/ (accessed on 30 October 2020).
- González, J.A.; Morcillo, M.; Escudero, E.; López, V.; Otero, E. Atmospheric corrosion of bare and anodized aluminum in a wide range of environmental conditions. Part I: Visual observations and gravimetric results. Surf. Coat. Technol. 2002, 153, 225–234. [Google Scholar] [CrossRef]
- Abrahami, S.T.; Hauffman, T.; de Kok, J.M.M.; Mol, J.M.C.; Terryn, H. XPS analysis of the surface chemistry and interfacial bonding of barrier-type Cr(VI)-free anodic oxides. J. Phys. Chem. C 2015, 119, 19967–19975. [Google Scholar] [CrossRef]
- Cramer, S.D.; Covino, B.S., Jr. (Eds.) Corrosion: Fundamentals, Testing, and Protection; ASM International: Cleveland, OH, USA, 2003; ISBN 978-1-62708-182-5. [Google Scholar]
- Habazaki, H.; Zhou, X.; Shimizu, K.; Skeldon, P.; Thompson, G.E.; Wood, G.C. Mobility of copper ions in anodic alumina films. Electrochim. Acta 1997, 42, 2627–2635. [Google Scholar] [CrossRef]
- Bononi, M.; Giovanardi, R.; Bozza, A.; Mattioli, P. Pulsed current effect on hard anodizing process of 2024-T3 aluminum alloy. Surf. Coat. Technol. 2016, 289, 110–117. [Google Scholar] [CrossRef]
- Torrescano-Alvarez, J.M.; Curioni, M.; Zhou, X.; Skeldon, P. Effect of anodizing conditions on the cell morphology of anodic films on AA2024-T3 alloy. Surf. Interface Anal. 2018, 51. [Google Scholar] [CrossRef]
- Cavezza, F.; Boehm, M.; Terryn, H.; Hauffman, T. A review on adhesively bonded aluminum joints in the automotive industry. Metals 2020, 10, 730. [Google Scholar] [CrossRef]
- Pletincx, S.; Fockaert, L.L.I.; Mol, J.M.C.; Hauffman, T.; Terryn, H. Probing the formation and degradation of chemical interactions from model molecule/metal oxide to buried polymer/metal oxide interfaces. npj Mater. Degrad. 2019, 3, 23. [Google Scholar] [CrossRef] [Green Version]
- Mohseni, M.; Mirabedini, M.; Hashemi, M.; Thompson, G.E. Adhesion performance of an epoxy clear coat on aluminum alloy in the presence of vinyl and amino-silane primers. Prog. Org. Coat. 2006, 57, 307–313. [Google Scholar] [CrossRef]
- Fowkes, F.M. Acid-Base Interactions in Polymer Adhesion. In Tribology Series; Georges, J.M., Ed.; Elsevier: Amsterdam, The Netherlands, 1981; Volume 7, pp. 119–137. ISBN 0167-8922. [Google Scholar]
- Sitnikov, P.A.; Vaseneva, I.N.; Belyy, V.A.; Kenzhin, R.M.; Volodin, A.M.; Vedyagin, A.A. Study on the interfacial interactions of sulfated alumina with epoxy polymer. Ceram. Int. 2019, 45, 8919–8925. [Google Scholar]
- Abrahami, S.T.; Hauffman, T.; de Kok, J.M.M.; Mol, J.M.C.; Terryn, H. Effect of anodic aluminum oxide chemistry on adhesive bonding of epoxy. J. Phys. Chem. C 2016, 120, 19670–19677. [Google Scholar] [CrossRef]
- Abrahami, S.T.; Hauffman, T.; de Kok, J.M.M.; Terryn, H.; Mol, J.M.C. The role of acid-base properties in the interactions across the oxide-primer interface in aerospace applications. Surf. Interface Anal. 2016, 48, 712–720. [Google Scholar] [CrossRef]
- Pizzi, A.; Mittal, K.L. Handbook of Adhesive Technology, Revised and Expanded; Taylor & Francis: Abingdon-on-Thames, UK, 2003; ISBN 978-0-203-91222-5. [Google Scholar]
- Awaja, F.; Gilbert, M.; Kelly, G.; Fox, B.; Pigram, P.J. Adhesion of polymers. Prog. Polym. Sci. 2009, 34, 948–968. [Google Scholar] [CrossRef]
- Abrahami, S.T.; de Kok, J.M.M.; Gudla, V.C.; Ambat, R.; Terryn, H.; Mol, J.M.C. Interface strength and degradation of adhesively bonded porous aluminum oxides. npj Mater. Degrad. 2017, 1, 8. [Google Scholar] [CrossRef] [Green Version]
- Maege, I.; Jaehne, E.; Henke, A.; Adler, H.-J.P.; Bram, C.; Jung, C.; Stratmann, M. Self-assembling adhesion promoters for corrosion resistant metal polymer interfaces. Prog. Org. Coat. 1998, 34, 1–12. [Google Scholar] [CrossRef]
- Hua, D.; Lin, J.; Zhang, B. Effects of salt spray on the mechanical properties of aluminum-epoxy adhesive joints. J. Adhes. Sci. Technol. 2013, 27, 1580–1589. [Google Scholar] [CrossRef]
- Funke, W. Improvement of Wet Adhesion of Organic Coatings by Thin Adhesion Layer. In Surface Phenomena and Lateyes in Waterborne Coatings and Printing Technology; Springer: Boston, MA, USA, 1995. [Google Scholar]
- Salstela, J.; Suvanto, M.; Pakkanen, T.T. Influence of hierarchical micro-micro patterning and chemical modifications on adhesion between aluminum and epoxy. Int. J. Adhes. Adhes. 2016, 66, 128–137. [Google Scholar] [CrossRef]
- Abrahami, S.T.; Hauffman, T.; de Kok, J.M.; Terryn, H.; Mol, J.M.C. Adhesive bonding and corrosion performance investigated as a function of aluminum oxide chemistry and adhesives. Corrosion 2017, 73, 903–914. [Google Scholar] [CrossRef] [Green Version]
- Al-Khaldi, T.A.; Lyon, S.B. The effect of interfacial chemistry on coating adhesion and performance: A mechanistic study using aminobutylphosphonic acid. Prog. Org. Coat. 2012, 75, 449–455. [Google Scholar] [CrossRef]
- Dalmoro, V.; Alemán, C.; Ferreira, C.A.; dos Santos, J.H.Z.; Azambuja, D.S.; Armelin, E. The influence of organophosphonic acid and conducting polymer on the adhesion and protection of epoxy coating on aluminum alloy. Prog. Org. Coat. 2015, 88, 181–190. [Google Scholar] [CrossRef]
- Hauffman, T.; Hubin, A.; Terryn, H. Study of the self-assembling of n-octylphosphonic acid layers on aluminum oxide from ethanolic solutions. Surf. Interface Anal. 2013, 45, 1435–1440. [Google Scholar] [CrossRef]
- Cree, A.M.; Weidmann, G.W. The fracture and fatigue properties of anodised aluminum alloy. Trans. IMF 1997, 75, 199–202. [Google Scholar] [CrossRef]
- Cree, A.M.; Weidmann, G.W. Effect of anodised coatings on fatigue crack growth rates in aluminum alloy. Surf. Eng. 1997, 13, 51–55. [Google Scholar] [CrossRef]
- Cirik, E.; Genel, K. Effect of anodic oxidation on fatigue performance of 7075-T6 alloy. Surf. Coat. Technol. 2008, 202, 5190–5201. [Google Scholar] [CrossRef]
- Lonyuk, B.; Apachitei, I.; Duszczyk, J. The effect of oxide coatings on fatigue properties of 7475-T6 aluminum alloy. Surf. Coat. Technol. 2007, 201, 8688–8694. [Google Scholar] [CrossRef]
- Sadeler, R. Effect of a commercial hard anodizing on the fatigue property of a 2014-T6 aluminum alloy. J. Mater. Sci. 2006, 41, 5803–5809. [Google Scholar] [CrossRef]
- Rateick, R.G.; Binkowski, T.C.; Boray, B.C. Effect of hard anodize thickness on the fatigue of AA6061 and C355 aluminum. J. Mater. Sci. Lett. 1996, 15, 1321–1323. [Google Scholar] [CrossRef]
- Lee, E.; Jeong, Y.; Kim, S. S-N Fatigue Behavior of Anodized 7050-T7451 Produced in Different Electrolytes. Metall. Mater. Trans. A 2012, 43, 2002–2011. [Google Scholar] [CrossRef]
- Savas, T.P.; Earthman, J.C. Surface characterization of 7075-T73 aluminum exposed to anodizing pretreatment solutions. J. Mater. Eng. Perform. 2008, 17, 674–681. [Google Scholar] [CrossRef]
- de Camargo, J.A.M.; Cornelis, H.J.; Cioffi, V.M.O.H.; Costa, M.Y.P. Coating residual stress effects on fatigue performance of 7050-T7451 aluminum alloy. Surf. Coat. Technol. 2007, 201, 9448–9455. [Google Scholar] [CrossRef]
- Merati, A.; Eastaugh, G. Determination of fatigue related discontinuity state of 7000 series of aerospace aluminum alloys. Eng. Fail. Anal. 2007, 14, 673–685. [Google Scholar] [CrossRef]
- Critchlow, G.W.; Yendall, K.A.; Bahrani, D.; Quinn, A.; Andrews, F. Strategies for the replacement of chromic acid anodising for the structural bonding of aluminum alloys. Int. J. Adhes. Adhes. 2006, 26, 419–453. [Google Scholar] [CrossRef] [Green Version]
- Critchlow, G.W.; Brewis, D.M. Review of surface pretreatments for aluminum alloys. Int. J. Adhes. Adhes. 1996, 16, 255–275. [Google Scholar] [CrossRef]
- Kock, E. Tartaric Sulfuric Acid Anodising; Airbus: Bremen, Germany, 2006. [Google Scholar]
- Park, S.Y.; Choi, W.J.; Choi, H.S.; Kwon, H.; Kim, S.H. Recent trends in surface treatment technologies for airframe adhesive bonding processing: A review (1995–2008). J. Adhes. 2010, 86, 192–221. [Google Scholar] [CrossRef]
- Arrowsmith, D.J.; Clifford, A.W. Morphology of anodic oxide for adhesive bonding of aluminum. Int. J. Adhes. Adhes. 1983, 3, 193–196. [Google Scholar] [CrossRef]
- Kape, J. Unusual anodizing processes and their practical applications. Electroplat. Met. Finish. 1961, 14, 407. [Google Scholar]
- Kape, J.M. Anodizing in aqueous solutions of organic carboxylic acids. Trans. Imf 1967, 45, 34–42. [Google Scholar] [CrossRef]
- Sulka, G.D.; Parkoła, K.G. Temperature influence on well-ordered nanopore structures grown by anodization of aluminum in sulfuric acid. Electrochim. Acta 2007, 52, 1880–1888. [Google Scholar] [CrossRef]
- Elaish, R.; Curioni, M.; Gowers, K.; Kasuga, A.; Habazaki, H.; Hashimoto, T.; Skeldon, P. Effect of fluorozirconic acid on anodizing of aluminum and AA 2024-T3 alloy in sulfuric and tartaric-sulfuric acids. Surf. Coat. Technol. 2018, 342, 233–243. [Google Scholar] [CrossRef]
- Kondo, R.; Nakajima, D.; Kikuchi, T.; Natsui, S.; Suzuki, R.O. Superhydrophilic and superhydrophobic aluminum alloys fabricated via pyrophosphoric acid anodizing and fluorinated SAM modification. J. Alloy. Compd. 2017, 725, 379–387. [Google Scholar] [CrossRef]
- Hua, L.; Liu, J.; Li, S.; Yu, M. Effect of Adipic Acid on DEIS Characteristics during the Aluminum Anodizing Process in Sulfuric Acid Bath. Int. J. Electrochem. Sci. 2015, 10, 2194–2205. [Google Scholar]
- Shingubara, S.; Morimoto, K.; Sakaue, H.; Takahagi, T. Self-organization of a porous alumina nanohole array using a sulfuric/oxalic acid mixture as electrolyte. Electrochem. Solid-State Lett. 2004, 7, E15. [Google Scholar] [CrossRef]
- Ukaji, Y.; Soeta, T. 3.6 Acetogenin (Polypriopionate) Derived Auxiliaries: Tartaric Acid. In Comprehensive Chirality; Carreira, E.M., Yamamoto, H., Eds.; Elsevier: Amsterdam, The Netherlands, 2012; pp. 176–201. ISBN 978-0-08-095168-3. [Google Scholar]
- Dattilo, A.; Tamiro, S.; Romano, C. Anodizing process, with low environmental impact, for a woodpiece of aluminum or aluminum alloys 2002. U.S. Patent 20020157961A1, 31 October 2002. [Google Scholar]
- Boisier, G.; Pébère, N.; Druez, C.; Villatte, M.; Suel, S. FESEM and EIS study of sealed AA2024 T3 anodized in sulfuric acid electrolytes: Influence of tartaric acid. J. Electrochem. Soc. 2008, 155, C521–C529. [Google Scholar]
- Ma, Y.; Zhou, X.; Thompson, G.E.; Zhang, X.; Luo, C.; Curioni, M.; Liu, H. Microstructural modification arising from alkaline etching and its effect on anodizing behavior of Al–Li–Cu Alloy. J. Electrochem. Soc. 2013, 160, C111–C118. [Google Scholar] [CrossRef]
- Arenas, M.A.; Conde, A.; de Damborenea, J.J. Effect of acid traces on hydrothermal sealing of anodising layers on 2024 aluminum alloy. Electrochim. Acta 2010, 55, 8704–8708. [Google Scholar] [CrossRef]
- González-Rovira, L.; González-Souto, L.; Astola, P.J.; Bravo-Benítez, C.; Botana, F.J. Assessment of the corrosion resistance of self-ordered anodic aluminum oxide (AAO) obtained in tartaric-sulfuric acid (TSA). Surf. Coat. Technol. 2020, 399, 126131. [Google Scholar] [CrossRef]
- Wang, R.; Wang, L.; He, C.; Lu, M.; Sun, L. Studies on the sealing processes of corrosion resistant coatings formed on 2024 aluminum alloy with tartaric-sulfuric anodizing. Surf. Coat. Technol. 2019, 360, 369–375. [Google Scholar] [CrossRef]
- Bensalah, W.; Elleuch, K.; Feki, M.; Depetris-Wery, M.; Ayedi, H.F. Optimization of tartaric/sulfuric acid anodizing process using Doehlert design. Surf. Coat. Technol. 2012, 207, 123–129. [Google Scholar] [CrossRef]
- Setianto, M.H.; Korda, A. Characterization of tartaric-sulfuric acid anodized 2024-T3 aluminum alloys with anodizing potential variation. J. Phys. Conf. Ser. 2019, 1204, 012039. [Google Scholar] [CrossRef]
- Ma, Y.; Zhou, X.; Liao, Y.; Chen, X.; Zhang, C.; Wu, H.; Wang, Z.; Huang, W. Effect of anodizing parameters on film morphology and corrosion resistance of AA2099 aluminum-lithium alloy. J. Electrochem. Soc. 2016, 163, C369–C376. [Google Scholar] [CrossRef] [Green Version]
- Leth-Olsen, H.; Nordlien, J.H.; Nisancioglu, K. Filiform corrosion of aluminum sheet. iii. microstructure of reactive surfaces. Corros. Sci. 1998, 40, 2051–2063. [Google Scholar] [CrossRef]
- Ambat, R.; Davenport, A.J.; Afseth, A.; Scamans, G. Electrochemical behavior of the active surface layer on rolled aluminum alloy sheet. J. Electrochem. Soc. 2004, 151, B53. [Google Scholar] [CrossRef]
- Zhou, X.; Liu, Y.; Thompson, G.E.; Scamans, G.M.; Skeldon, P.; Hunter, J.A. Near-surface deformed layers on rolled aluminum alloys. Metall. Mater. Trans. A 2011, 42, 1373–1385. [Google Scholar] [CrossRef]
- Ma, Y.; Zhou, X.; Thompson, G.E.; Skeldon, P. Surface texture formed on AA2099 Al–Li–Cu alloy during alkaline etching. Corros. Sci. 2013, 66, 292–299. [Google Scholar] [CrossRef]
- Koroleva, E.V.; Thompson, G.E.; Hollrigl, G.; Bloeck, M. Surface morphological changes of aluminum alloys in alkaline solution:: Effect of second phase material. Corros. Sci. 1999, 41, 1475–1495. [Google Scholar] [CrossRef]
- Moffitt, C.E.; Wieliczka, D.M.; Yasuda, H.K. An XPS study of the elemental enrichment on aluminum alloy surfaces from chemical cleaning. Surf. Coat. Technol. 2001, 137, 188–196. [Google Scholar] [CrossRef]
- Yasakau, K.A.; Zheludkevich, M.L.; Lamaka, S.V.; Ferreira, M.G.S. Role of intermetallic phases in localized corrosion of AA5083. Electrochim. Acta 2007, 52, 7651–7659. [Google Scholar] [CrossRef]
- Guillaumin, V.; Mankowski, G. Localized corrosion of 2024 T351 aluminum alloy in chloride media. Corros. Sci. 1998, 41, 421–438. [Google Scholar] [CrossRef]
- Czurgelies, J. Bewertung und modifikation von chromatfreien oberflächenschutzsystemen auf kupferhaltigen aluminumlegierungen. Master Thesis, Hochschule Esslingen/Hochschule Aalen, Esslingen am Neckar, Germany, 2013. [Google Scholar]
- Carangelo, A.; Curioni, M.; Acquesta, A.; Monetta, T.; Bellucci, F. Cerium-based sealing of anodic films on AA2024T3: Effect of pore morphology on anticorrosion performance. J. Electrochem. Soc. 2016, 163, C907–C916. [Google Scholar]
- Arrowsmith, D.J.; Moth, D.A.; Vickery, C.M. Etching aluminum for adhesive bonding. Trans. IMF 1988, 66, 112–115. [Google Scholar] [CrossRef]
- Wegman, R.F.; Van Twisk, J. 2-Aluminum and Aluminum Alloys. In Surface Preparation Techniques for Adhesive Bonding, 2nd ed.; Wegman, R.F., Van Twisk, J., Eds.; William Andrew Publishing: Amsterdam, The Netherlands, 2013; pp. 9–37. ISBN 978-1-4557-3126-8. [Google Scholar]
- Farrell, R.; Horner, E. Metal cleaning. Met. Finish. 2007, 105, 86–97. [Google Scholar] [CrossRef]
- Bijlmer, P.F.A. Pickling of Aluminum. U.S. Patent 4042475A, 16 August 1977. [Google Scholar]
- Exalto, R.; Kwakernaak, A. Pickling of Aluminum. U.S. Patent 4397721A, 9 August 1983. [Google Scholar]
- Exalto, R.; Kwakernaak, A. Pickling of Aluminum. U.S. Patent 4394232A, 19 July 1983. [Google Scholar]
- Visser, P.; Terryn, H.; Mol, J.M.C. On the importance of irreversibility of corrosion inhibitors for active coating protection of AA2024-T3. Corros. Sci. 2018, 140, 272–285. [Google Scholar] [CrossRef]
- Ofoegbu, S.U.; Fernandes, F.A.; Pereira, A.B. The sealing step in aluminum anodizing: A focus on sustainable strategies for enhancing both energy efficiency and corrosion resistance. Coatings 2020, 10, 226. [Google Scholar]














Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paz Martínez-Viademonte, M.; Abrahami, S.T.; Hack, T.; Burchardt, M.; Terryn, H. A Review on Anodizing of Aerospace Aluminum Alloys for Corrosion Protection. Coatings 2020, 10, 1106. https://doi.org/10.3390/coatings10111106
Paz Martínez-Viademonte M, Abrahami ST, Hack T, Burchardt M, Terryn H. A Review on Anodizing of Aerospace Aluminum Alloys for Corrosion Protection. Coatings. 2020; 10(11):1106. https://doi.org/10.3390/coatings10111106
Chicago/Turabian StylePaz Martínez-Viademonte, Mariana, Shoshan T. Abrahami, Theodor Hack, Malte Burchardt, and Herman Terryn. 2020. "A Review on Anodizing of Aerospace Aluminum Alloys for Corrosion Protection" Coatings 10, no. 11: 1106. https://doi.org/10.3390/coatings10111106
APA StylePaz Martínez-Viademonte, M., Abrahami, S. T., Hack, T., Burchardt, M., & Terryn, H. (2020). A Review on Anodizing of Aerospace Aluminum Alloys for Corrosion Protection. Coatings, 10(11), 1106. https://doi.org/10.3390/coatings10111106