1. Introduction
Anisotropic micro- and nano-objects enable dissimilar and multiple properties in physical, chemical, and biological aspects, thereby offering unrivaled synergistic multifunctional properties compared to the simple functions usually found in conventional isotropic materials. These advanced anisotropic materials have been shown to be useful in a wide range of applications as energy materials, optical materials, and biomaterials [
1,
2,
3]. Many review articles have outlined recent progress in the fabrication processes and the promising applications of these anisotropy materials [
4,
5,
6,
7,
8,
9].
Exciting combinations used to access anisotropy across the building blocks of distinct materials have generated complexity with regard to a new dimension of combined physical (geometrical) properties and chemical (material) functionalities. Although successfully predicted [
2], there are challenges regarding different thermodynamic complexities, such as intermolecular interactions between diverse molecules in composited systems [
10,
11], unfavorable and phase-separated boundaries, compromised functionality due to structural disorders, and limited experimental methods for the fabrication of sophisticated and sensitive anisotropic materials. Currently, existing methods for producing these materials include colloidal assembly [
12], the application of postmachinal force to reshape structures [
13,
14], the application of regional modifications to create directional patches [
15,
16], and electrified jetting with combined jetting solutions (cojetting) [
17,
18]. Specific limitations, however, include restricted sizes and low yields, problematic intermolecular interactions, e.g., chemical reactions or physical bindings, causing various forms of aggregations, and uncontrollable diffusion or phase separation, resulting in undesired cross-boundary issues in each component composed of sensitive structures, which are subjected to irreversible deconstruction and denaturation during the fabrication process. These problems still limit the construction of anisotropic materials composed of sensitive structures and delicate functionalities, and novel approaches to produce these anisotropic materials easily with high yields are still in great demand. Chemical vapor sublimation and deposition (CVSD) is a unique and versatile technique to construct porous materials with sizes ranging from centimeters to nanometers [
19,
20]. This technique has been successfully employed for vapor-depositing a poly-
p-xylylene polymer system on an iced template substrate. Compared to the stationary substrates used in conventional vapor deposition, the CVSD deposition occurred on a dynamic sublimation substrate, where shrinkage in the volume and perimeter of the sublimating substrate caused a mobile surface in the direction of shrinkage. The resulting poly-
p-xylylene deposition on this mobile substrate enabled not only the planar coverage of poly-
p-xylylene on the substrate surface, but also allowed for deposition into the third depth dimension due to the mobile substrate, resulting in a three-dimensional bulk material of poly-
p-xylylene. Whereas past studies have exploited a refined CVSD approach to form nanometer-sized objects [
19,
21], we found that CVSD on a mixed solution of a solid template containing nonvolatile components can result in the construction of an anisotropic material final product (
Figure 1). Two stages were proposed to fabricate the anisotropic object by CVSD and were controlled by a programmable time (
) parameter to regulate the sublimation and deposition in the stripping stage and the encapsulation stage during the CVSD process, i.e., an iced template was subjected to sublimation to evaporate water molecules, and the same template was subjected to vapor-deposited poly-
p-xylylene to transform the template into a poly-
p-xylylene matrix for encapsulation of the SBA protein tubes. CVSD offers the same advantages as the conventional chemical vapor deposition process, i.e., no solvent, dry process, and conformal deposition with respect to the substrate topology and geometry [
22,
23,
24]. Additionally, it provides (1) controlled unsteady-state mass transport by using a sublimation substrate, avoiding problematic intermolecular and phase separation issues, (2) a time-dependent control parameter to independently regulate the mass transport of each component in the sublimation phase and the deposition phase during CVSD, enabling the construction of the final anisotropic object with tunable bulk size and/or geometric shape, and (3) the predetermined distribution of the solute phases (one or multiple), eliminating stresses from interphase and intermolecular interactions due to changes in concentrations and rehydration, which frequently occur in conventional solution-based systems [
25,
26] and freeze-dried products [
27,
28], enabling the preservation of the delicate function and structure of sensitive molecules.
2. Materials and Methods
2.1. Preparation Process of SBA Protein Tubes
First, 4-(2-Hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffer was prepared by dissolving 0.952 g HEPES and 0.468 g NaCl into 200 mL pure water ([HEPES] = 20 mM and [NaCl] = 40 mM). The pH of the buffer was adjusted by adding 1 mM NaOH solution until a value of 7.2 was achieved. The SBA protein, CaCl2, MnCl2 and the ligands were dissolved in buffer separately and kept at 4 °C for one day. All solutions were filtered through a 0.22 µm membrane before use. The SBA tube sample was generated by simply mixing these solutions and adjusting the final concentration of the sample to [SBA] = 0.1 mM, [ligand] = 0.1 mM, [CaCl2] = 5 mM, [MnCl2] = 5 mM), [HEPES] = 20 mM and [NaCl] = 40 mM. The formation of these SBA protein assemblies was induced by dual-supramolecular interactions: carbohydrate−protein interactions occurred first between the protein and N-acetyl-α-D-galactosamine on the ligand, followed by rhodamine dimerization between two ligands. The protein tubes were formed in 48 h, and their length gradually increased with increasing time. These protein tubes remained stable for more than one year at 4 °C in a mild neutral pH = 7.2 buffer solution.
2.2. Fabrication Process
The synthesized SBA protein tube solution with a concentration of 0.1 mmol/L was used to prepare the ice-particle templates. Droplets of the solution were formed by spraying the solution onto a hydrophobic poly(tetrafluoroethylene) (PTEF) substrate. A solidification procedure using a liquid nitrogen bath was performed to transform the solution droplets into iced particles. These ice particles served as templates for the subsequent CVSD process. The ice particles were placed in a homemade vapor deposition chamber [
23,
24] for the sublimation and deposition processes. The processing conditions were a pressure of 150 mTorr and at a deposition temperature of 4 °C. For the vapor deposition of poly-
p-xylylene, polymerization from a precursor dichloro-[2,2]-paracyclophane (Galxyl C, Galentis, Marcon, Italy) was performed by vaporizing the precursor at approximately 120 °C, followed by a higher temperature of 650 °C to pyrolyze the vapor precursor into quinodimethane radicals. The deposition and polymerization finally occurred in a cooled sample holder at 4 °C. Argon carrier gas at a flow rate of 15 sccm was used during the process to deliver the vapor precursors and the radicals. A deposition rate of 0.5 Å/s was monitored by in situ quartz crystal microbalance (QCM) equipment (STM-100/MF, Sycon Instruments, East Syracuse, New York, NY, USA) mounted on the deposition chamber.
2.3. Characterizations
The vapor compositions during the CVSD fabrication process were characterized by real-time mass spectrometry with a residual gas analyzer (RGA, HAL RC 511, Hiden Analytical, Warrington, UK). The operation conditions were a pressure of 10−7 mbar, an electron ionization energy of 70 eV and an ionization emission current of 20 μA. The mass detection range was from 0 amu to 400 amu. SEM images were recorded with a NovaTM NanoSEM (FEI, Hillsboro, OR, USA) at a primary voltage of 10 eV and emission current of 204 μA with an Everhart-Thornley detector (ETD). Negative TEM images were obtained by staining the samples with 1 wt% uranyl acetate for 10 s. The TEM experiments were then performed with an H-7650 TEM (Hitachi, Tokyo, Japan) at 25 kV, and the images were captured by a Veleta TEM camera (Olympus, Tokyo, Japan). Cryo-TEM samples were obtained using Vitrobot (Thermo Fisher, Waltham, MA, USA) then transferred to Fischione 2550 Cryo transfer holder (Fischione, Export, PA, USA) and images were captured under 200 kV by a Tecnai TF20 TEM (FEI, Hillsboro, OR, USA). AFM experiments were acquired by a nanoscope IIIa (Veeco, Edina, MN, USA) in tapping mode using silicon nitride tips (Bruker, Billerica, MA, USA) with a tip radius of 12 nm and a spring constant of 0.04 N/m. FT-IR spectra of the fabricated particle samples were recorded by a Spectrum 100 spectrometer (PerkinElmer, Waltham, MA, USA) equipped with an advanced grazing angle specular reflectance accessory (AGA, PIKE Technologies, Fitchburg, WI, USA) equipped with a liquid nitrogen-cooled MCT detector. The recorded spectra ranged from 600 to 4500 cm−1 with 16 scan times at 4 cm−1 resolution. An IX71 fluorescence microscope (Olympus, Center Valley, PA, USA) equipped with a Lumen200 200 W fluorescence lamp (Prior, Cambridge, UK) was used to detect the ligand within the SBA proteins, and a VK-9500 3D profile microscope (Keyence, Osaka, Japan) was also used to analyze the morphology and height of the fabricated particle samples.
2.4. Simulations
A simulation of the sublimation of an ice particle was performed and constructed with COMSOL Multiphysics, and two-dimensional (2D) and three-dimensional (3D) finite element analysis were applied. The deformed geometry method was employed to trace the sublimation interface. The model framework was constructed based on the assumption of homogeneous and isotropic composition of the ice particle with a symmetric and spherical geometry. The velocity of the deformed interface was derived based on mass and heat balance and was given by:
where
N is the mass flux of water vapor,
is the latent heat of sublimation of ice, which is determined by the Clapeyron equation [
29] since we assumed that the frozen ice phase is in equilibrium with the water vapor at the ice-air interface,
is the interface velocity,
is the density of ice, and
is the heat flux at the interface. The interface velocity was determined by the Stefan condition [
30] and arbitrary Lagrangian–Eulerian (ALE) formulation [
31]. The saturation vapor pressure at the boundary was obtained from the real-time mass detector, which is measured to be
torr. The general and reasonable diffusion coefficient assumed a constant dependence on temperature [
32]. The bulk and interface temperatures were room temperature (20 °C) and at a phase change temperature of water at 150 mTorr, respectively.
3. Results and Discussion
Delicate soybean agglutinin (SBA) protein assemblies were fabricated by the ligand strategy following previously reported procedures, and were used for the generation of the proposed anisotropic object due to their unique structure, which is symmetric and tubular in three dimensions. Briefly, SBA assemblies were constructed utilizing the specific sugar-binding capability of its four monomers in the presence of Mn
2+ and Ca
2+ [
33]. Through the assistance of a developed ligand composed of N-acetyl-α-D-galactosamine, ethylene oxide spacer and rhodamine B, a self-assembled SBA tetramer structure with a helical tube with a diameter of approximately 20 nm and a length of 100–2000 nm was generated. The synthesized SBA was characterized with dynamic light scattering (DLS) and Cryo-TEM (cryogenic transmission electron microscopy), and the results showed an SBA tube approximately 250 nm in size and 26 nm in diameter (
Figure 2a,b). Suspension solutions of the synthesized SBA protein tubes with a defined composition were then prepared to form droplets on hydrophobic surfaces [
21] and transformed into solidified protein solution particles by a temperature solidification process, e.g., liquid nitrogen or dry ice bath, resulting in a sublimation template for the CVSD process used in this study. The thermodynamic conditions (approximately 0.2 mbar and 20 °C) of the CVSD operation favored the transformation of the solid-phase water component (ice) of the solidified protein solution into water vapor, similar to the generation of CO
2 vapor from dry ice in ambient conditions [
34] or to the freeze-drying process to evaporate the water (or solvent) phase from a solution mixture used in many purification and processing operations [
35,
36]. Under the CVSD conditions, water molecules were evaporated by sublimation from the solid iced particles, whereas the nonsublimating protein molecules retained their controlled distribution on the substrate and were seamlessly encapsulated into the deposited polymer structure [
20,
21].
The construction of the proposed anisotropic objects was therefore established by CVSD with a controllable two-stage process: (i) stripping stage, (ii) encapsulating stage, where the time-dependent parameter, Δ
t, was used to control the sublimation degree of the sublimated ice template, and the elapsed time was propositional to the sublimated volume of shrinkage. More specifically, in the stripping stage, the protein molecules were used as a nonsublimation-solute system in the ice particles with the sublimating water molecules (solvent system). The sublimation of water molecules resulted in a smaller volume and receding interface perimeters of the ice particles, where the protein molecules were located towards the outer particle surfaces, while in the encapsulating stage, the vapor deposition of poly-
p-xylylene occurred stagewise (after the same programmed time), and the remaining volume (mass) of the sublimating ice particles was transformed into porous structures of poly-
p-xylylene, which was used a matrix for the encapsulation of the protein molecules without affecting their configuration and structure. Compared to the stripping stage, during which mass transport flux only occurred in one (outward) direction through sublimation, the encapsulating stage additionally involved the mass flux of deposited poly-
p-xylylene into the system onto the sublimating template/substrate, which led to a transformation through the replacement of vaporized water molecules with the deposited poly-
p-xylylene molecules. The stagewise stripping and encapsulating processes finally resulted in the proposed anisotropic nanoparticle product, which was composed of poly-
p-xylylene as the matrix and distributed SBA proteins within the poly-
p-xylylene structure. In the experiments, symmetrical sphere ice particles of the described protein solution were prepared by forming droplets on hydrophobic surfaces (water contact angle approximately 137.3 ± 6.5 degrees,
Figure 2c) followed by a solidification process using a liquid nitrogen bath. The ice particles served as sublimation templates, and the operation time (Δ
t1, Δ
t2, Δ
t3,
…) was controlled and resulted in emerging and eventually exposed nonvolatile components of the protein solutes by the time-dependent shrinkage of the ice perimeter due to sublimation in the two-stage process of the fabrication process. The controllable Δ
t parameter regulated the timing of the sequences (i) and (ii) during the fabrication process and determined the aspect ratio and shape of the finalized (hybrid-) nanoparticle products. A real-time mass spectroscopic gas analyzer was used to monitor the vapor composition (
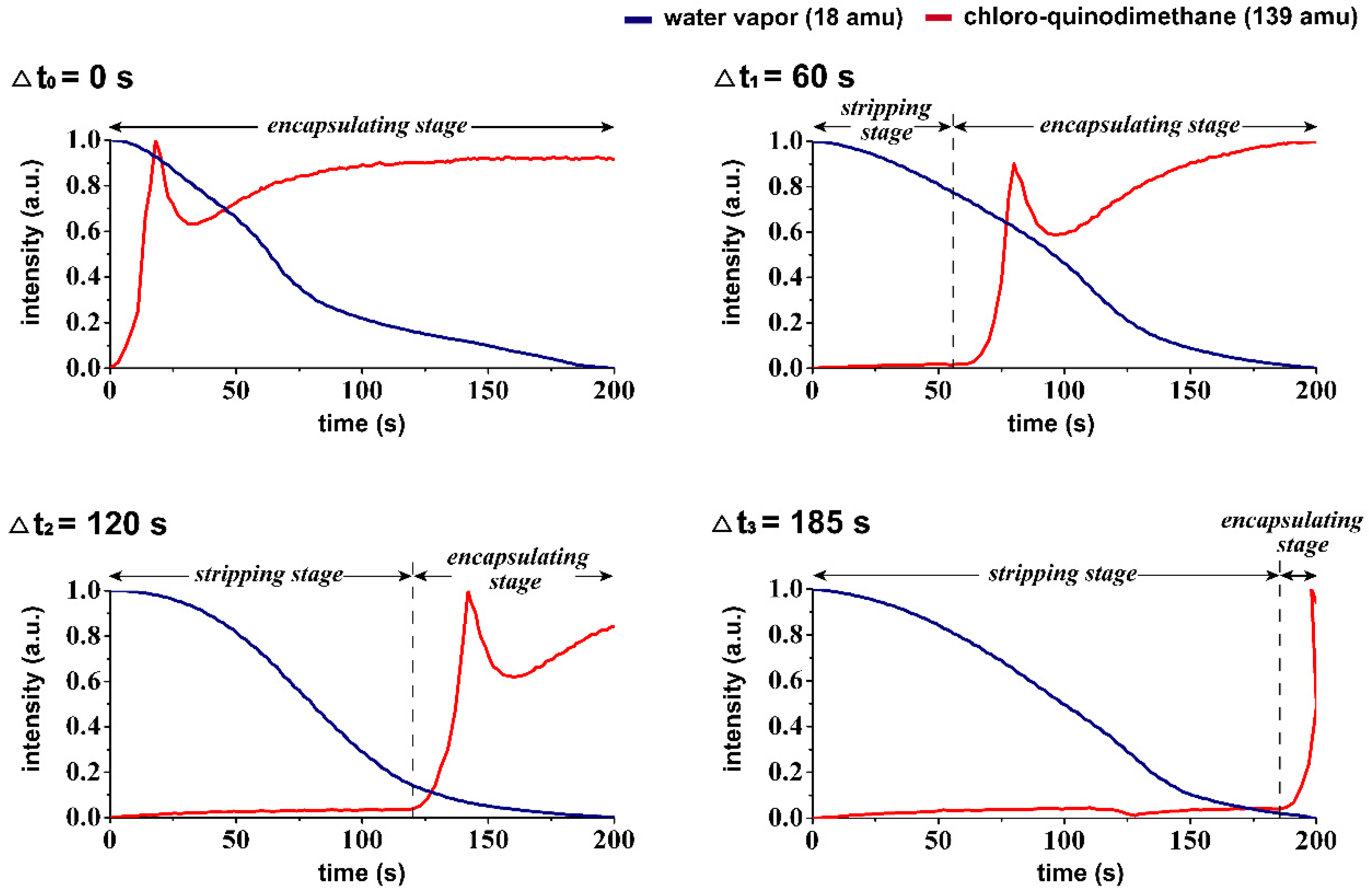
Figure 2d), and the correspondent species including water vapor (18 amu), carrier gas of argon (40 amu), derivatives of quinodimethanes (104 amu and 139 amu) were detected in the studied vapor system of the CVSD fabrication process. A more systematic analysis of the changes in mass was further performed to verify the controllability of time during the stripping and encapsulating stages in the CVSD process. As revealed in
Figure 3, the detection of an increased level of water vapor (18 amu) indicated the occurrence of sublimation in the stripping stage, and similarly, a vaporized chloro-quinodimethane precursor (139 amu) was detected to indicate the initiation of deposition in the encapsulation stage. The timing parameter, with values of Δ
t1 = 60 s, Δ
t2 = 120 s, and Δ
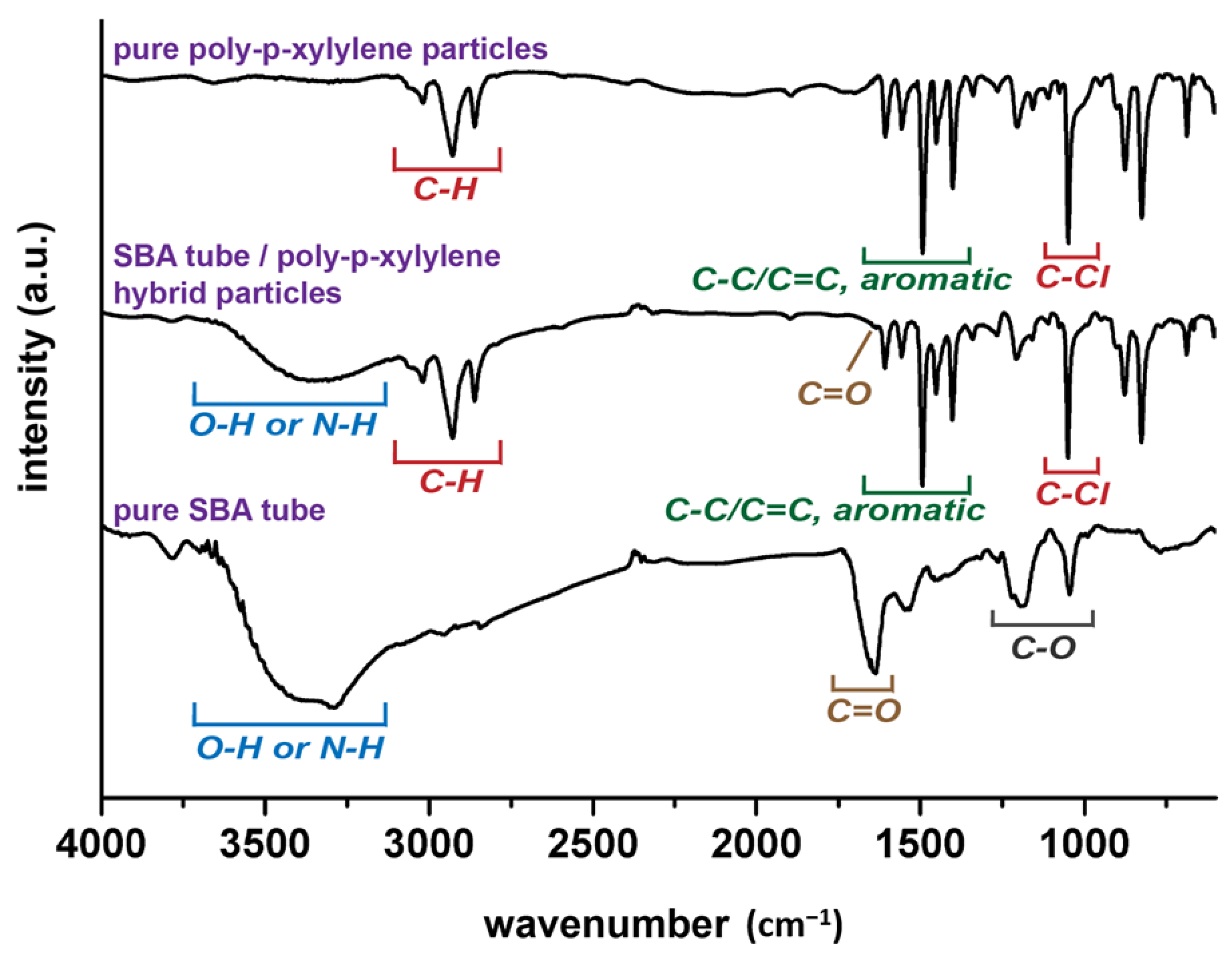
t3 = 185 s, was shown to control and program both the stripping stage and the encapsulating stage, resulting in changes in the vapor compositions at the selected and designated times. From a chemical point of view, characterizations for the resultant hybrid particles products were also performed by using FT-IR analysis. As shown in
Figure 4, the recorded spectra showed the characteristic –C–Cl and –C–H bands from poly-
p-xylylene, similar to other reported poly-
p-xylylene systems. For the hybrid product comprised both SBA and poly-
p-xylylene, despite possible overlapped peaks of –C=O and –C–O with the –C–Cl peaks, additional –N–H and –O–H bands from solely SBA were detected and confirmed the successful fabrication of the hybrid materials of poly-
p-xylylene/SBA.
A simulation analysis was then performed to understand the timing mechanism during the CVSD and to rationalize the controllability of the geometrical properties vs. the Δ
t parameters. Taking advantage of the similarity of a freeze-drying process with the ice-template sublimation process of the current system during the proposed stripping stage, a reported mathematical freeze-drying model was used in the simulations [
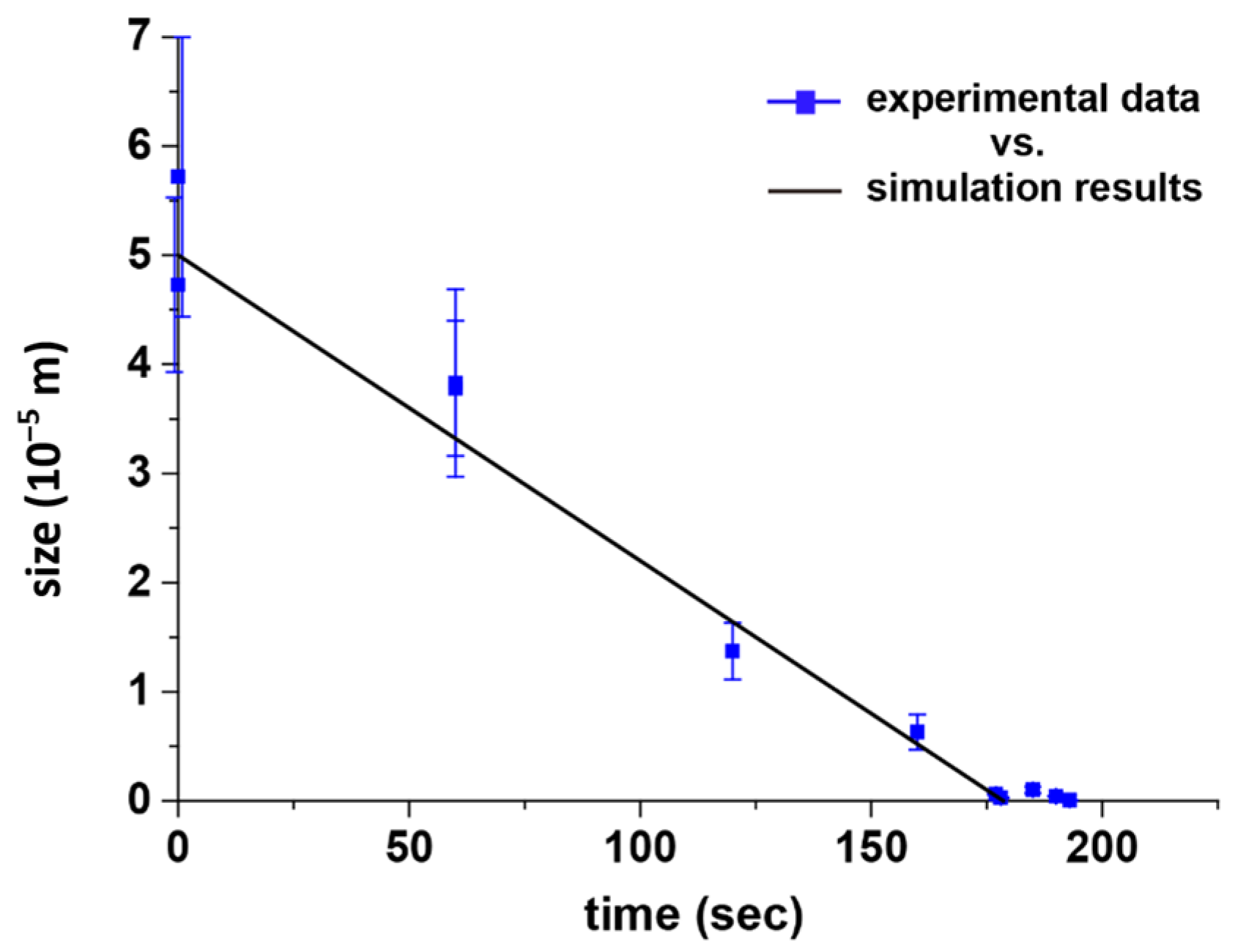
37]. Our model framework was designed according to the proposed dimensional scale of an ice particle system, and the simulated dimensional and temporal information was collected. Briefly, a spherical ice particle was initiated and coupled through heat and mass transfer mechanisms in its sublimation process, and the moving sublimation boundary was treated as a sharp interface. The interfacial temperature was obtained by the saturation vapor pressure, while the interface velocity was determined by the normal heat flux difference at the interface. The processing pressure of the CVSD process was used as the pressure of the external environment of the ice sphere. Although the SBA tubes were described as nonsublimating solutes within the ice template, they were sparsely distributed and nonreactive during the CVSD process. Therefore, the SBA proteins should have a limited effect on the sublimation process in the simulation. The simulation results were consistent with the experimental results, for example, a Δ
t = 60 s reduced the size of an ice particle from the initial 50 μm to 32 μm, 160 s resulted in a size decrease to 5 μm, 177 s to 500 nm, and 178 s to 250 nm, as shown in
Figure 5a. After the stripping stage, with the determined dimension configurations according to the set Δ
t, the subsequent encapsulating stage initiated the transformation by inducing mass transfer of poly-
p-xylylene through vapor deposition (in contrast to retrograding mass transfer by sublimation) onto the sublimating ice particle. The final asymmetrical nanoparticles were obtained by replacement of the remaining ice/water molecules with the aforementioned polymerized poly-
p-xylylene molecules. As revealed in
Figure 5b, combinations of characterization techniques, including optical microscopy, scanning electron microscopy (SEM), transmission electron microscopy (TEM), and AFM, showed that the encapsulation and poly-
p-xylylene deposition occurred by retrograding the outer surface of the ice particles, thereby exposing the protein to the particle surface, and a time-dependent configuration of the size and the exposed protein-assembly dimensions was found, which was consistent with the simulation results. Particles with 38.3 ± 8.6 μm size were fabricated when Δ
t = 60 s, 6.3 ± 1.6 μm when Δ
t = 160 s, 608.3 ± 56.8 nm when Δ
t = 177 s, and 294.3 ± 38.6 nm when Δ
t = 178 s; a first-order linear correlation of Δ
t vs. length (
) was found, which supported the simulation data and the reported results [
19]. The fabricated anisotropic particle structure was preserved without compromising the delicate SBA tube structure, and a tube diameter of approximately 40 nm were found to be consistent with the prepared SBA tube dimension in the solution phase. The approximately 14 nm-increased diameter thickness (from 26 nm of a bare SBA tube to 40 nm) was found to be comparable to the deposited thickness of poly-
p-xylylene and was due to the deposition on the nonsublimating SBA tube, analogous to conventional poly-
p-xylylene depositions on a solid substrate [
22,
38]. To compare the results, the time-dependent particle sizes based on the simulation trajectory and the fabricated particles are plotted in
Figure 6, and a recorded simulation video showing a sublimating ice particle template with respect to the elapsed time is included in the supporting information.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





