When planning for an XRD experiment on organic thin films, one needs to consider three questions: (1) if there is a necessity to use a synchrotron, (2) what quantities are going to be analyzed, and (3) what measurement geometry should be used.
The first question is rather unique to organic thin films. Organic materials such as polymers have much lower diffraction strength compared to similar inorganic samples at the same X-ray flux. Combine this with the thin nature of our samples, and we are posed with the necessity for very long data integration time. Lab-scale X-ray systems are typically not bright enough for a timely high-quality data collection of organic samples thinner than ∼100 nm regardless of measurement geometry, and, therefore, a synchrotron-based experiment is usually required. Thicker samples can be measured with lab-scale systems, although they often need to be measured in grazing-incidence geometries for best results. Additionally, depending on the sample, the investigator often need to weigh in possible sample damage due to X-ray radiation or exposure to ambient air. An inert measurement chamber is typically used to reduce potential degradation in ambient air, while translating the sample stage in between measurements has been used to reduce X-ray damage to the sample.
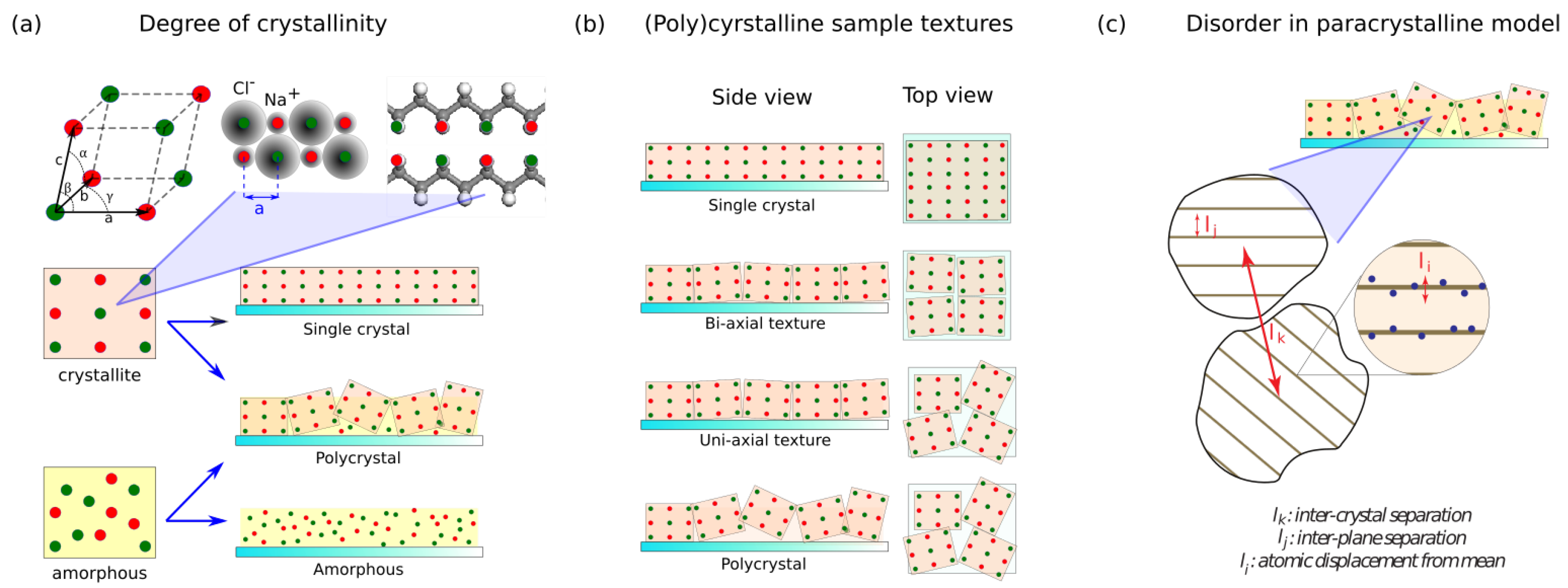
There are three quantities that can be analyzed: (1) peak location, (2) peak intensity, and (3) peak shape. From Bragg’s law, peak location gives information on lattice constants and can therefore be used to examine chemical composition or crystal symmetry. Peak intensity is related to the amount of crystallites that are reflecting X-rays into the detector (i.e., crystallites with correct orientation) and may shed light on, for example, degree of crystallinity. The dependence of peak intensity on the crystallite orientation will also be important in investigating crystallographic texture of the sample. Lastly, peak shape is a convolution of various factors such as crystallite size, strain, and paracrystallinity. It is therefore related to, but not exclusively, the degree of disorder in the material. Note that peak shape analyses typically require multiple harmonics of the XRD peak of interest and therefore require ample X-ray dosing and high resolution.
The basic rules on measurement geometry are as follows: (1) From Bragg’s Law, changing the difference in X-ray angles determines the lattice constant that is being sampled. On the other hand, (2) changing the X-ray angles with respect to the sample angles determines which crystallite orientation is being sampled and will be useful in determining sample texture. (3) Consider the goniometer axes’ range and precision when choosing the goniometer angle that is to be scanned. Rotate the sample if necessary. (4) Lower incident angle means higher sampling area, possibly lower background signal from the substrate, and reduced beam damage on the sample. While this is appropriate for thin films, it comes with significant trade-offs that will be discussed in the subsequent subchapters. Lastly, (5) when variable wavelength X-ray source is available, consider optimizing the wavelength for your application.
4.1. Common Peak Intensity Corrections and Background Subtraction
When analyzing XRD peak intensity and peak shape, it is important to apply the relevant intensity corrections and do background subtraction. While most of these corrections depend on the geometry of the measurement system, the following two correction factors are not geometry-dependent (peak intensity is to be divided by correction factor). Additional corrections specific to special measurement configurations will be discussed in the appropriate subsections.
The first correction factor is the scattering factor of different atoms. Higher atomic number corresponds to a higher number of electrons and thus higher X-ray scattering intensity. For most polymeric systems, this factor is not very important since most of the scattering centers are carbon. However, it does explain why polymeric systems have low X-ray peak intensity. Should one deals with analysis of Bragg planes that consist of other elements, and the table for atomic scattering factor as a function of atomic number and X-ray energy can be found in most synchrotron resources.
The second correction factor is applicable to X-ray sources that are partially polarized. When the X-ray source is partially polarized, the following polarization correction needs to be applied:
where
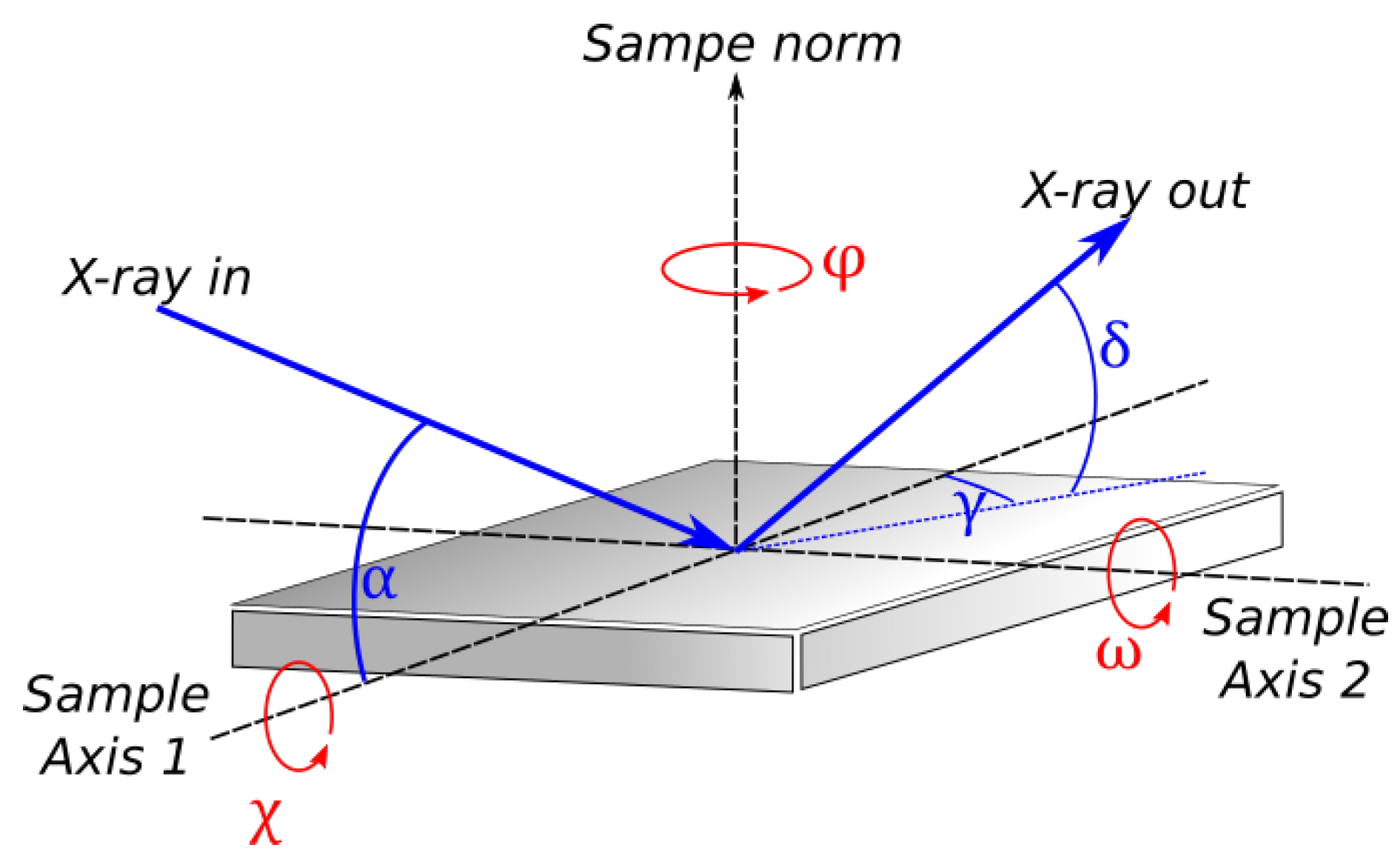
is the fraction of light that is polarized parallel to the sample surface,
is the exit angle perpendicular to the sample surface, and
is the exit angle parallel to the sample surface (
Figure 3). Lab-scale sources tend to be randomly polarized, while synchrotron radiation tends to be highly polarized.
For further reading, references [
36,
37,
38] discuss these corrections in great detail and touch upon a few other corrections that may be important for other material systems.
Background signals may arise from diffuse scattering from the air along the beam path or the sample substrate [
37]. While this can be very complex to model, in most cases the background as a function of
q can be sufficiently fitted with a decaying exponential function at low
q and power function at higher
q. Note that when multiple peaks are present, all peaks and the two background functions must be fitted simultaneously. The isolated peak, therefore, equals the signal minus the background and all other peaks.
4.2. Bragg–Brentano Geometry
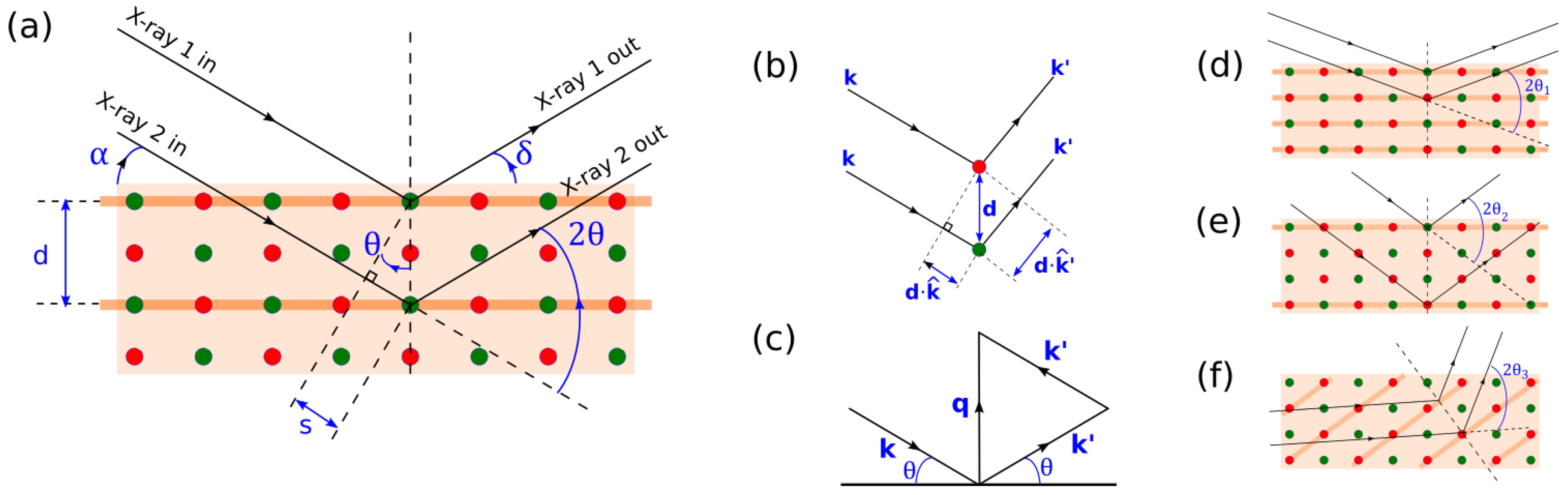
The most commonly used XRD measurement geometry is called Bragg–Brentano geometry. In this technique,
is fixed at zero while
and
are scanned equally.
Figure 2a illustrates this geometry (note that
always equal
in this geometry). This way, the angle between
and
is always equal 2
, allowing us to easily identify the lattice constant.
Because
, Bragg–Brentano geometry only samples Bragg planes whose norm is perpendicular to
(
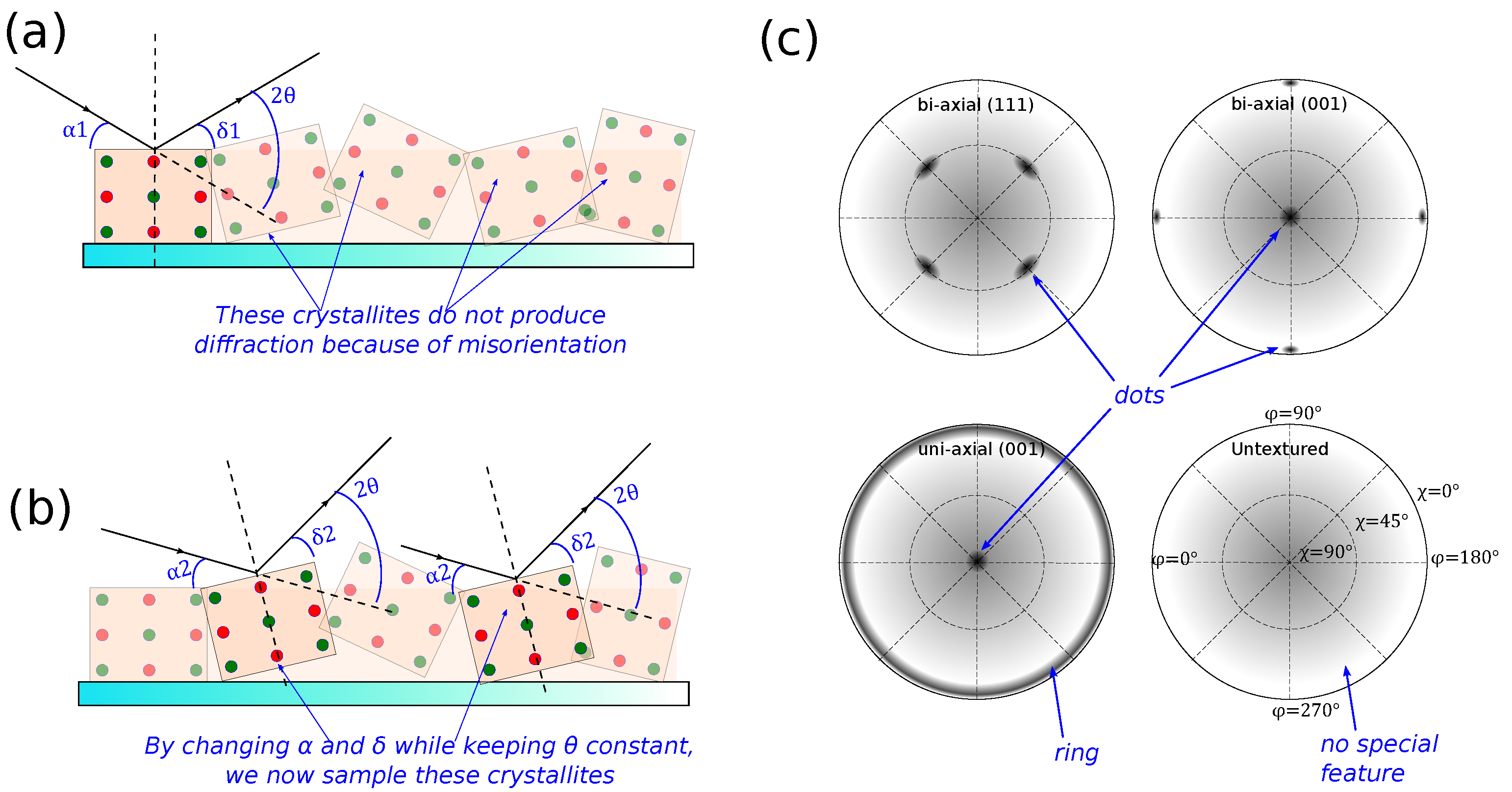
Figure 2a,d,e). This means that if the sample is textured and the crystallites are facing other directions (or slightly misoriented), the peak height may be dramatically reduced or the measurement may not even pick up any signal at all, potentially leading to a false negative if not done carefully. Some measurement techniques introduced in the next subsections will address this major disadvantage.
Another important point regarding analyses on measurement results from Bragg–Brentano geometry is that when analyzing XRD data involving a scan in
, the Lorentz correction need to be applied:
This correction takes into account the amount of time each diffracting crystallite remains in the diffracting position, thus correcting for the different integration time of different Bragg angles [
36,
39]. Usually, this correction factor combines with the polarization correction and is collectively known as the
Lorentz-polarization factor. However, it is important to note that, unlike the polarization correction, the Lorentz correction will change the shape of diffraction peaks due to its
-dependence and is therefore important in peak shape analyses.
When all correction and background subtraction has been properly applied, Bragg–Brentano geometry is suitable to be used for peak shape analysis. The general procedure for Warren–Averbach analysis goes as follows: first, high quality data where multiple peaks are clearly resolved with ample data points are taken (Reference [
10] suggests at least 100 data points over full-width-at-half-max).
Proper correction and background subtraction is then applied to the data. Equation (
7) is then simultaneously fitted to the Fourier transform of multiple isolated XRD peaks. Note that each peak will have different diffraction order
m and that the fit must be done simultaneously across all peaks. Estimation of crystallite size in the direction normal to the sample (i.e., in
), rms non-cumulative disorder, and the degree of paracrystallinity can then be obtained from the fit results. Reference [
10] provides a rigorous example of Warren–Averbach analysis to quantify disorder in organic films. A more basic example on clays is explained in Reference [
30]. Furthermore, Reference [
10] also demonstrates the utilization of pseudo-Voigt fit to get a qualitative understanding of the various disorders in the sample.
4.3. Pole Figure
When the sample is textured, it is often essential to be able to quantify the distribution of crystallite orientation. For example, if one wishes to accurately measure XRD peak height or shape, in general, one needs to first find the direction of the reference Bragg planes, and then precisely orient the sample such that is perpendicular to the Bragg planes before starting the measurement.
A
pole figure is a visualization of the distribution of crystallite orientations in the sample, made by rotating the sample axes (
and
) while fixing
and
corresponding to the Bragg reflection of interest. Take, for example, a pole figure of a Si (111) planes from a <100> wafer. To produce the pole figure, we first set the sample and goniometer in Bragg–Brentano geometry, but with
fixed at the value corresponding to Si (111) planes (this value is dependent on the X-ray wavelength. For the commonly used
sources,
). Instead of scanning
, we will now rotate
, and incrementally increase
. In this way, we essentially map all directions at which Si (111) crystallites in the sample are oriented. Because of the crystal symmetry, we will only see data at four points at
. This, and examples of how the data should look for a few other cases are shown in
Figure 4c. The figures also illustrate the effect of the background from an amorphous substrate (e.g., glass), which will be common when investigating a polymeric thin film because, most likely, the X-ray would penetrate the sample.
A pole figure is important in investigation of organic materials considering the low symmetry of the material; however, sometimes there is a high chance of uni-axial texture due to the preferred orientation in molecular packing. This method, for example, has been used to show that certain deposition conditions of poly-3-hexylthiophene (P3HT) may result in highly-textured nanofilms with high electronic transport properties along the substrate plane [
9].
4.4. Glancing Incidence X-ray Diffraction (GIXD)
Thin organic materials, including polymeric thin films, pose a rather unique challenge to X-ray experiments. The low scattering factor of carbon combined with the low sample volume in thin film samples often means long measurement time. This is especially true for sub-micron films. In cases where the polymeric film is not chemically stable (e.g., degradation in ambient atmosphere, or X-ray damage), measurement time can be a very important consideration. The most direct workaround is to enclose the sample in an inert chamber or to modify the beam intensity. Another way to tackle this issue is by modifying the measurement geometry.
Grazing Incidence XRD, or Glancing Incidence XRD (GIXD) refers to an XRD measurement done with a fixed, very low incidence angle . In the case of measurements with 1D detectors, the detector arm may be scanned out of the sample plane or in the sample plane. This measurement geometry is particularly useful for increasing signal yield by increasing the irradiated volume, especially in the case of thin film samples. It also has the added benefit of reducing irradiated substrate volume.
In the simplest case, this added benefit can be explained using pure geometry. Consider a sample that is thinner than the absorption length of the X-ray (∼
m), and that the X-ray is incident at a high angle (
Figure 5a). In this case, potentially a large portion of the incident X-ray is actually absorbed by the substrate rather than the sample, leading to signal loss. Reducing the incident angle will allow more X-rays to be absorbed by the sample rather than the substrate, thus increasing signal yield. Additionally, the X-ray flux on the sample is also reduced as the X-ray is now spread over a larger sample area, reducing X-ray damage.
Another important consideration when measuring GIXD is the critical angle for total X-ray external reflection on the sample, which is typically a very low angle, around the range of incident angle of interest. Below this angle, X-rays will be totally reflected off the sample surface and will only couple with the sample through the evanescent (waveguide) modes. Therefore, setting the incident angle above and below the critical angle will further allow the investigator to sample from, respectively, the “bulk” of the thin film sample or just the surface.
When combined, these benefits may reduce data collection time by orders of magnitude, while reducing beam damage on the sample. However, there are a few additional subtleties related to interpreting and post-processing GIXD data that need to be addressed [
38,
40,
41]:
Due to the fixed incidence angle, the orientation of the Bragg planes is necessarily different for different ,
Blind area due to forbidden Bragg reflection;
Absorption correction which depends on film thickness, increased irradiated area, and the position of the sample;
Shift in measured due to refraction.
The first point can be understood with the help of
Figure 2c while fixing the incoming angle. Let
be fixed, and the magnitude of
be varied. As discussed in the previous section, the conditions for constructive interference require (1) magnitude of
equal to that of
, and (2)
being the bisector of
and
. In order to satisfy both conditions, the direction of
must swing closer to
as its magnitude increases. Since
also points in the direction of the diffracting crystallites, it follows that the orientation of the crystallites that are being sampled, and, therefore, the associated Bragg planes must also be different for different
.
The second point is related, but important only when investigating texture of the film. Suppose that we restrict the incoming angle at a low angle
, and investigate a fixed
q (
Figure 5). Equation (
4) dictates that a diffraction spot will only be produced when orientation of the crystallite is parallel to
. Remember that
always bisects the incoming and outgoing beam. Suppose the outgoing beam does not deviate left or right (
). Then,
will be akin to
in
Figure 5c. Likewise, if the outgoing beam is parallel to the sample surface, it will be like
in
Figure 5c. Directions of vectors
and
represent the extreme crystallite orientation that can be sampled at a fixed
. Note that any
that lies inside the cone subtended by
is not within these extremes. In other words, crystallites whose orientation lies within the cone
from the normal will never satisfy Equation
4 and gives no diffraction peak. In the limit that
, the validity limit of the data becomes
[
9].
The absorption correction corrects for the change in scattering volume due to the geometrical effects. It depends on film thickness and is important for thin films (intensity data are to be divided by the correction factors):
where
and
are the incidence and exit angles (
Figure 3),
t is the film thickness, and
is the extinction coefficient of the X-ray in the material. In the case where the sample size is finite, the beam may spill over and diffraction intensity is limited by the sample size. Intensity might also be limited by the effective detector size, which is set by the X-ray optics.
The last correction is important when
is close to the critical angle for total internal reflection of the X-ray inside the sample. At an incidence angle close to the critical angle for total external reflection of the X-ray, the position of the measured Bragg angle shifts by a few tenths of a degree to a smaller angle due to refraction [
41]:
where
is the critical angle for total external reflection of X-ray, and
is the imaginary part of the material’s index of refraction.
This technique has become more frequently used in organic film samples [
3,
9,
10] because it greatly compensates for the low structure factor of carbon. However, the fact that in this geometry the direction of
varies makes it unsuitable for rigorous peak-shape analysis unless it is known that the sample is not textured.
4.5. GIXD and Pole Figure with a 2D-Detector
Another way to greatly improve measurement time is by replacing the point detector with a two-dimensional detector [
42], which offers much more information at the expense of resolution. Unlike in inorganic materials, polymeric thin films tend to exhibit broad peaks, so lower data resolution is often not a limiting factor. Furthermore, data resolution can be improved at the expense of data range by moving the detector closer to the sample. Since the main interest in the polymeric thin film community is typically limited to phase identification, texture, and semi-quantitative analysis of degree of crystallinity, GIXD with a 2D-detector is often the preferred way of doing XRD on thin polymeric films. Much of the development of this technique has been in support of the organic semiconductor community [
12,
13,
14]; nevertheless, undoubtedly the techniques and analysis methods are readily applicable to any polymeric thin films.
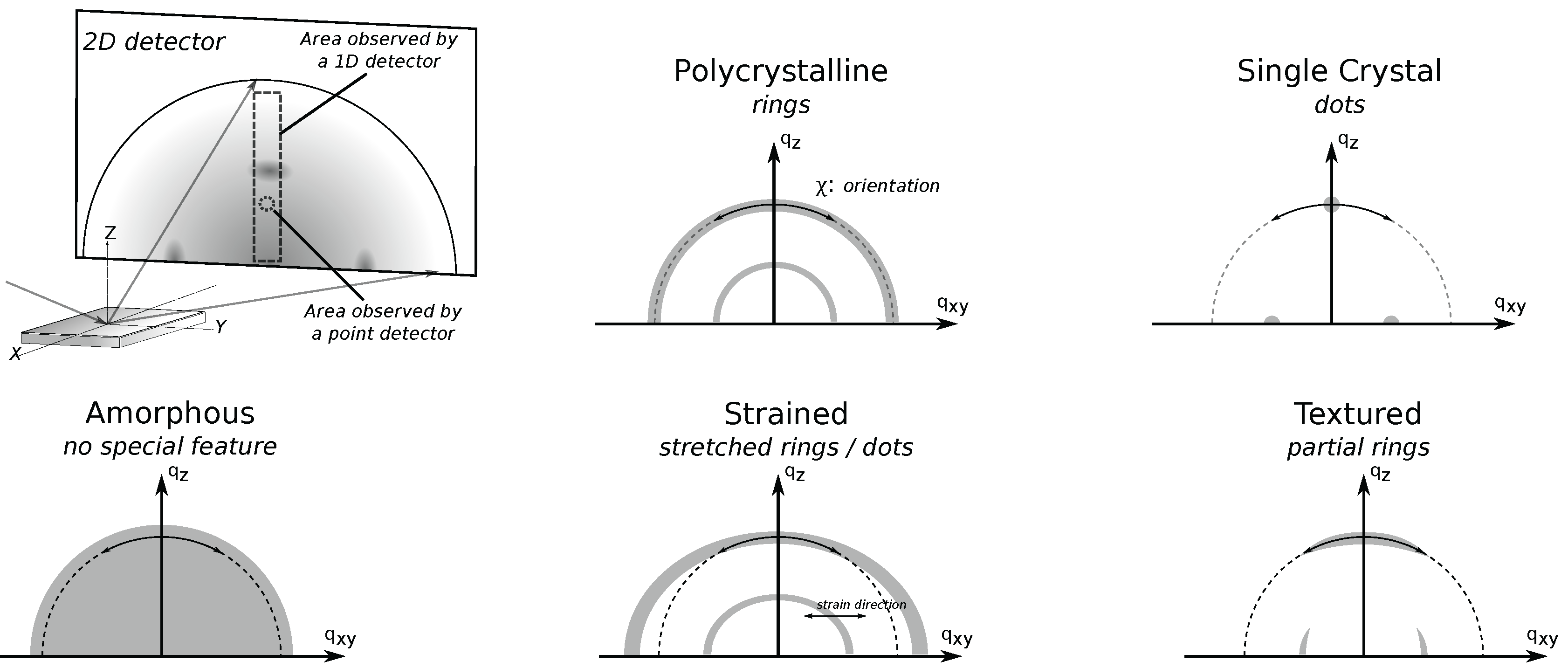
Figure 6 illustrates the richness in XRD data taken using a 2D-detector, which takes a “picture” of the XRD pattern (Debye rings). Data taken using lower-dimension detectors are essentially a subset of the data taken using a 2D detector. In 2D XRD data, radial direction is the direction of increasing
, while the azimuthal direction is much like
in pole figures. However, because of the forbidden Bragg reflection (“hole”) in GIXD discussed in the previous subsection , the direction perpendicular to sample norm on the detector corresponds to
instead of
, and additional specular data is needed if one wishes for data from this forbidden
-range.
Sample texture is readily assessed from 2D GIXD data (
Figure 6). For example, a perfect polycrystalline sample displays full Debye rings, while single crystals appear as dots. Likewise, data from samples with texture somewhere in-between these extremes will appear as partial rings and macrostrains readily result in a shifted dot or a stretched ring in 2D XRD data. On the other hand, data from amorphous materials will be lacking any special feature.
Three points are worth mentioning about this measurement geometry. First, like in the case of GIXD, since the incidence angle is fixed, the different Bragg reflections (different
or
) must necessarily come from Bragg planes with different orientation. This means that 2D detectors should not be considered as a silver bullet for examining structural properties of highly-textured samples such as single crystals. In such cases, the chance of missing data is still very high if the sample is misoriented. Second, since the image detectors are usually flat while diffraction is spherically symmetric, there is a non-linear relationship between
(or
) and pixel position on the detector. A correction can be applied when proper calibration is done as per Reference [
43]. Lastly, this also means that
(or
) resolution of the detector also changes radially, with the highest resolution achieved at the positions furthest from the detector origin. Consequently, the sample-detector distance can be adjusted to get the optimum resolution for the
-range of interest [
43].
With the proper normalizations, this rich data may offer much insight into the sample. For example, one may compare relative degree of crystallinity, texture, and crystallite size estimation of different samples. References [
3,
9,
44] introduce one such technique that is now commonly used in quantitative texture and degree of crystallinity comparison of polymeric thin films. The technique involves merging of data collected from three synchrotron measurements, and, despite it being developed for electronic polymeric ultra-thin films, is applicable to any uniaxially-textured organic thin films. The essence of the experiment goes as follows: assume that the sample has a uniaxial texture along the sample norm (
Figure 1b and
Figure 3) and has a known XRD signal at
. Then, a radial slice of the 2D GIXD data at constant
will uniquely describe the texture of the sample—essentially a
pole-figure—and can be used to compare the degree of texture among samples. Relative degree of crystallinity of the samples can be obtained from the area under the curve and can also be compared. The complexity in the technique goes into the details on how to get a complete data set, background subtraction, and normalization, which we shall briefly discuss with the help of
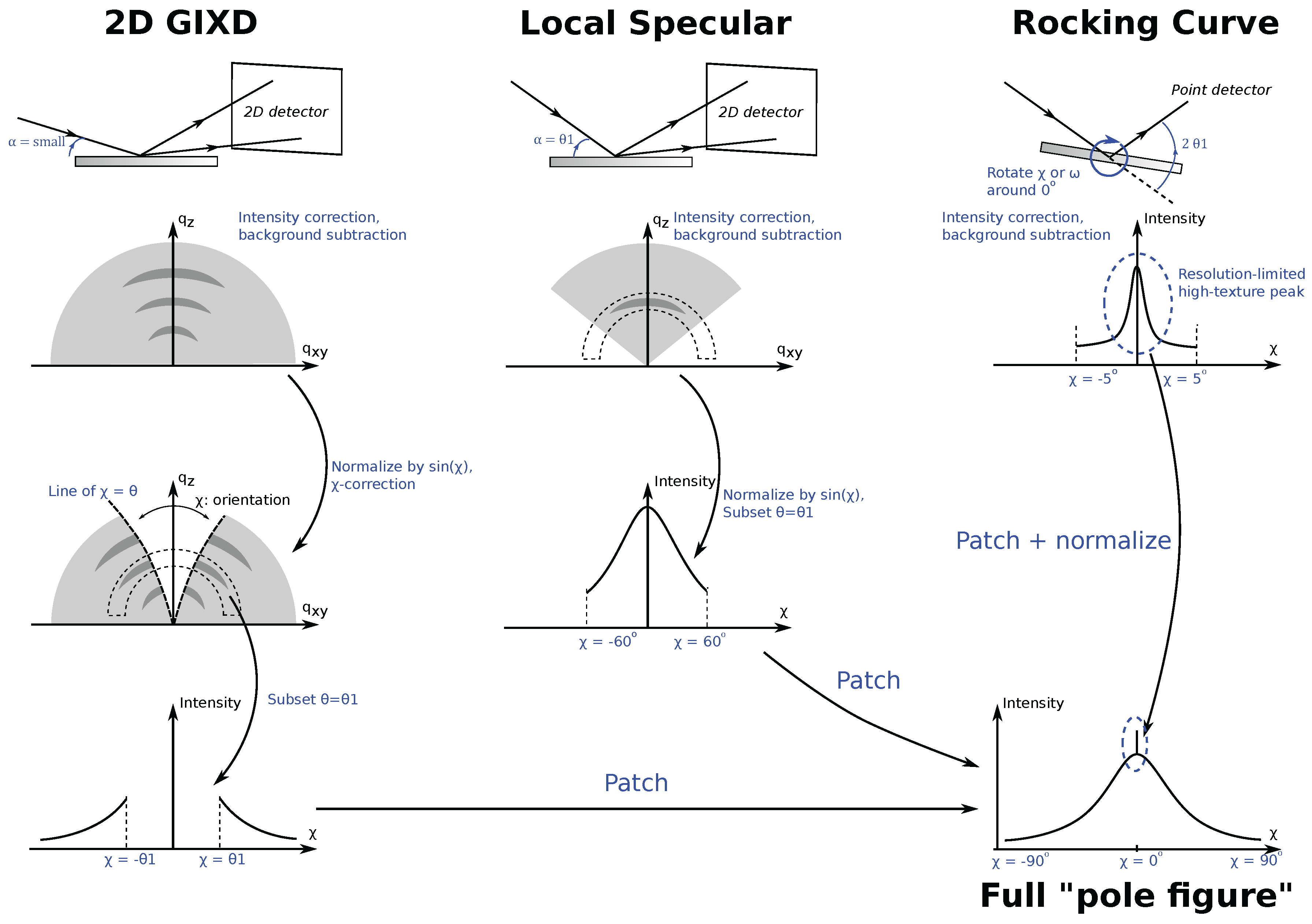
Figure 7.
The first data collection, 2D GIXD at slightly above the polymer critical angle, makes up most of the data. This data alone is incomplete because of the forbidden Bragg reflection, causing a “hole” in the data (
Section 4.4). Consequently, the first correction is to transform the detector radial angle in the 2D data properly into the actual sample axis
. To fill in the hole in GIXD measurements, a specular 2D XRD data is taken at
corresponding to the
of interest. This second measurement, called the “local specular measurement”, is valid from
, except very near
. The last measurement, a rocking curve with a high-resolution point detector, is done by setting
to the relevant
and varying
(in this case, equivalent to
due to the sample’s uniaxial texture along the sample norm,
Figure 3) between
and
. This measurement is aimed to fill in the small patch near
. In References [
3,
9,
44], it has two additional purposes: (1) to investigate the possible presence of resolution-limited super-texture due to vertical spatial confinement of the film, and (2) intensity normalization. The latter is important especially in Reference [
3] because the experiment involved various film thicknesses and the authors varied the data integration time for the first two measurements to achieve a good signal-to-noise ratio. The authors then used the normalized photon counts from the last data collection to normalize intensity data in the pole figures that allow for sample-to-sample comparison.
Each individual data must first be background subtracted, 2D data sliced at constant
, patched together, and then normalized. Furthermore, data slices from 2D GIXD need to be corrected by a factor of
to account for geometrical factors prior to patching [
3,
9,
44]. Patching slices from 2D GIXD and local specular data can be done as follows: first, identify a range in
where the data overlap. The two intensity pieces of data should be proportional to each other, the coefficient of which can be obtained from a linear fit of the data from the overlapping region. Therefore, after background subtraction, scaling one of the data sets by (and adding the small constant from) the fit result will ensure a smooth patching. Patching the rocking curve data is done similarly; however, this time, the peak intensity is set to equal the normalized intensity of the rocking curve data.
From this
pole figure, one may readily compare the relative degree of crystallinity of the samples as obtained from the area under the intensity curve, and the degree of uni-axial texture of the sample, which is inversely related to the peak width in
. These data can then be correlated to, for example, the film’s anisotropic conductivity [
8,
9,
44]. Alternatively, when the samples consist of films of the same material but different thicknesses, this kind of data may offer a significant semi-qualitative insight into the kinetics of film deposition [
3,
17].
GIXD with a 2D-detector, however, is not a solve-all technique. Measurements such as Warren–Averbach analysis require both much higher resolution than what is offered by 2D-detectors and a long scan range that spans multiple diffraction orders. In this case, the approriate approach is to start with a preliminary measurement using a 2D-detector to locate the peaks, followed by a high-resolution scan [
10].


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






