3.1. Starting Materials
Tung oil and its corresponding fatty acids and methyl esters were analyzed by FTIR,
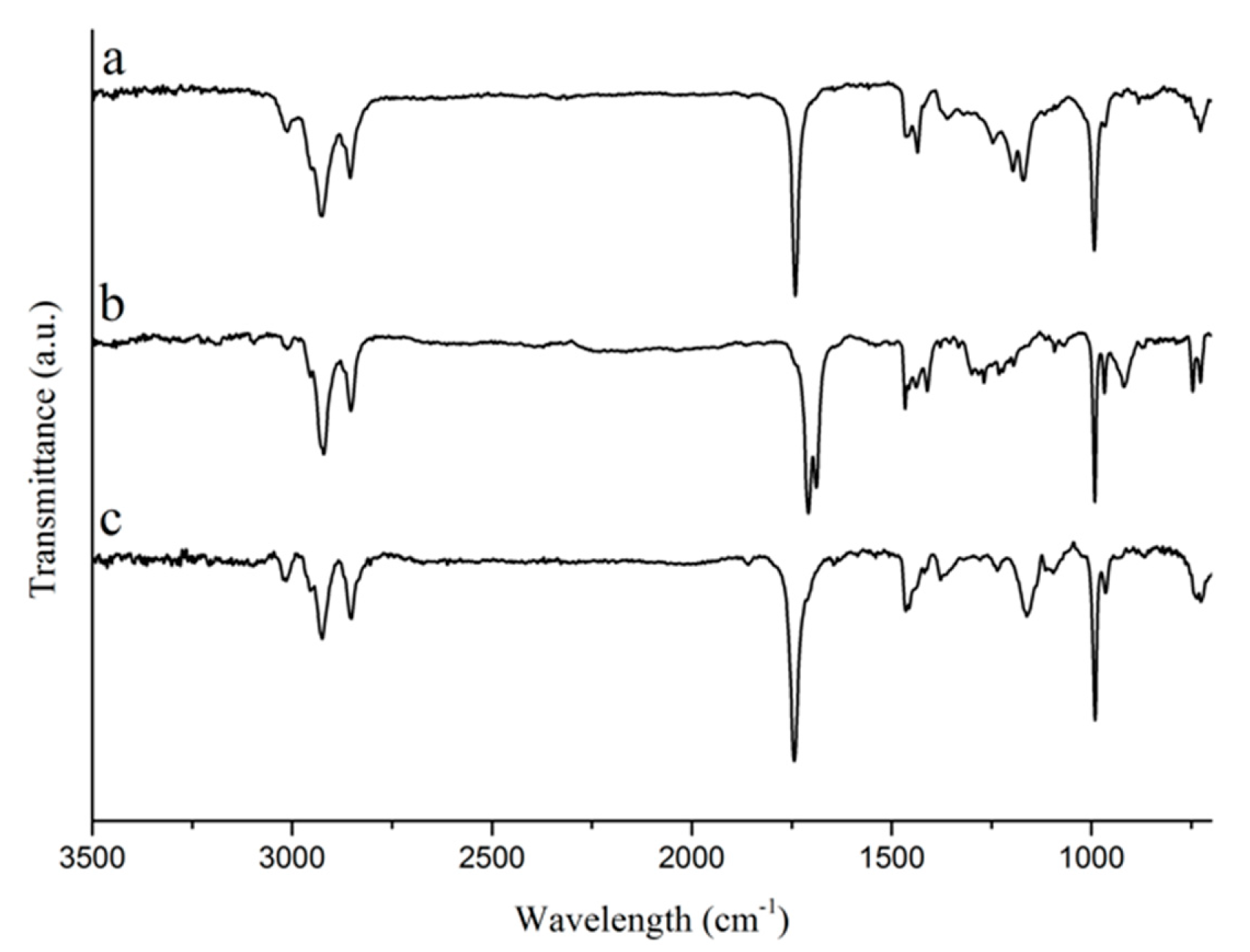
1H NMR, and Raman spectroscopies, and by GC-MS in order to check for purity and to confirm the structure of the bio-based starting materials used for the preparation of polymers and composites in this work. When comparing the FTIR spectra of tung oil, isolated α-eleostearic acid, and tung oil methyl esters (
Figure 1), it can be observed that the main structural features of the fatty acid chain in tung oil are unaltered after saponification and transesterification. Signals, such as the strong peak at approximately 1750 cm
−1, representing the carbonyl C=O stretch, and the strong peak at approximately 1000 cm
−1, which corresponds to the C–O stretch in the ester (tung oil and methyl esters) or carboxyl (α-eleostearic acid) functional groups, can be seen in all three spectra (
Figure 1). Likewise, the peak at 990–980 cm
−1, representing the C–H wagging vibrations of conjugated carbon–carbon double bonds are also observed in all three compounds (
Figure 1) [
20]. It’s interesting to note the absence of the typical signal for the O–H stretch in the spectrum of isolated α-eleostearic acid (
Figure 1). It is believed that, due to the basic reaction conditions employed during saponification, the final fatty acid is obtained in its salt form as a sodium carboxylate, which does not affect its subsequent polymerization and/or future interactions with the reinforcement.
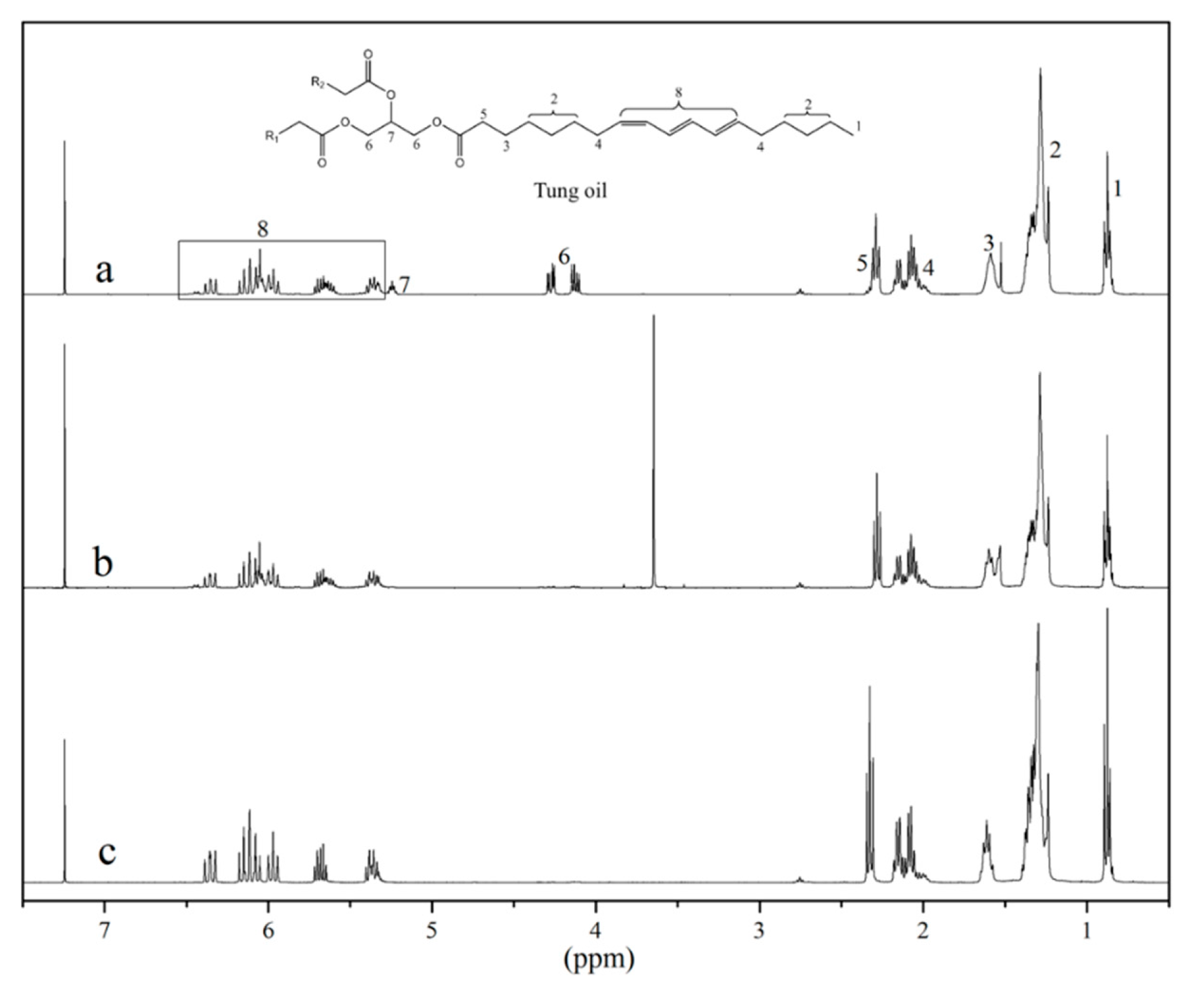
The
1H NMR spectra of the bio-based starting materials (
Figure 2) and the peak integration of their corresponding key signals provide further insight into reaction completion and product purity. The characteristic signals for the methylene (4.1–4.3 ppm, signal 6) and the methyne (~5.25 ppm, signal 7) hydrogens of the glycerol backbone can be clearly seen in the
1H NMR spectrum of tung oil (
Figure 2a). Their absence from the spectra of tung oil methyl esters and isolated α-eleostearic acid (
Figure 2b,c, respectively) confirms successful separation of any residual glycerol from the desired products. The intense singlet at 3.5 ppm in the spectrum of the tung oil methyl ester (
Figure 2b) corresponds to its newly formed methyl group. The absence of such peak from the spectrum of isolated α-eleostearic acid (
Figure 2c) suggests successful synthesis, purification, and drying of the desired fatty acid. As explained previously, the fatty acid is obtained in its salt form, therefore, the proton of the carboxyl group isn’t detected for the fatty acid.
It is important to note that all peaks related to the fatty acid carbon backbone of tung oil (signals 1–5 and 8,
Figure 2a) are virtually unchanged after either transesterification (
Figure 2b), or saponification (
Figure 2c), indicating that the carbon chain is intact, including the set of three naturally conjugated carbon–carbon double bonds. Indeed, the integration of vinylic hydrogens (5.3–6.4 ppm, signal 8) with respect to the terminal methyl group (~0.8 ppm, signal 1) in each spectrum confirms that the unsaturations were not affected by the reactions performed. In each case, the integration indicates a total of 6 vinylic hydrogens per fatty acid chain. Another indication that the unsaturations are not compromised by either transesterification or saponification of tung oil can be obtained by a quick analysis of the Raman spectra of the three samples (
Figure 3). Indeed, the intense peak at approximately 1630 cm
−1, associated to C=C vibrations, is clearly observed in all three spectra (
Figure 3).
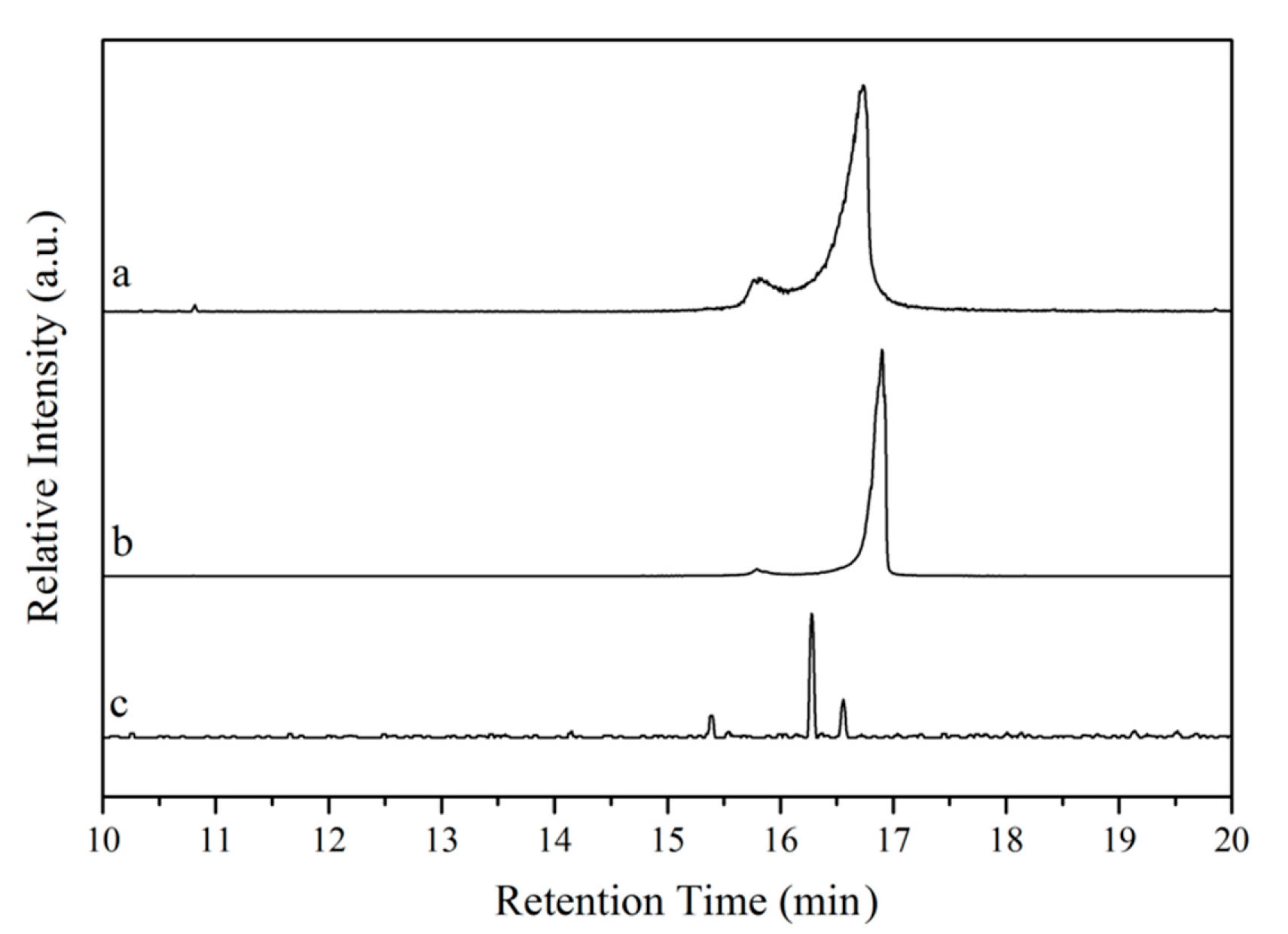
The purity of the bio-based starting materials was verified by GC-MS. The chromatograms obtained are presented in
Figure 4. In the chromatograms of tung oil and isolated α-eleostearic acid (
Figure 4a,b, respectively), two signals are observed at 15–17 min. The existence of these signals in the chromatogram of tung oil (
Figure 4a) is believed to relate to the various possible triglyceride structures of the oil based on its fatty acid composition (~83% of α-eleostearic acid, 9% of linoleic acid, 4% of oleic acid, and 4%–6% of palmitic acid) [
21], with a predominance of triglycerides bearing α-eleostearic acid accounting for the major signal. A similar scenario is reflected in the chromatogram of isolated α-eleostearic acid (
Figure 4b). The absence of other relevant signals suggests that, besides the mixture of fatty acids, no other by-products are detected. For the methyl esters (
Figure 4c), a better separation of the different methyl ester chains is achieved, most likely due to their independent structure when compared to triglycerides, and their lower polarity in comparison to free fatty acids. Three signals are indeed obtained in the 15–17 min. range with one major signal attributed to the methyl ester of α-eleostearic acid (
Figure 4c).
When comparing the retention times of the major signal in each one of the chromatograms (
Figure 4a–c), a clear and logical trend in retention times can be observed. On one hand, while tung oil’s major signal displays a retention time of 16 min and 45 s (
Figure 4a), the major signal for the fatty acid sample exhibits a slightly longer retention time (16 min and 54 s,
Figure 4b) that can be attributed to its higher polarity and, therefore, better affinity for the GC column. On the other hand, the methyl ester’s major signal exhibits a shorter retention time (16 min and 16 s,
Figure 4c) in comparison to that of tung oil’s major signal. Despite their similar polarities, independent methyl ester chains move more freely through the column than triglycerides, resulting in the observed shorter retention time. Based on the FT-IR, 1H NMR, Raman, and GC-MS results, it can be concluded that the preparation of methyl esters and the isolation of fatty acids from tung oil were successful.
3.2. Cure Evaluation
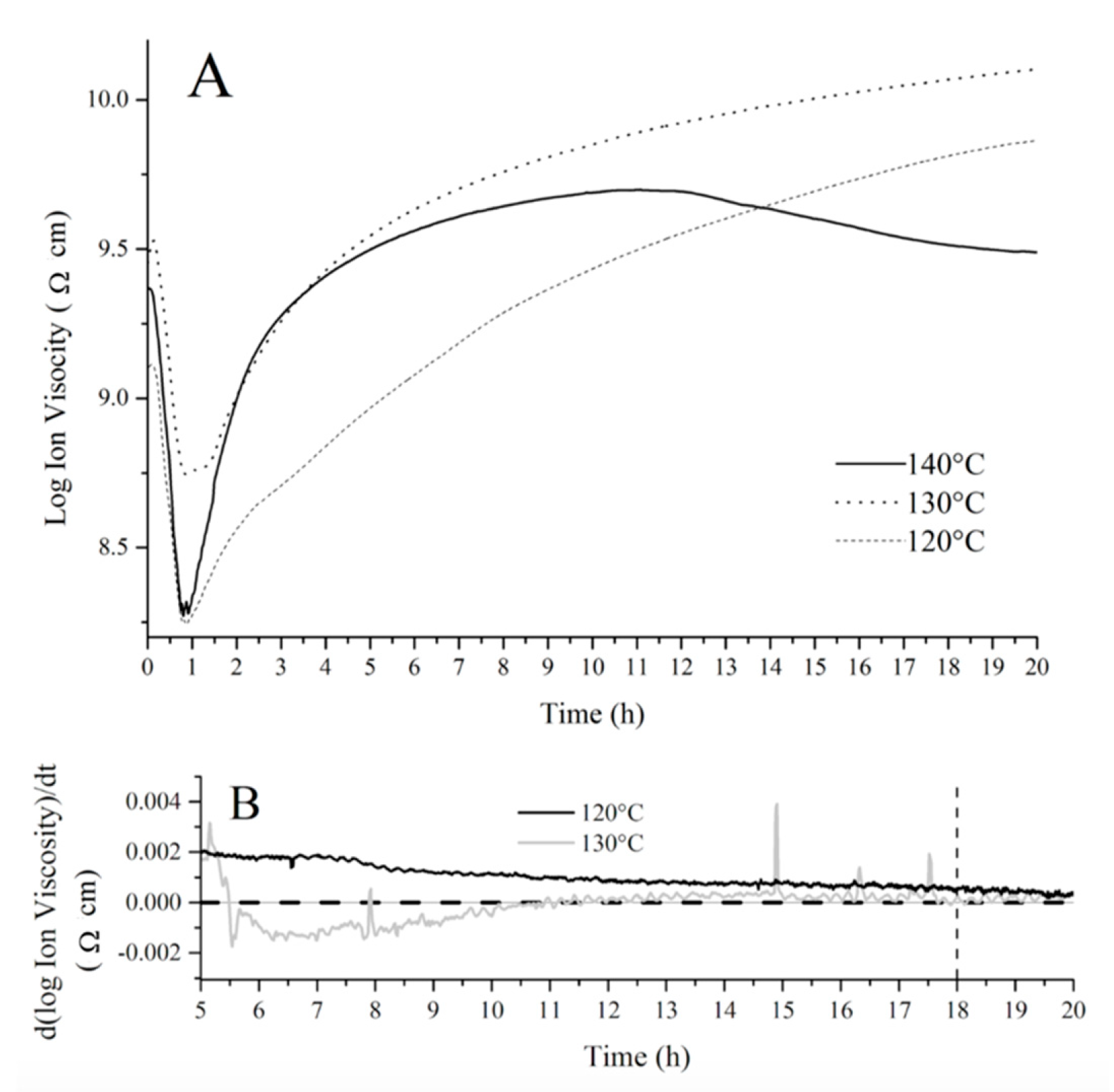
In order to establish an optimum cure schedule for the fatty acid resins prepared, the cure evolution of a crude resin containing 50 wt % of tung oil fatty acids, 30 wt % of BMA, and 20 wt % of DVB was monitored through dielectric analysis (DEA) at different temperatures as shown in
Figure 5A, where the log of ion viscosity (an artificial property derived from the permittivity of the resin) is plotted against time at each chosen temperature. This resin composition was selected based on previous studies with similar bio-based systems [
12]. The initial decrease in ion viscosity observed in the first 50 min of each run (
Figure 5A) corresponds to the initial increase in temperature before polymerization starts. Indeed, the increase in temperature prior to extensive polymerization leads to greater molecular mobility, which translates into a decrease in ion viscosity (
Figure 5A). At approximately 50 min, the polymerization process reaches a significant rate, resulting in an increase in ion viscosity due to a decrease in molecular mobility, as the crosslinked polymer network grows. Once the ion viscosity plateaus, it is accepted that no further changes occur in the material’s electrical and physical properties, indicating cure completion. When the fatty acid resin is cured at 140 °C, a significant amount of shrink cracks can be easily noticed in the sample, indicating an inappropriate cure temperature. Additionally, the DEA curve obtained at 140 °C (
Figure 5A) displayed a secondary decrease in ion viscosity at approximately 12 h, which could be attributed to exposure of the DEA sensor to air after formation of shrink cracks in the polymer, affecting the permittivity measured.
For the DEA curve of the fatty acid resin cured at 120°C (
Figure 5A), the ion viscosity continuously increases throughout the duration of the experiment (20 h). In order to obtain a clearer comparison between the cure at 120 and 130 °C, the derivative of ion viscosity with respect to time was plotted against cure time in
Figure 5B. Close inspection of
Figure 5B reveals that the derivative approaches zero (indicating the change in electrical and physical properties is zero and the cure is completed) at approximately 18 h (dashed line added as a guide to the eye,
Figure 5B) when the resin is heated at 130 °C. When the resin is heated at 120 °C, the derivative of the DEA curve never reaches zero for the duration of the experiment, indicating an incomplete cure after 20 h (
Figure 5B).
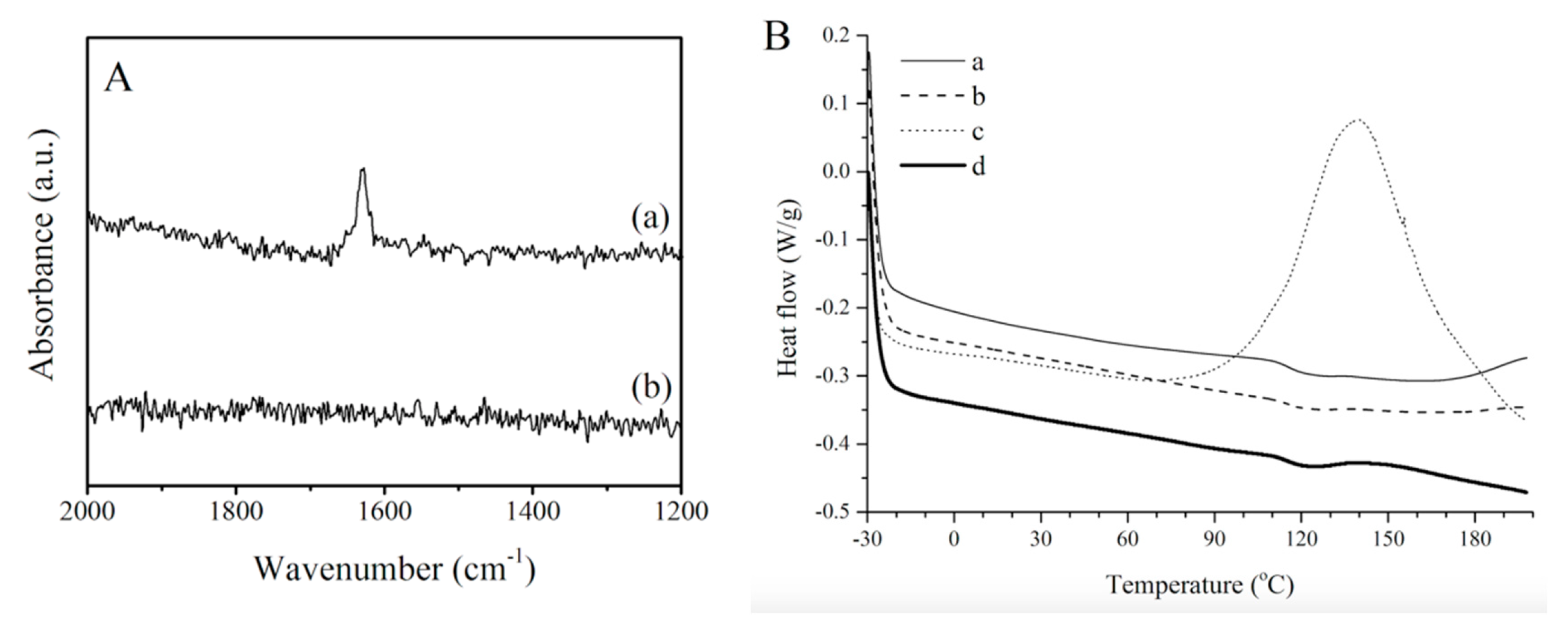
A comparison of the Raman spectra of a crude fatty acid resin and a cured sample (
Figure 6A) provides further evidence that cure completion was achieved for the fatty acid resin after heating at 130 °C for 18 h. Indeed, the presence of a signal at approximately 1630 cm
−1 in the spectrum of the crude resin (
Figure 6A) is indicative of unreacted carbon–carbon double bonds from the various resin components. That signal is absent in the spectrum of the cured resin (
Figure 6A), indicating that the polymer was fully cured as previously supported by the DEA results.
In order to validate the DEA and Raman results and to confirm cure completion, a tung oil fatty acid resin cured at 130 °C for 18 h was subjected to a DSC experiment (
Figure 6B). The same cure schedule was applied to similar resins prepared from tung oil and from tung oil methyl esters. The DSC curves of all three samples are presented in
Figure 6B(a–c). The DSC results indicate that the cure schedule used is adequate for tung oil and fatty acid resins since no exothermic peaks are observed for these two samples (
Figure 6B). However, the methyl ester resin is not fully cured when subjected to that same cure schedule, as indicated by a large exothermic peak centered at approximately 140 °C (
Figure 6B). In order to ensure cure completion, the tung oil methyl ester resin was subjected to a longer (24 h) cure schedule at 130 °C. The increase in cure time by an additional 6 h resulted in a fully cured sample as evidenced by the absence of any exothermic peaks in the corresponding DSC curve (
Figure 6B). In view of these results, the cure schedule at 130 °C for 18 h is considered to be optimal for the fatty acid and tung oil resins, whereas the tung oil methyl ester resin requires heating at 130 °C for 24 h. These cure schedules will, therefore, be employed with their corresponding resins in the remainder of this work.
3.5. Thermo-Mechanical Properties
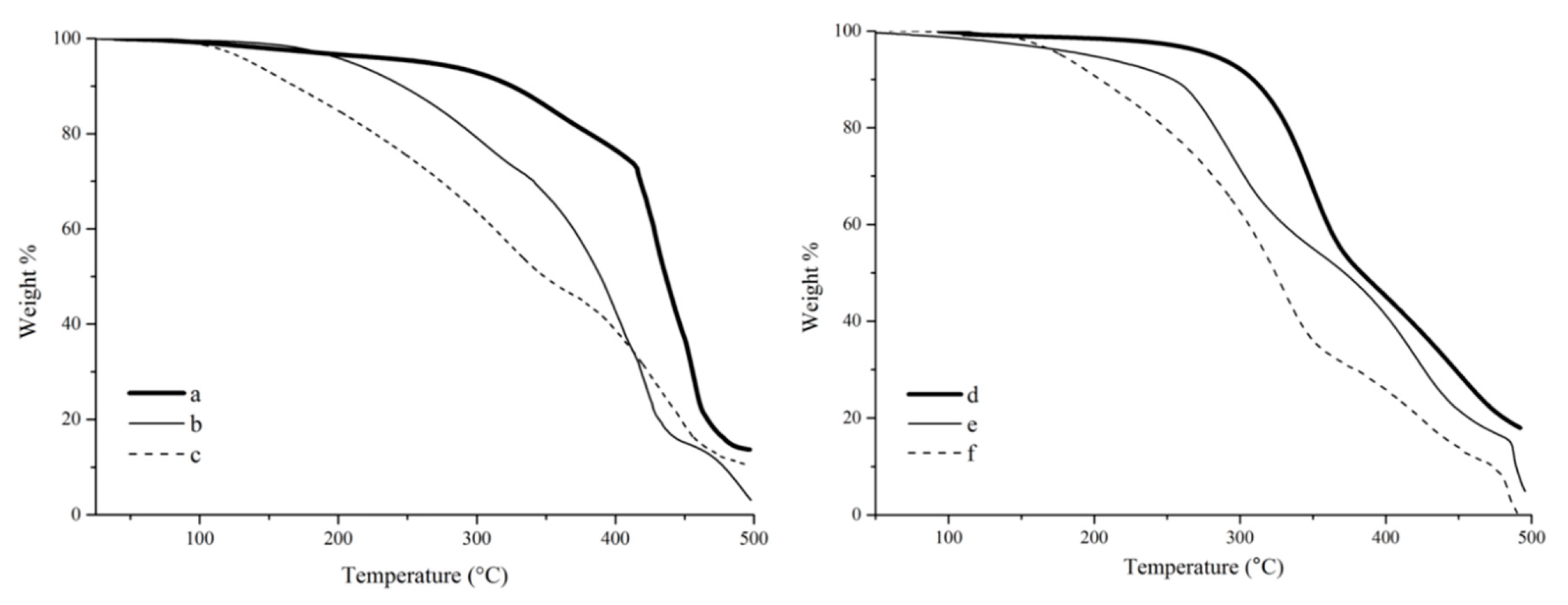
The thermogravimetric analysis (TGA) of tung oil, tung oil fatty acid, and tung oil methyl ester resins (
Figure 8a–c) shows a clear trend for their thermal stability. It is indeed obvious that the onset degradation temperature (
Tonset) for the tung oil methyl ester resin (~100 °C) is much lower than that of samples prepared with tung oil fatty acids (~180 °C) or tung oil triglycerides (~300 °C). This trend can be explained as a consequence of the chemical structure of the major resin components. For example, the markedly higher
Tonset observed for the tung oil resin is the result of a highly crosslinked structure, due to the fact that the tung oil triglyceride monomer consists of three fatty acid esters connected to a glycerol backbone. In both fatty acids and methyl esters, the polymerizable fatty chains are independent from each other, resulting in a much less crosslinked polymer network. The difference observed in the
Tonset of fatty acid and methyl ester polymers can be associated to their polarity. The polar fatty acid resin exhibits stronger hydrogen bonding interactions between polymer chains, resulting in a more compact arrangement of the overall polymer structure and, therefore, constituting a more thermally stable material in comparison to its methyl ester analogue.
It is also apparent, from the TGA results (
Figure 8a–c), that the bulk of the thermal degradation process occurs from
Tonset to approximately 480 °C for the resins prepared with tung oil and methyl esters (
Figure 8a,c), while the resin prepared with fatty acids clearly exhibits an additional secondary step starting at approximately 450 °C (
Figure 8b). The major degradation process is believed to correspond to the breakdown of the cross-link network of the thermoset. The additional degradation step observed for the resin prepared with fatty acids could be associated to the final oxidation of residues and char formation, but its definitive origin can’t be reliably inferred from the TGA results alone, lying outside the scope of the work described in this manuscript.
A very similar trend in thermal stability can be observed for the composites (
Figure 8d–f) compared to their corresponding resins (
Figure 8a–c). All composites exhibit a very clear thermal degradation profile, with two distinct stages that can be associated with the degradation of the resin and the degradation of the reinforcement, although it is unavoidable that the degradation of these two components (resin and reinforcement) occur with some overlap. When correlating the results observed with fiber-matrix interactions, one would expect that a stronger binding between resin and reinforcement would result in a better fiber protection, and ultimately in a delayed degradation, especially towards the end of the thermal degradation process. Indeed, when comparing the temperature at 80 wt % degradation (
T80) for resins and their corresponding composites, respectively, an increase from ~430 to ~450 °C can be seen for samples prepared with fatty acids, whereas a decrease from ~445 to ~425 °C is observed for samples prepared with methyl esters. This behavior is reflective of the better fiber-matrix interactions obtained with fatty acids in comparison to methyl esters. In the case of tung oil triglyceride, the increase in
T80 from ~455 to ~475 °C, for resin and composite, respectively, can be associated to the highly crosslinked structure of the oil offering thermal protection to the fiber despite the expected poor fiber-matrix interaction.
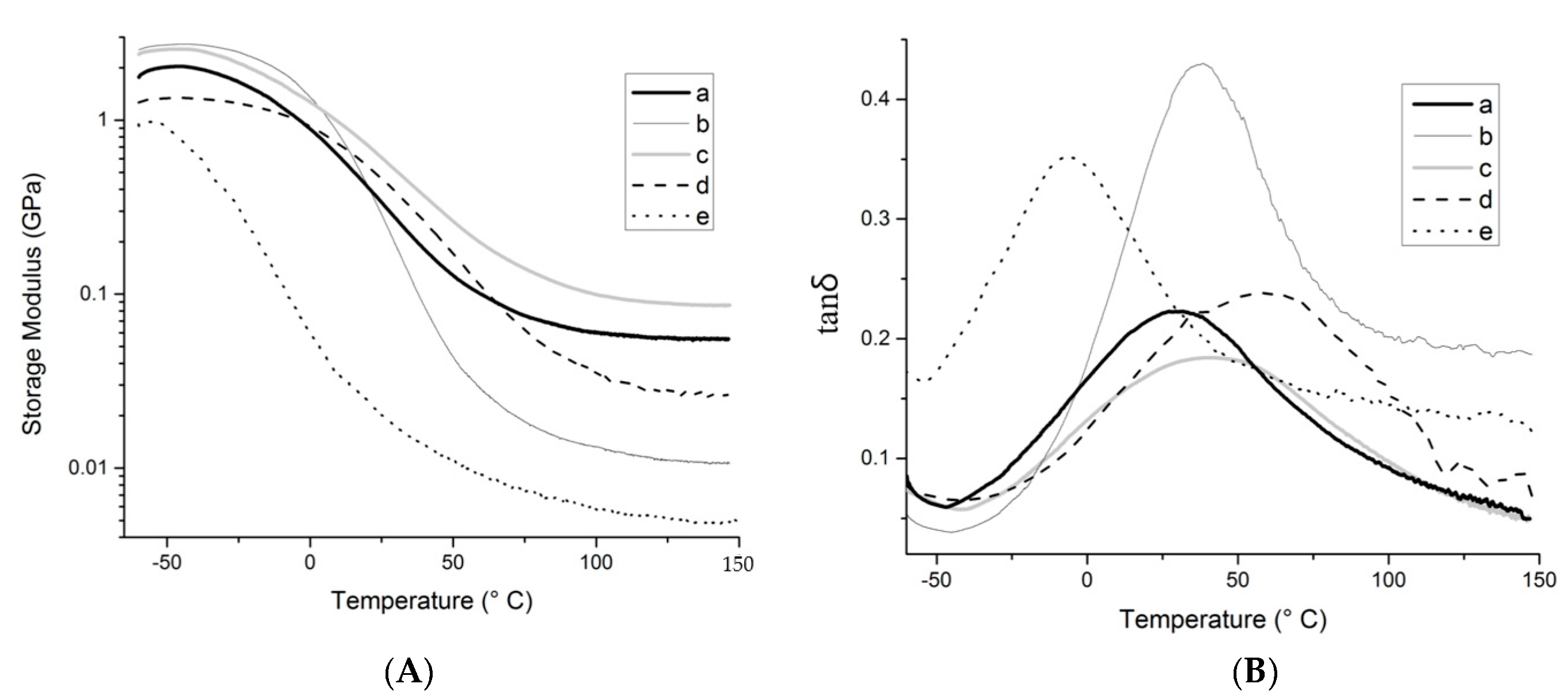
DMA was used to assess thermo-mechanical properties of the resins and composites studied. These results are summarized in
Figure 9 and
Table 2. At temperatures below 20 °C, the resin prepared with fatty acids displays a higher storage modulus than the resin prepared with tung oil triglycerides (
Figure 9a,b), exhibiting a behavior that is reflective of the expected, more prevalent hydrogen bonding. Above 20 °C, it is believed that chain mobility overcomes hydrogen bonding, and that the more crosslinked structure of the resin prepared with tung oil accounts for the higher storage modulus observed in comparison to the resin prepared with fatty acids. At the rubbery plateau (
Tg + 50 °C), above all phase transitions of the materials, the storage modulus for the resin prepared with tung oil is approximately 4.7 times higher than that of the resin prepared with fatty acids (68.9 MPa vs. 14.6 MPa, respectively,
Table 2). Indeed, that same trend is also clear in the calculated crosslink density, with 16.2 × 10
−4 mol/cm
3 for the fatty acid resin and 77.7 × 10
−4 mol/cm
3 for the tung oil resin (
Table 2). Cross-link densities were calculated based on the rubbery elasticity theory, according to Equation (1) [
24,
25]:
where
E’ is the storage modulus at
Tg + 50 °C, R is the gas constant, and
T is the absolute temperature (K). Crosslink density was not calculated for composites (
Table 2) because the numbers would be artificially inflated by the natural increase in
E’ resulting from the addition of a reinforcement, therefore masking the true crosslink density value. It is also worth noting that DMA specimens could not be produced from the resin prepared with methyl esters due to its exceeding fragility.
In the presence of the cellulose reinforcement, the composite prepared with methyl esters exhibits significant lower storage modulus than the composite made from fatty acids (
Figure 9d,e), indicating that a stronger reinforcing effect is obtained for the more polar resin, as expected due to its increased affinity with the reinforcement. For the composite prepared with tung oil triglyceride (
Figure 9c), the storage modulus is consistently higher than all other composites, revealing that, contrary to expected, the crosslink density of the resin is a greater contributor towards final mechanical properties than fiber-matrix interactions. It is interesting to note that at lower temperatures, the fatty acid resin (
Figure 9b) exhibits higher storage modulus than the other systems, surpassing the tung oil resin (
Figure 9a) below 24 °C as discussed previously, the fatty acid composite (
Figure 9d) below 23 °C, and the tung oil composite (
Figure 9c) below 3 °C. This trend indicates that below the
Tg of the polar resin, hydrogen bonding between the resin and reinforcement is limited, therefore, the ligno-cellulosic filler particles represent weak points within the sample, contributing to the lower mechanical performance observed at lower temperatures in comparison to the pure resin.
For all samples investigated, the tanδ curves (
Figure 9) exhibit a single major peak, representing the glass transition of the system evaluated. The presence of a single transition is indicative of a homogeneous polymer with no evident phase separation. It is worth noting that the use of vegetable oils with less carbon–carbon double bonds typically results in polymers with more than one
Tg [
8,
9], and this behavior has been attributed to a micro-phase separation within the polymer network due to a significant difference in reactivity of the co-monomers used. The presence of a single tanδ peak for the composite samples (
Figure 9c–e) also suggests that cellulose fibers have been evenly distributed throughout the polymer, providing universal thermo-mechanical properties for the entire sample. Another indicator of resin heterogeneity is the breadth of the tanδ peak. Indeed, the narrower the peak, the more homogeneous the system. It is interesting to notice how the addition of reinforcement changes the breadth of the tanδ peak for the samples prepared with fatty acids (
Figure 9b,d). The same effect is not observed for the other resins, therefore revealing greater interaction between resin and reinforcement on the composite prepared with fatty acids. Indeed, as observed previously in similar systems, a greater interaction of one of the resin components with the reinforcement contributes to composite heterogeneity at the molecular level [
8].
The greater interaction between fatty acids and cellulose is also reflected in the
Tg values of the samples (
Table 2). Indeed, there is only a small change in the
Tg of samples prepared with tung oil upon addition of cellulose, revealing that the presence of cellulose has little effect on the polymerization of the resin and its intrinsic properties. For the fatty acid resin, a significant increase of the
Tg from 39 to 56 °C (
Table 2) is observed upon the addition of cellulose. This increase in
Tg suggests a stronger affinity between resin and reinforcement, resulting in more restricted chain mobility. Among all samples, the composite prepared with methyl esters exhibits the lowest
Tg (−6 °C,
Table 2), indicating the combination of a weak resin with a poor adhesion between the resin and reinforcement.
Finally, the maximum value in the tanδ curve (tanδ
max) can be associated to the damping properties of the material. It is noteworthy that all composites exhibit tanδ
max values lower than their corresponding unreinforced resins (
Figure 9), with a more pronounced decrease (from approximately 0.44 to approximately 0.23,
Figure 9b,d) for the fatty acid system. This greater decrease is most likely the result of limited ability of the composite in dispersing shock/sound waves into molecular/chain movements, due to the greater interaction between resin and reinforcement, effectively interlocking the polymer chains through hydrogen bonding. When comparing the different composites, it is clear that the sample prepared with methyl esters exhibits the highest tanδ
max, which can be associated to a better ability to dampen vibration through dissipation of the energy into polymer chain movement.












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}