
FT-IR Examination of the Development of Secondary Cell Wall in Cotton Fibers †


Abstract
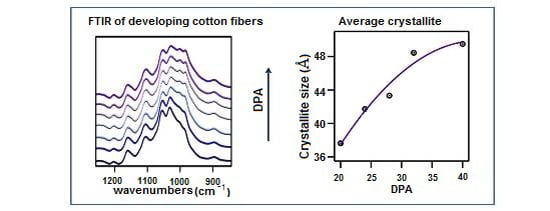
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
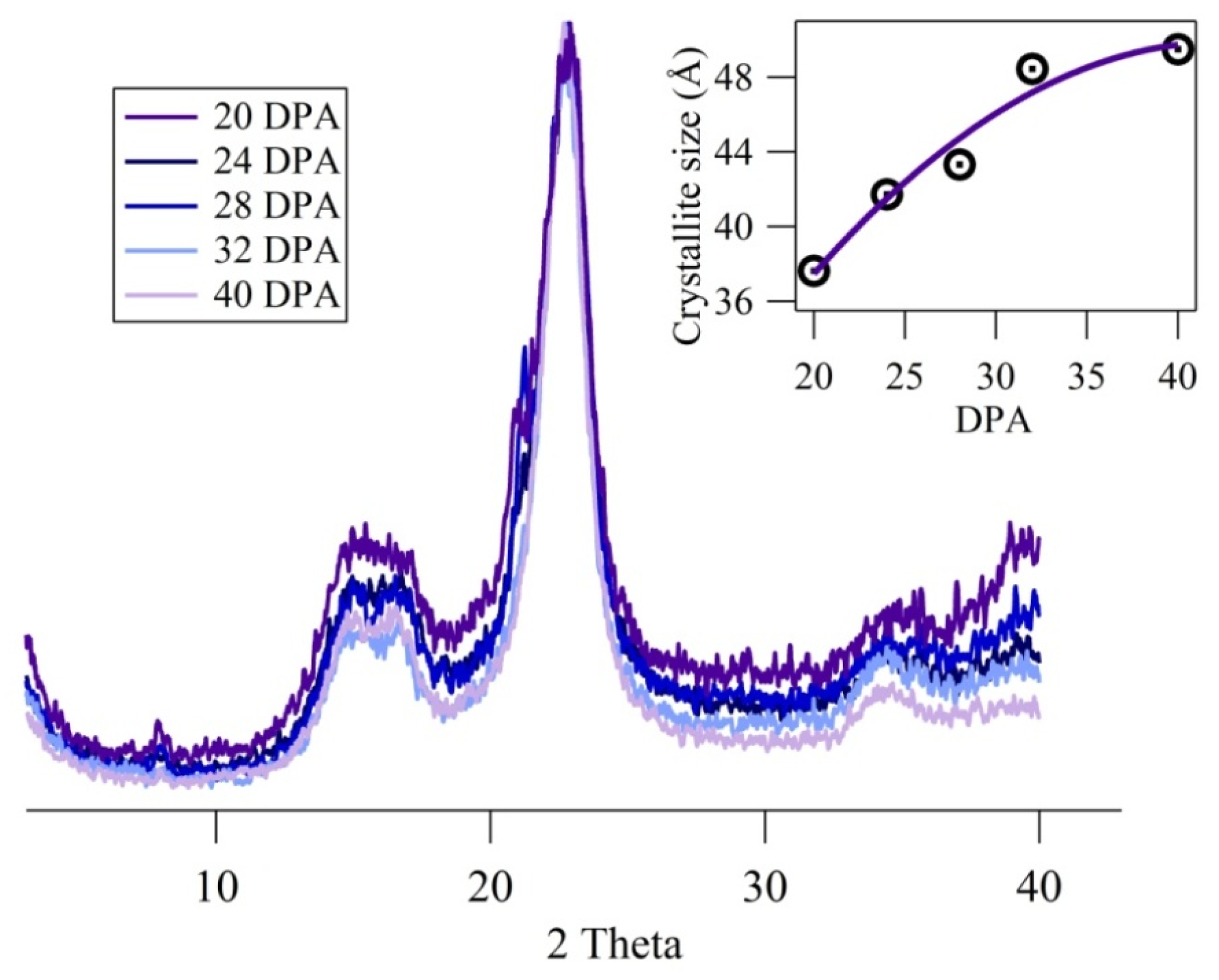{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Experimental Section
2.1. Plant Material and Field Collection
2.2. FT-IR Analysis
2.3. Cotton Fiber Deuteration
2.4. X-Ray Powder Diffraction
3. Results and Discussion
3.1. ATR FT-IR Spectroscopy of the O–H Stretching Region


3.2. O–H Bending Region


3.3. Fingerprint Region; C–H and O–H Bending

3.4. Fingerprint Region; C–O Vibrations

4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wakelyn, P.J.; Bertoniere, N.R.; French, A.D.; Thibodeaux, D.P.; Triplett, B.A.; Rouselle, M.A.; Goynes, W.R.; Edwards, J.V.; Hunter, L.; McAllister, D.D.; et al. Cotton Fiber Chemistry and Technology; CRC Press: New York, NY, USA, 2007. [Google Scholar]
- Tripp, V.W.; Moore, A.T.; Rollins, M.L. Some observations on the constitution of the primary wall of the cotton fiber. Text. Res. J. 1951, 21, 886–894. [Google Scholar] [CrossRef]
- Tripp, V.W.; Rollins, M.L. Morphology and chemical composition of certain components of cotton fiber cell wall. Anal. Chem. 1952, 24, 1721–1728. [Google Scholar] [CrossRef]
- Kim, H.J.; Triplett, B.A. Cotton fiber growth in planta and in vitro. Models for plant cell elongation and cell wall biogenesis. Plant Physiol. 2001, 127, 1361–1366. [Google Scholar] [CrossRef] [PubMed]
- Seagull, R.W.; Oliveri, V.; Murphy, K.; Binder, A.; Kothari, S. Cotton fiber growth and development 2. Changes in cell diameter and wall birefringence. J. Cotton Sci. 2000, 4, 97–104. [Google Scholar]
- Cosgrove, D.J. Growth of the plant cell wall. Nat. Rev. Mol. Cell Biol. 2005, 6, 850–861. [Google Scholar] [CrossRef] [PubMed]
- Gamble, G.R. Variation in surface chemical constituents of cotton (Gossypium hirsutum) fiber as a function of maturity. J. Agric. Food Chem. 2003, 51, 7995–7998. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, M.K.; Turley, R.B.; Kim, H.J.; Li, P.; Thyssen, G.; Tang, Y.; Delhom, C.D.; Naoumkina, M.; Fang, D.D. Transcript profiling by microarray and marker analysis of the short cotton (Gossypium hirsutum L.) fiber mutant Ligon lintless-1 (Li1). BMC Genomics 2013, 14, 403. [Google Scholar] [CrossRef] [PubMed]
- Abidi, N.; Hequet, E.; Cabrales, L.; Gannaway, J.; Wilkins, T.; Wells, L.W. Evaluating cell wall structure and composition of developing cotton fibers using Fourier transform infrared spectroscopy and thermogravimetric analysis. J. Appl. Polym. Sci. 2008, 107, 476–486. [Google Scholar] [CrossRef]
- Abidi, N.; Cabrales, L.; Hequet, E. Fourier transform infrared spectroscopic approach to the study of the secondary cell wall development in cotton fiber. Cellulose 2010, 17, 309–320. [Google Scholar] [CrossRef]
- Liu, Y.; Thibodeaux, D.; Gamble, G. Development of Fourier transform infrared spectroscopy in direct, non-destructive, and rapid determination of cotton fiber maturity. Text. Res. J. 2011, 81, 1559–1567. [Google Scholar] [CrossRef]
- Abidi, N.; Hequet, E.; Cabrales, L. Applications of Fourier Transform Infrared Spectroscopy to study cotton fibers. In Fourier Transforms—New Analytical Approaches and FTIR Strategies; Nikolic, G., Ed.; InTech: Cairo, Egypt, 2011; pp. 89–114. [Google Scholar]
- Nelson, M.L.; Mares, T. Accessibility and lateral order distribution of cellulose in the developing cotton fiber. Text. Res. J. 1965, 35, 592–603. [Google Scholar] [CrossRef]
- Lokhande, H.T.; Daruwalla, E.H.; Padhye, M.R. Deuteration of cotton fibers. II. A novel method of deuteration-infrared study of cellulose in fiber form. J. Appl. Polym. Sci. 1977, 21, 2943–2952. [Google Scholar] [CrossRef]
- Hinchliffe, D.J.; Yeater, K.M.; Kim, H.J.; Woodward, A.W.; Chen, Z.J.; Meredith, W.R.; Triplett, B.A. Near-isogenic cotton germplasm lines that differ in fiber-bundle strength have temporal differences in fiber gene expression patterns as revealed by comparative high-throughput profiling. Theor. Appl. Genet. 2010, 120, 1347–1366. [Google Scholar] [CrossRef] [PubMed]
- Maréchal, Y.; Chanzy, H. The hydrogen bond network in Iβ cellulose as observed by infrared spectrometry. J. Mol. Struct. 2000, 523, 183–196. [Google Scholar] [CrossRef]
- Meredith, W.R. Minimum number of genes controlling cotton fiber strength in a backcross population. Crop Sci. 2005, 45, 1114–1119. [Google Scholar] [CrossRef]
- Meredith, W.R. Registration of MD 52ne high fiber quality cotton germplasm and recurrent parent MD 90ne. Crop Sci. 2005, 45, 807–808. [Google Scholar] [CrossRef]
- Scherrer, P. Bestimmung der Grösse und der inneren Struktur von Kolloidteilchen mittels Röntgenstrahlen. Nachr. Ges. Wiss. Göttingen 1918, 26, 98–100. (In German) [Google Scholar]
- Jeffries, R. The amorphous fraction of cellulose and its relation to moisture sorption. J. Appl. Polym. Sci. 1964, 8, 1213–1220. [Google Scholar] [CrossRef]
- Rousselle, M.A.; Nelson, M.L. Accessibility of cotton cellulose by deuterium exchange. Text. Res. J. 1971, 41, 599–604. [Google Scholar] [CrossRef]
- Fan, Q.G.; Lewis, D.M.; Tapley, K.N. Characterization of cellulose aldehyde using fourier transform infrared spectroscopy. J. Appl. Polym. Sci. 2001, 82, 1195–1202. [Google Scholar] [CrossRef]
- Tsuboi, M. Infrared spectrum and crystal structure of cellulose. J. Polym. Sci. 1957, 25, 159–171. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cintrón, M.S.; Hinchliffe, D.J. FT-IR Examination of the Development of Secondary Cell Wall in Cotton Fibers . Fibers 2015, 3, 30-40. https://doi.org/10.3390/fib3010030
Cintrón MS, Hinchliffe DJ. FT-IR Examination of the Development of Secondary Cell Wall in Cotton Fibers . Fibers. 2015; 3(1):30-40. https://doi.org/10.3390/fib3010030
Chicago/Turabian StyleCintrón, Michael Santiago, and Doug J. Hinchliffe. 2015. "FT-IR Examination of the Development of Secondary Cell Wall in Cotton Fibers " Fibers 3, no. 1: 30-40. https://doi.org/10.3390/fib3010030
APA StyleCintrón, M. S., & Hinchliffe, D. J. (2015). FT-IR Examination of the Development of Secondary Cell Wall in Cotton Fibers . Fibers, 3(1), 30-40. https://doi.org/10.3390/fib3010030