Chitosan as a Coupling Agent for Phosphate Glass Fibre/Polycaprolactone Composites

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Phosphate Glass and Fibre Preparation
2.2. Coating Application
2.3. Single-Fibre Composite (SFC) Preparation
2.4. Single Fibre Tensile Test (SFTT)
2.5. Single-Fibre Fragmentation Test (SFFT)
2.6. Statistical Analysis
2.7. SEM
2.8. Thermogravimetric Analysis (TGA)
2.9. FTIR and Microscope FTIR
2.10. Raman
2.11. XPS
3. Results
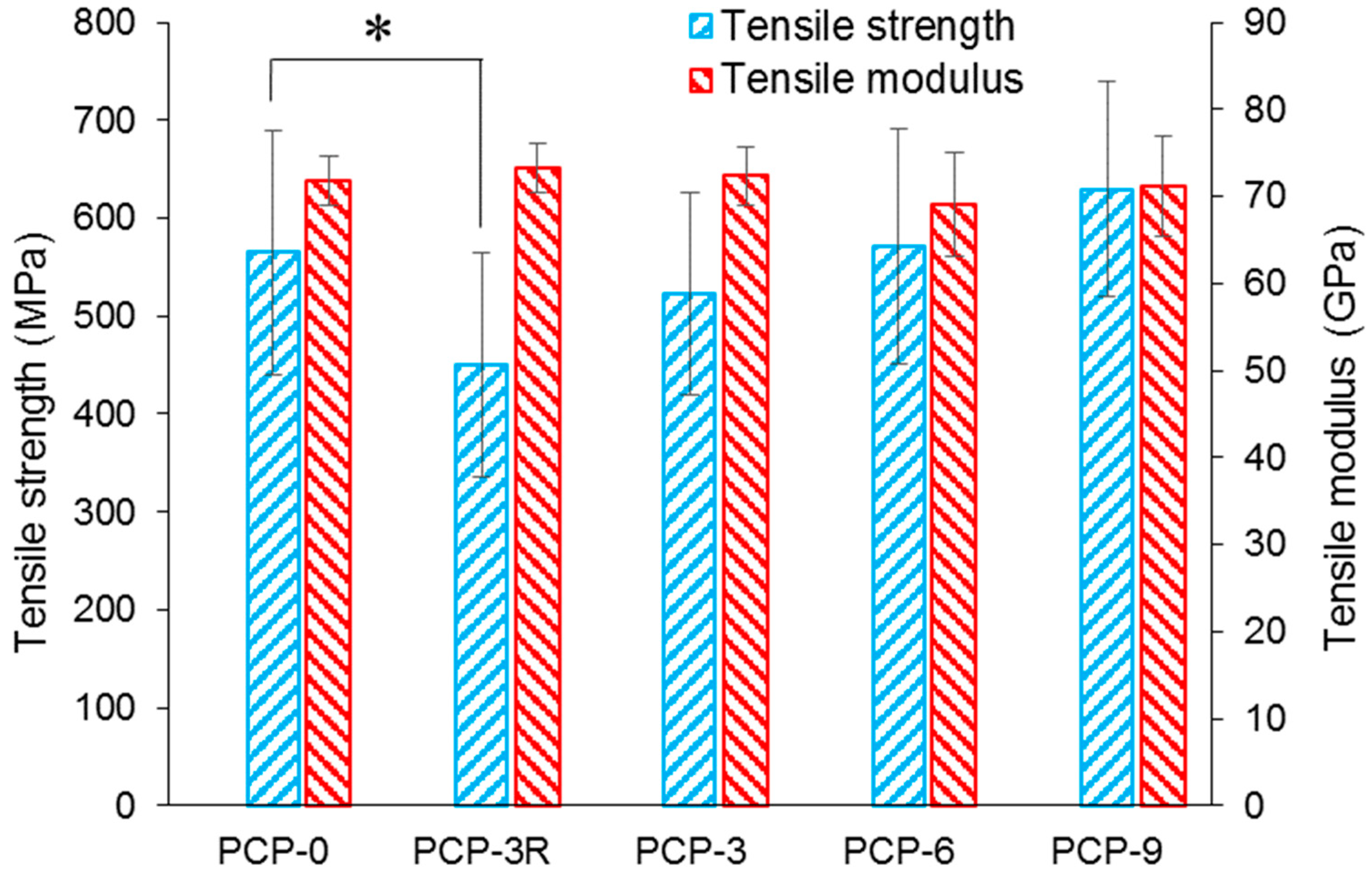
3.1. Mechanical Properties
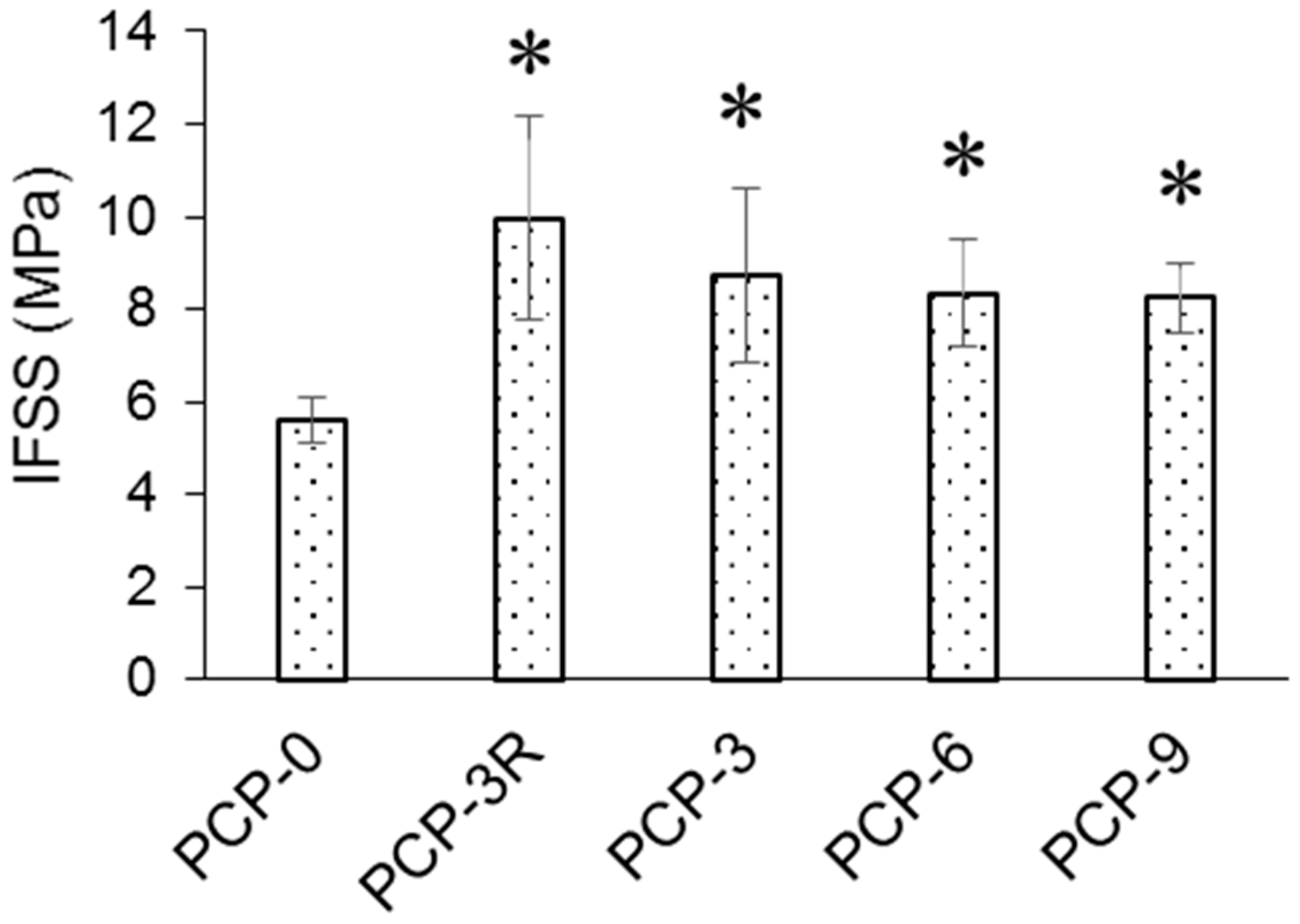
3.2. IFSS Analysis
3.3. SEM Analysis
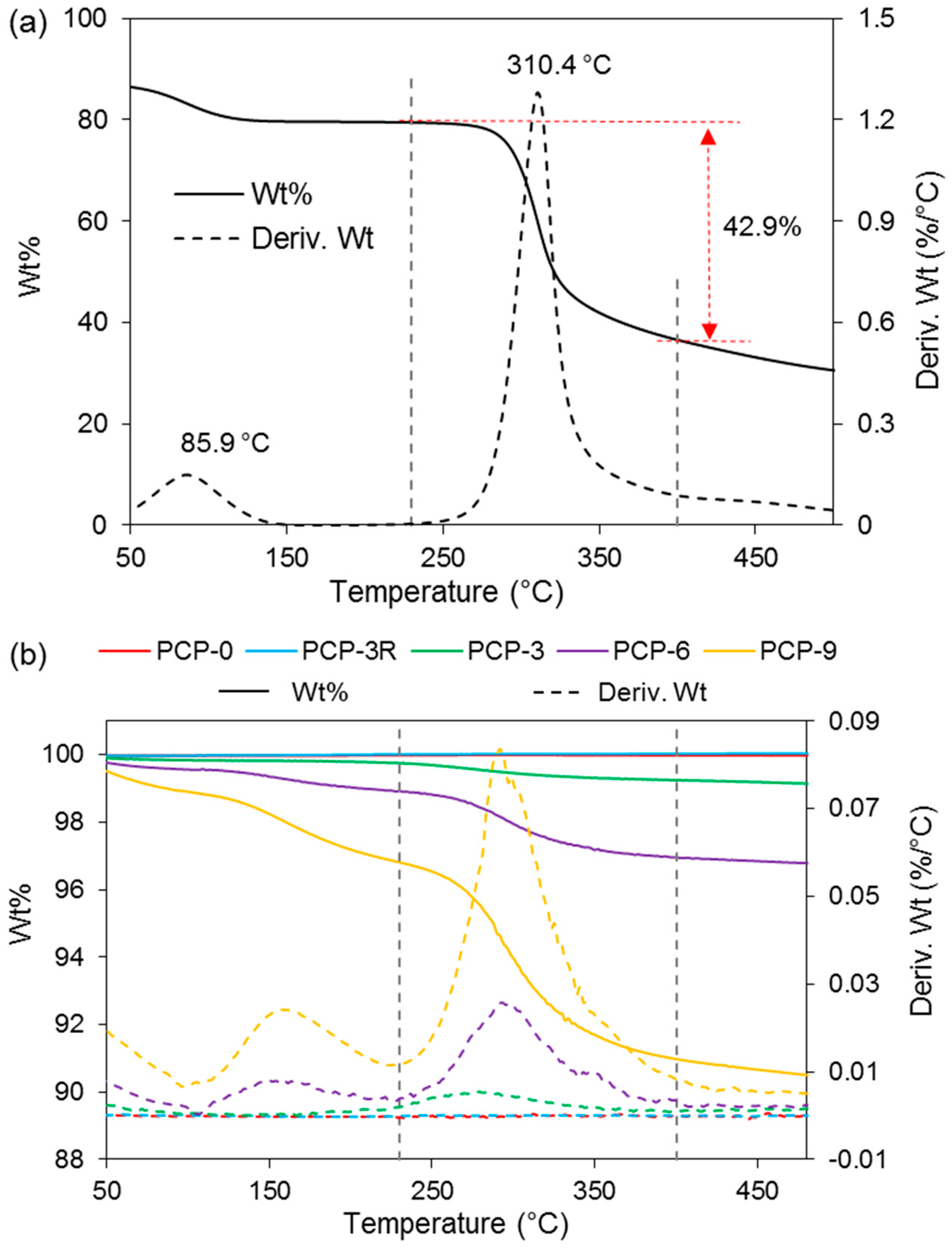
3.4. TGA
3.5. FTIR Analysis
3.5.1. Microscope-FTIR
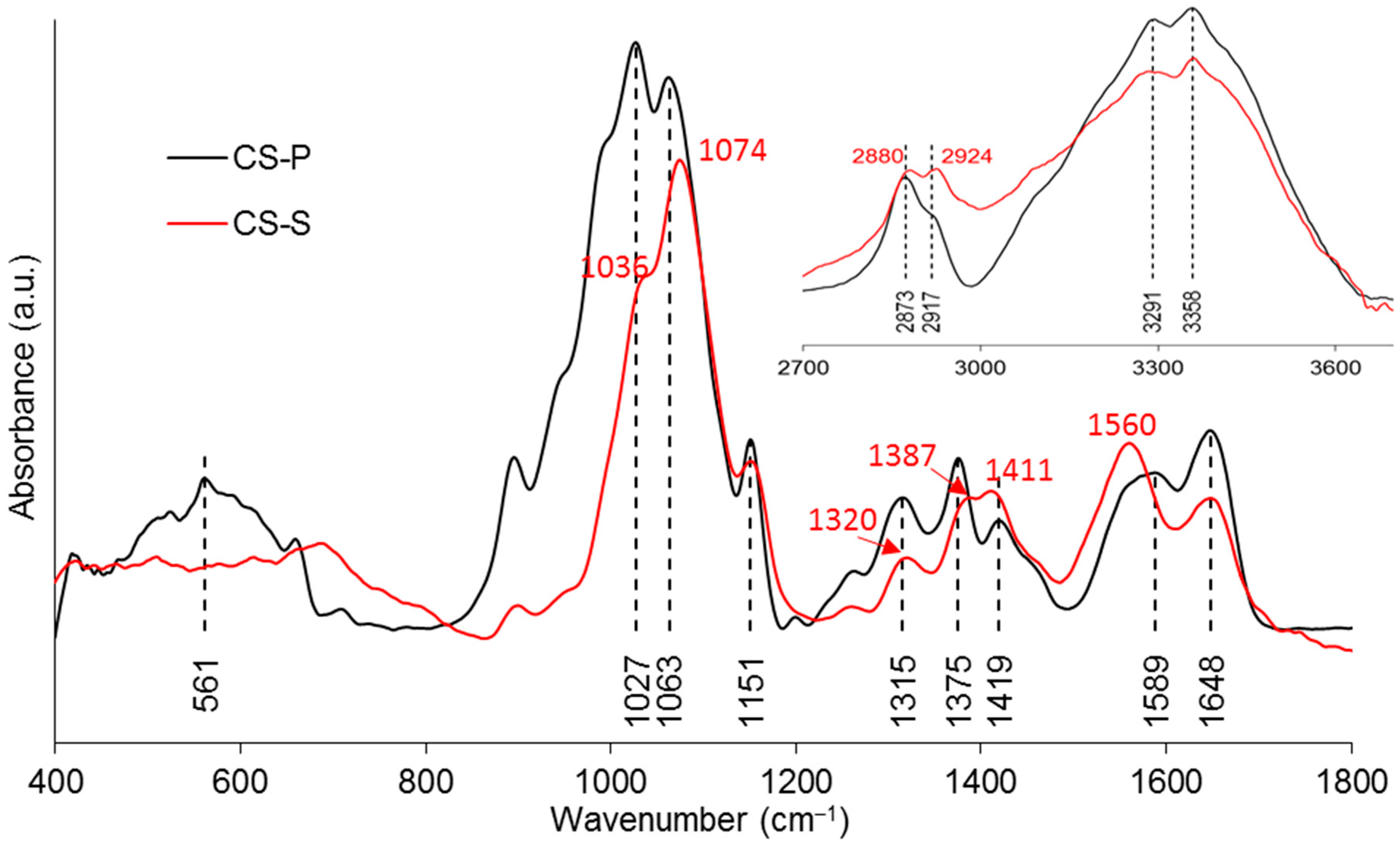
3.5.2. ATR-FTIR
3.6. Raman Analysis
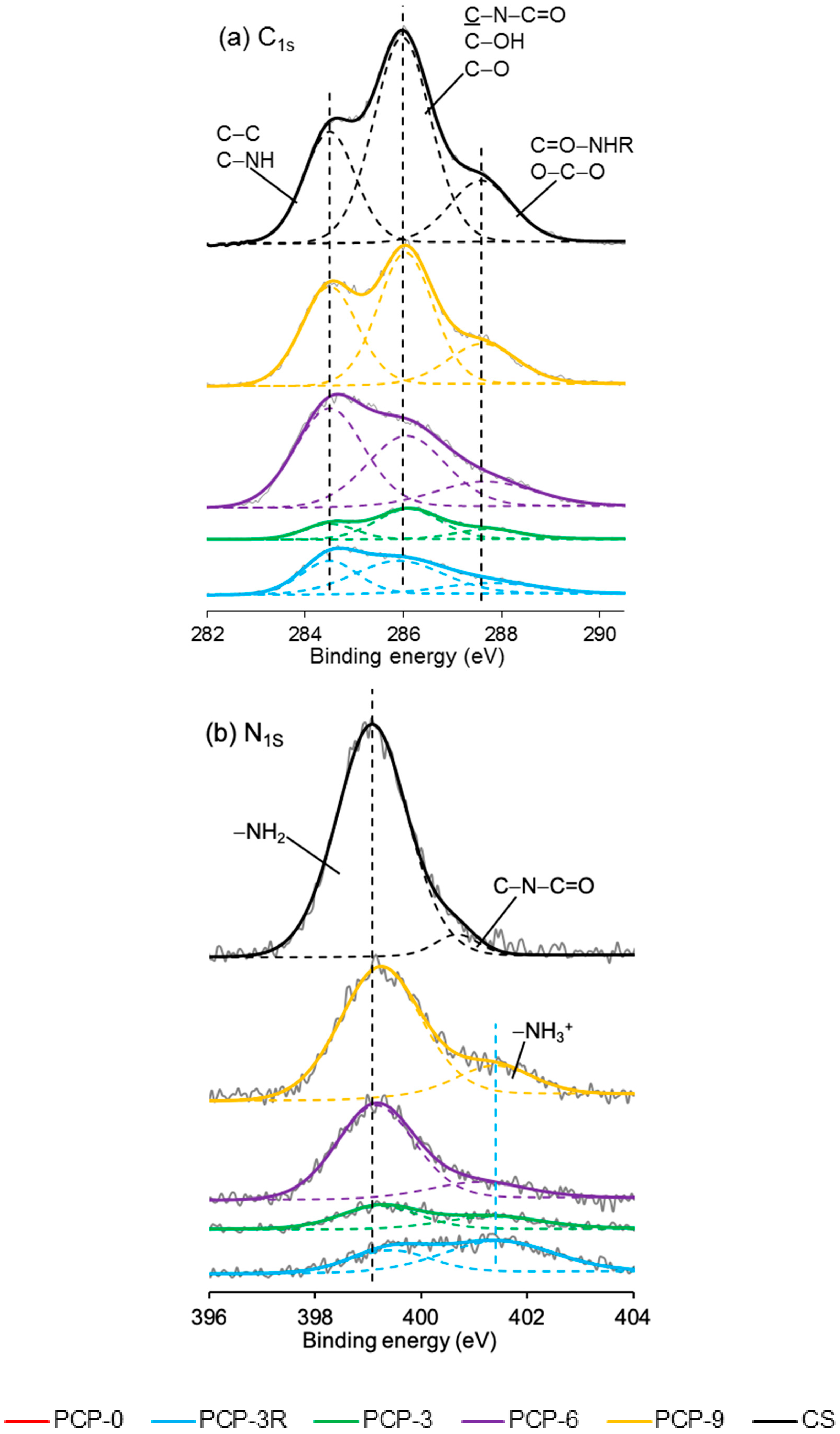
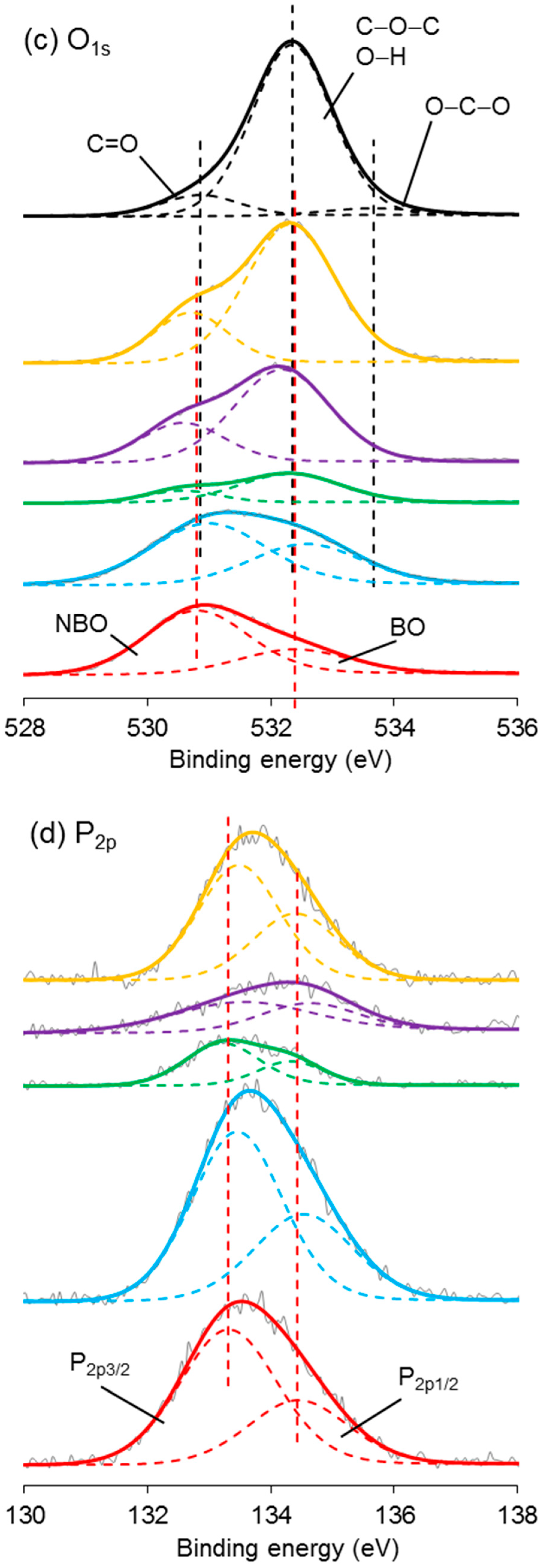
3.7. XPS Analysis
4. Discussion
4.1. Mechanical Properties
4.2. Interfacial Properties
4.3. SEM Analysis
4.4. TGA
4.5. FTIR and Raman Analyses
4.6. XPS Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ahmed, I.; Jones, I.A.; Parsons, A.J.; Bernard, J.; Farmer, J.; Scotchford, C.A.; Walker, G.S.; Rudd, C.D. Composites for bone repair: Phosphate glass fibre reinforced PLA with varying fibre architecture. J. Mater. Sci. Mater. Med. 2011, 22, 1825–1834. [Google Scholar] [CrossRef]
- Parsons, A.J.; Ahmed, I.; Haque, P.; Fitzpatrick, B.; Niazi, M.I.K.; Walker, G.S.; Rudd, C.D. Phosphate glass fibre composites for bone repair. J. Bionic Eng. 2009, 6, 318–323. [Google Scholar] [CrossRef]
- Bas, O.; De-Juan-Pardo, E.M.; Meinert, C.; D’Angella, D.; Baldwin, J.G.; Bray, L.J.; Wellard, R.M.; Kollmannsberger, S.; Rank, E.; Werner, C. Biofabricated soft network composites for cartilage tissue engineering. Biofabrication 2017, 9, 25014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scribante, A.; Vallittu, P.K.; Özcan, M. Fiber-Reinforced Composites for Dental Applications. Biomed. Res. Int. 2018, 2018. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.S.; Ahmed, I.; Parsons, A.J.; Walker, G.S.; Scotchford, C.A. The influence of coupling agents on mechanical property retention and long-term cytocompatibility of phosphate glass fibre reinforced PLA composites. J. Mech. Behav. Biomed. Mater. 2013, 28, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, M.S.; Ahmed, I.; Muja, N.; Rudd, C.D.; Bureau, M.N.; Nazhat, S.N. Effect of phosphate-based glass fibre surface properties on thermally produced poly(lactic acid) matrix composites. J. Mater. Sci. Mater. Med. 2011, 22, 2659–2672. [Google Scholar] [CrossRef] [PubMed]
- Haque, P.; Barker, I.A.; Parsons, A.; Thurecht, K.J.; Ahmed, I.; Walker, G.S.; Rudd, C.D.; Irvine, D.J. Influence of compatibilizing agent molecular structure on the mechanical properties of phosphate glass fiber-reinforced PLA composites. J. Polym. Sci. Polym. Chem. 2010, 48, 3082–3094. [Google Scholar] [CrossRef]
- Lee, D.W.; Lim, H.; Chong, H.N.; Shim, W.S. Advances in chitosan material and its hybrid derivatives: A review. Open Biomater. J. 2009, 1, 10–29. [Google Scholar] [CrossRef]
- Rinaudo, M. Chitin and chitosan: Properties and applications. Prog. Polym. Sci. 2006, 31, 603–632. [Google Scholar] [CrossRef]
- Pillai, C.K.S.; Paul, W.; Sharma, C.P. Chitin and chitosan polymers: Chemistry, solubility and fiber formation. Prog. Polym. Sci. 2009, 34, 641–678. [Google Scholar] [CrossRef]
- Ramya, R.; Sudha, P.N.; Mahalakshmi, J. Preparation and characterization of chitosan binary blend. Int. J. Sci. Res. Publ. 2012, 2, 1–9. [Google Scholar]
- Xu, K.; Li, K.; Zhong, T.; Guan, L.; Xie, C.; Li, S. Effects of chitosan as biopolymer coupling agent on the thermal and rheological properties of polyvinyl chloride/wood flour composites. Compos. Part B Eng. 2014, 58, 392–399. [Google Scholar] [CrossRef]
- Cai, X.; Tong, H.; Shen, X.; Chen, W.; Yan, J.; Hu, J. Preparation and characterization of homogeneous chitosan-polylactic acid/hydroxyapatite nanocomposite for bone tissue engineering and evaluation of its mechanical properties. Acta Biomater. 2009, 5, 2693–2703. [Google Scholar] [CrossRef] [PubMed]
- Min, D.H.; Kim, M.J.; Yun, J.H.; Kim, C.S.; Lee, Y.K.; Choi, S.H.; Kim, K.N.; Kim, C.K. Effect of calcium phosphate glass scaffold with chitosan membrane on the healing of alveolar bone in 1 wall intrabony defect in the beagle dogs. Key Eng. Mater. 2005, 284–286, 851–854. [Google Scholar] [CrossRef]
- Cooper, A.; Bhattarai, N.; Zhang, M. Fabrication and cellular compatibility of aligned chitosan—PCL fibers for nerve tissue regeneration. Carbohydr. Polym. 2011, 85, 149–156. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, M. Microstructural and mechanical characterization of chitosan scaffolds reinforced by calcium phosphates. J. Non-Cryst. Solids 2001, 282, 159–164. [Google Scholar] [CrossRef]
- Felfel, R.M.; Ahmed, I.; Parsons, A.J.; Harper, L.T.; Rudd, C.D. Initial mechanical properties of phosphate-glass fibre-reinforced rods for use as resorbable intramedullary nails. J. Mater. Sci. 2012, 47, 4884–4894. [Google Scholar] [CrossRef]
- Pacaphol, K.; Aht-Ong, D. The influences of silanes on interfacial adhesion and surface properties of nanocellulose film coating on glass and aluminum substrates. Surf. Coat. Technol. 2017, 320, 70–81. [Google Scholar] [CrossRef]
- Gunanathan, C.; Ben-David, Y.; Milstein, D. Direct synthesis of amides from alcohols and amines with liberation of H2. Science 2007, 317, 790–792. [Google Scholar] [CrossRef]
- El-Hefian, E.A.; Nasef, M.M.; Yahaya, A.H. The preparation and characterization of chitosan/poly (vinyl alcohol) blended films. J. Chem. 2010, 7, 1212–1219. [Google Scholar] [CrossRef]
- Tan, C.; Ahmed, I.; Parsons, A.J.; Sharmin, N.; Zhu, C.; Liu, J.; Rudd, C.D.; Liu, X. Structural, thermal and dissolution properties of MgO- and CaO-containing borophosphate glasses: Effect of Fe2O3 addition. J. Mater. Sci. 2017, 52, 7489–7502. [Google Scholar] [CrossRef]
- Tan, C.; Ahmed, I.; Parsons, A.J.; Zhu, C.; Betanzos, F.B.; Rudd, C.D.; Liu, X. Effects of Fe2O3 addition and annealing on the mechanical and dissolution properties of MgO- and CaO-containing phosphate glass fibres for bio-applications. Biomed. Glasses 2018, 4, 57–71. [Google Scholar] [CrossRef]
- Jin, R.M.; Sultana, N.; Baba, S.; Hamdan, S.; Ismail, A.F. Porous PCL/chitosan and nHA/PCL/chitosan scaffolds for tissue engineering applications: Fabrication and evaluation. J. Nanomater. 2015, 2015, 1–8. [Google Scholar] [CrossRef]
- Ahmed, I.; Parsons, A.J.; Palmer, G.; Knowles, J.C.; Walker, G.S.; Rudd, C.D. Weight loss, ion release and initial mechanical properties of a binary calcium phosphate glass fibre/PCL composite. Acta Biomater. 2008, 4, 1307–1314. [Google Scholar] [CrossRef] [PubMed]
- Haque, P.; Parsons, A.J.; Barker, I.A.; Ahmed, I.; Irvine, D.J.; Walker, G.S.; Rudd, C.D. Interfacial properties of phosphate glass fibres/PLA composites: Effect of the end functionalities of oligomeric PLA coupling agents. Compos. Sci. Technol. 2010, 70, 1854–1860. [Google Scholar] [CrossRef] [Green Version]
- Sharmin, N.; Rudd, C.D.; Parsons, A.J.; Ahmed, I. Structure, viscosity and fibre drawing properties of phosphate-based glasses: Effect of boron and iron oxide addition. J. Mater. Sci. 2016, 51, 7523–7535. [Google Scholar] [CrossRef]
- Zawadzki, J.; Kaczmarek, H. Thermal treatment of chitosan in various conditions. Carbohydr. Polym. 2010, 80, 394–400. [Google Scholar] [CrossRef]
- Lu, M.; Wang, F.; Liao, Q.; Chen, K.; Qin, J.; Pan, S. FTIR spectra and thermal properties of TiO2-doped iron phosphate glasses. J. Mol. Struct. 2015, 1081, 187–192. [Google Scholar] [CrossRef]
- Qian, B.; Yang, S.; Liang, X.; Lai, Y.; Gao, L.; Yin, G. Structural and thermal properties of La2O3-Fe2O3-P2O5 glasses. J. Mol. Struct. 2012, 1011, 153–157. [Google Scholar] [CrossRef]
- Ciceo-Lucacel, R.; Radu, T.; Ponta, O.; Simon, V. Novel selenium containing boro-phosphate glasses: Preparation and structural study. Mater. Sci. Eng. C Mater. Biol. Appl. 2014, 39, 61–66. [Google Scholar] [CrossRef]
- Sharmin, N.; Hasan, M.S.; Rudd, C.D.; Boyd, D.; Werner-Zwanziger, U.; Ahmed, I.; Parsons, A.J. Effect of boron oxide addition on the viscosity-temperature behaviour and structure of phosphate-based glasses. J. Biomed. Mater. Res. B 2016, 105, 754–777. [Google Scholar] [CrossRef] [PubMed]
- Kweon, H.; Um, I.C.; Park, Y.H. Structural and thermal characteristics of Antheraea pernyi silk fibroin/chitosan blend film. Polymer 2001, 42, 6651–6656. [Google Scholar] [CrossRef]
- Pawlak, A.; Mucha, M. Thermogravimetric and FTIR studies of chitosan blends. Thermochim. Acta 2003, 396, 153–166. [Google Scholar] [CrossRef]
- Zheng, H.; Du, Y.; Yu, J.; Huang, R.; Zhang, L. Preparation and Characterization of Chitosan/Poly(vinyl alcohol) Blend Fibers. J. Appl. Polym. Sci. 2001, 80, 2558–2565. [Google Scholar] [CrossRef]
- Mohammadi, Z.; Mesgar, A.S.; Rasouli-Disfani, F. Reinforcement of freeze-dried chitosan scaffolds with multiphasic calcium phosphate short fibers. J. Mech. Behav. Biomed. Mater. 2016, 61, 590–599. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.M.; Liang, X.F.; Yang, S.Y.; Wang, J.X.; Cao, L.H.; Dai, B. Raman and FTIR spectra of iron phosphate glasses containing cerium. J. Mol. Struct. 2011, 992, 84–88. [Google Scholar] [CrossRef]
- Saranti, A.; Koutselas, I.; Karakassides, M.A. Bioactive glasses in the system CaO-B2O3-P2O5: Preparation, structural study and in vitro evaluation. J. Non-Cryst. Solids 2006, 352, 390–398. [Google Scholar] [CrossRef]
- Ma, L.; Brow, R.K.; Choudhury, A. Structural study of Na2O-FeO-Fe2O3-P2O5 glasses by Raman and Mössbauer spectroscopy. J. Non-Cryst. Solids 2014, 402, 64–73. [Google Scholar] [CrossRef]
- Joseph, K.; Premila, M.; Amarendra, G.; Kutty, K.V.G.; Sundar, C.S.; Rao, P.R.V. Structure of cesium loaded iron phosphate glasses: An infrared and Raman spectroscopy study. J. Nucl. Mater. 2012, 420, 49–53. [Google Scholar] [CrossRef]
- Mikhailov, G.P.; Tuchkov, S.V.; Lazarev, V.V.; Kulish, E.I. Complexation of chitosan with acetic acid according to Fourier transform Raman spectroscopy data. Russ. J. Phys. Chem. A 2014, 88, 936–941. [Google Scholar] [CrossRef]
- Ren, X.D.; Liu, Q.S.; Feng, H.; Yin, X.Y. The Characterization of Chitosan Nanoparticles by Raman Spectroscopy. Appl. Mech. Mater. 2014, 665, 367–370. [Google Scholar] [CrossRef]
- Reinas, A.E.; Hoscheid, J.; Outuki, P.M.; Cardoso, M.L.C. Preparation and characterization of microcapsules of Pterodon pubescens Benth. by using natural polymers. Braz. J. Pharm. Sci. 2014, 50, 919–930. [Google Scholar] [CrossRef] [Green Version]
- Retamal, M.J.; Cisternas, M.A.; Gutierrez-Maldonado, S.E.; Perez-Acle, T.; Seifert, B.; Busch, M.; Huber, P.; Volkmann, U.G. Towards bio-silicon interfaces: Formation of an ultra-thin self-hydrated artificial membrane composed of dipalmitoylphosphatidylcholine (DPPC) and chitosan deposited in high vacuum from the gas-phase. J. Chem. Phys. 2014, 141, 104201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zajac, A.; Hanuza, J.; Wandas, M.; Dyminska, L. Determination of N-acetylation degree in chitosan using Raman spectroscopy. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2015, 134, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Orrego, C.E.; Salgado, N.; Valencia, J.S.; Giraldo, G.I.; Giraldo, O.H.; Cardona, C.A. Novel chitosan membranes as support for lipases immobilization: Characterization aspects. Carbohydr. Polym. 2010, 79, 9–16. [Google Scholar] [CrossRef]
- Bunker, B.C.; Casey, W.H. The Aqueous Chemistry of Oxides; Oxford University Press: Oxford, UK, 2016. [Google Scholar]
- Gao, S.-L.; Zhang, J.; Liu, J.-W.; Zhuang, R.-C.; Plonka, R.; Mäder, E. Healing Microcracks and Early Warning Composite Fractures. In Proceedings of the 6th Colloquium on Textile Reinforced Structures (CTRS6), Berlin, Germany, 19–20 September 2011. [Google Scholar]
- Perrin, C.L. The Logic behind a Physical-Organic Chemist’s Research Topics. J. Org. Chem. 2017, 82, 819–838. [Google Scholar] [CrossRef]
- Kohl, J.G.; Malicky, D.M.; Jones, A.M. Adhesion of epoxy (pseudobarnacles) to glass that has been treated with hydrophobic carbosilane-based coatings. Prog. Org. Coat. 2017, 107, 1–4. [Google Scholar] [CrossRef]
- Shah, B.L.; Matuana, L.M.; Heiden, P.A. Novel coupling agents for PVC/wood-flour composites. J. Vinyl Addit. Technol 2005, 11, 160–165. [Google Scholar] [CrossRef]
- Huang, C.L.; Chen, Y.B.; Lo, Y.L.; Lin, Y.H. Development of chitosan/beta-glycerophosphate/glycerol hydrogel as a thermosensitive coupling agent. Carbohydr. Polym. 2016, 147, 409–414. [Google Scholar] [CrossRef]
- Chenite, A.; Chaput, C.; Wang, D.; Combes, C.; Buschmann, M.D.; Hoemann, C.D.; Leroux, J.C.; Atkinson, B.L.; Binette, F.; Selmani, A. Novel injectable neutral solutions of chitosan form biodegradable gels in situ. Biomaterials 2000, 21, 2155–2161. [Google Scholar] [CrossRef]
- Amaral, I.F.; Granja, P.L.; Barbosa, M.A. Chemical modification of chitosan by phosphorylation: An XPS, FT-IR and SEM study. J. Biomater. Sci. Polym. Ed. 2005, 16, 1575–1593. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.S.; Ahmed, I.; Parsons, A.J.; Rudd, C.D.; Walker, G.S.; Scotchford, C.A. Investigating the use of coupling agents to improve the interfacial properties between a resorbable phosphate glass and polylactic acid matrix. J. Biomater. Appl. 2013, 28, 354–366. [Google Scholar] [CrossRef] [PubMed]
- Bhattarai, N.; Li, Z.; Gunn, J.; Leung, M.; Cooper, A.; Edmondson, D.; Veiseh, O.; Chen, M.; Zhang, Y.; Ellenbogen, R.G.; et al. Natural-synthetic polyblend nanofibers for biomedical applications. Adv. Mater. 2009, 21, 2792–2797. [Google Scholar] [CrossRef]
- Berger, J.; Reist, M.; Mayer, J.M.; Felt, O.; Peppas, N.A.; Gurny, R. Structure and interactions in covalently and ionically crosslinked chitosan hydrogels for biomedical applications. Eur. J. Pharm. Biopharm. 2004, 57, 19–34. [Google Scholar] [CrossRef]
- Tanigawa, J.; Miyoshi, N.; Sakurai, K. Characterization of chitosan/citrate and chitosan/acetate films and applications for wound healing. J. Appl. Polym. Sci. 2008, 110, 608–615. [Google Scholar] [CrossRef]
- Lawrie, G.; Keen, I.; Drew, B.; Chandler-Temple, A.; Rintoul, L.; Fredericks, P.; Grøndahl, L. Interactions between Alginate and Chitosan Biopolymers Characterized Using FTIR and XPS. Biomacromolecules 2007, 8, 2533–2541. [Google Scholar] [CrossRef] [PubMed]
- Bhumkar, D.R.; Pokharkar, V.B. Studies on Effect of pH on Cross-linking of Chitosan with Sodium Tripolyphosphate: A Technical Note. AAPS PharmSciTech 2006, 7, E138–E143. [Google Scholar] [CrossRef]
- Sayyar, S.; Murray, E.; Thompson, B.C.; Chung, J.; Officer, D.L.; Gambhir, S.; Spinks, G.M.; Wallace, G.G. Processable conducting graphene/chitosan hydrogels for tissue engineering. J. Mater. Chem. B 2015, 3, 481–490. [Google Scholar] [CrossRef] [Green Version]
- Yap, W.F.; Yunus, W.M.M.; Talib, Z.A.; Yusof, N.A. X-ray photoelectron spectroscopy and atomic force microscopy studies on crosslinked chitosan thin film. Int. J. Phys. Sci. 2011, 6, 2744–2749. [Google Scholar]
- Nikolenko, Y.M.; Kuryavyi, V.G.; Sheveleva, I.V.; Zemskova, L.A.; Sergienko, V.I. Atomic force microscopy and X-ray photoelectron spectroscopy study of chitosan-carbon fiber materials. Inorg. Mater. 2010, 46, 221–225. [Google Scholar] [CrossRef]
- Majjane, A.; Chahine, A.; Et-tabirou, M.; Echchahed, B.; Do, T.-O.; Breen, P.M. X-ray photoelectron spectroscopy (XPS) and FTIR studies of vanadium barium phosphate glasses. Mater. Chem. Phys. 2014, 143, 779–787. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
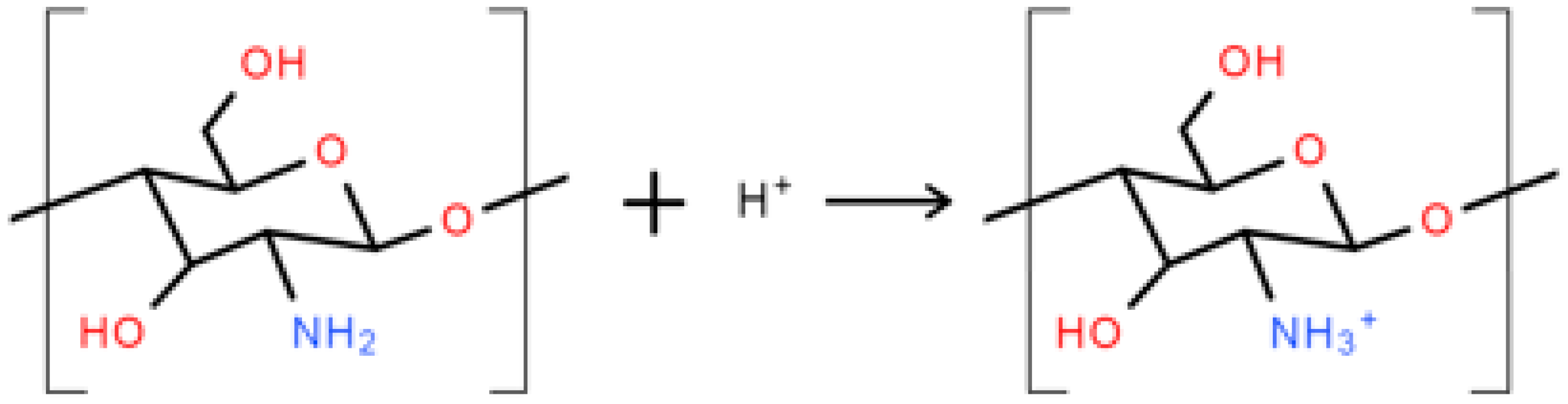{kind=link}
| Sample Codes | PCP-0 | PCP-3R | PCP-3 | PCP-6 | PCP-9 | |
|---|---|---|---|---|---|---|
| Acetic acid solution | mL | - | 100 | 100 | 100 | 100 |
| CS | g | - | 0.3 | 0.3 | 0.6 | 0.9 |
| Dip-coating at RT | min | - | 30 | 30 | 30 | 30 |
| Drying at RT | hour | - | 2 | 2 | 2 | 2 |
| Post-cleaning | min | - | 30 | - | - | - |
| Drying at 50 °C | hour | 24 | 24 | 24 | 24 | 24 |
| Sample Codes | Diameter (µm) | Tensile Strength (MPa) | Normalising Strength (MPa) | Weibull Modulus |
|---|---|---|---|---|
| PCP-0 | 26 ± 4 | 565 ± 124 | 613 | 5.5 |
| PCP-3R | 24 ± 3 | 451 ± 114 | 495 | 5.5 |
| PCP-3 | 25 ± 2 | 522 ± 104 | 565 | 5.3 |
| PCP-6 | 26 ± 3 | 571 ± 120 | 619 | 5.3 |
| PCP-9 | 25 ± 2 | 630 ± 111 | 675 | 6.7 |
| Sample Codes | PCP-0 | PCP-3R | PCP-3 | PCP-6 | PCP-9 | CS |
|---|---|---|---|---|---|---|
| Peaks on Deriv. Wt (°C) | - | - | <100 | <100 | <100 | 86 ± 0 |
| - | - | - | 159 ± 8 | 159 ± 2 | - | |
| - | - | 279 ± 1 | 293 ± 3 | 291 ± 4 | 308 ± 3 | |
| Wt loss at 230–400 °C (%) | 0 ± 0 | 0.02 ± 0.01 | 0.50 ± 0.01 | 1.98 ± 0.05 | 5.8 ± 0.2 | 43 ± 2 |
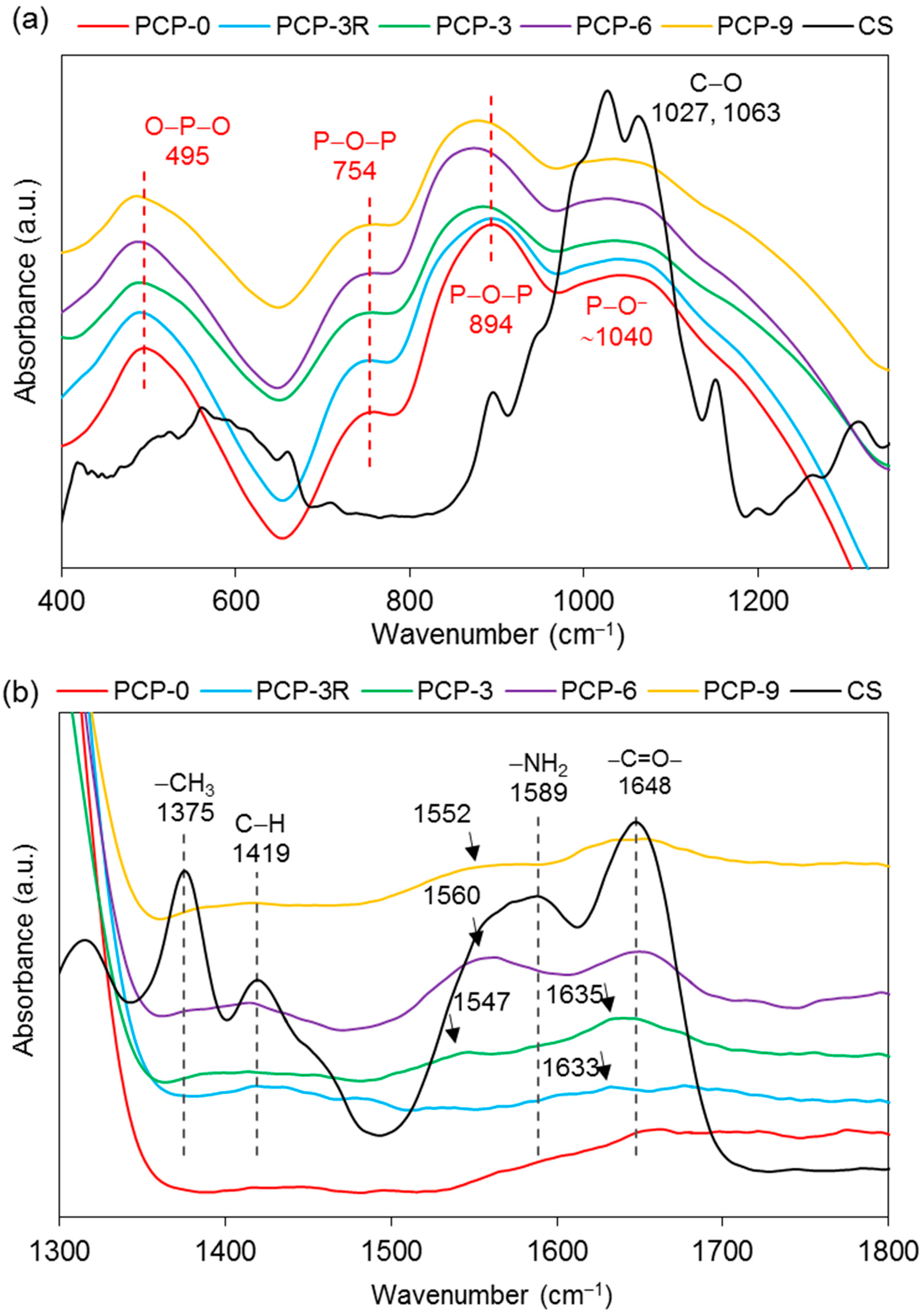
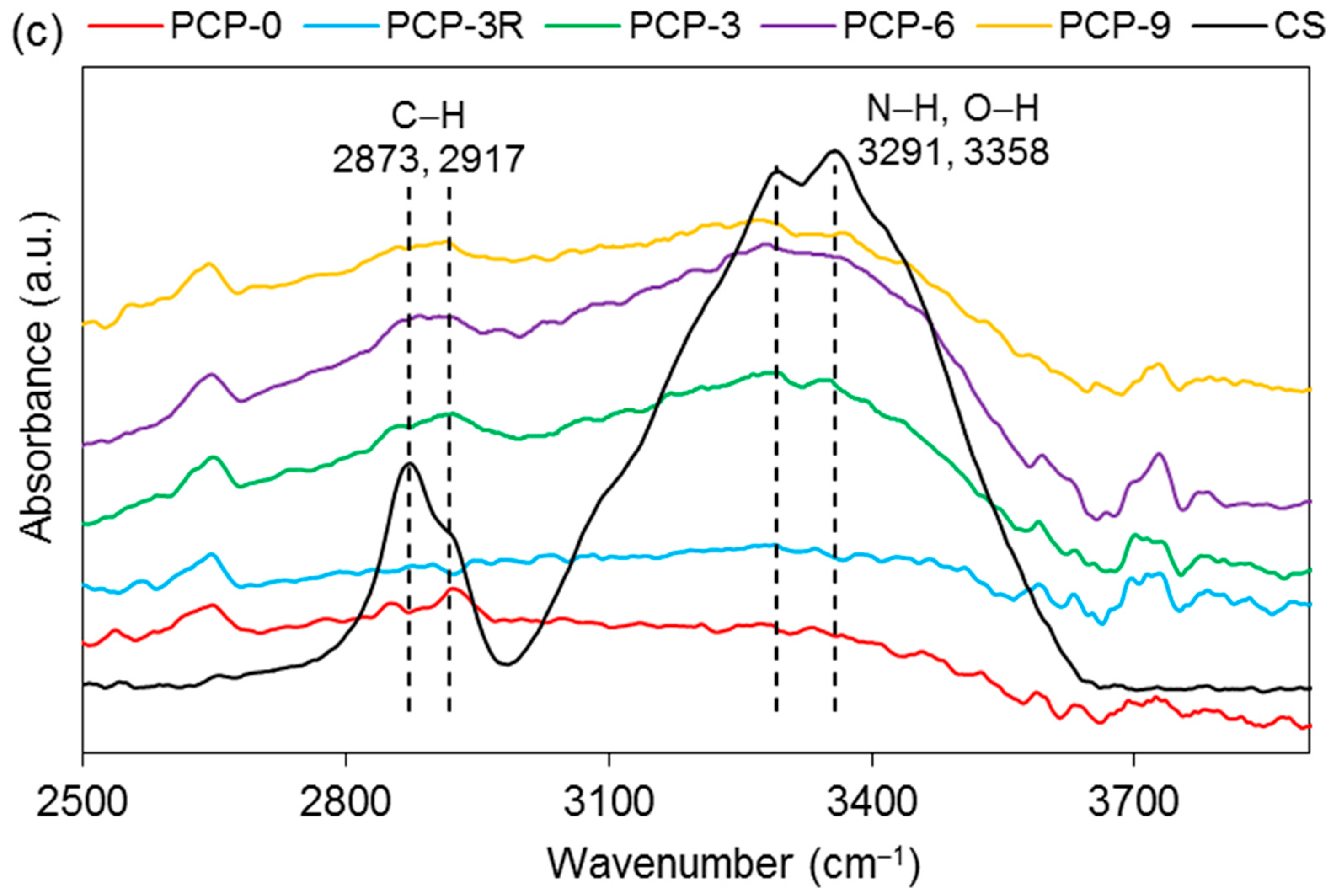
| Wavenumber (cm−1) | Assignments | Ref. |
|---|---|---|
| FTIR–PGF | ||
| 495 | δas(O–P–O) in Q1 species | [28,29] |
| 754, 894 | υs and υas(P–O–P) of the bridging oxygen in Q2 species | [29,30] |
| ~1040 | υs and υas(P–O−) in Q1 species | [29,30] |
| ~1200 | υas(PO2)− of the nonbridging oxygen in Q2 species | [28,31] |
| FTIR–CS | ||
| 1027, 1063 | υ(C–O) in the pyranose ring | [11,32] |
| 1151 | υas(C–O–C) in the glycosidic linkage | [11,32] |
| 1315 | υ(CH2) | [33] |
| 1375 | symmetric deformation mode of CH3 | [11,34] |
| 1419 | δ(C–H) | [33,35] |
| 1589 | δ(NH2) | [32,33] |
| 1648 | υ(–C=O–) in the amide group | [11,35] |
| 2873, 2917 | υ(C–H) | [11,33] |
| 3291, 3358 | υs(N–H) and υs(O–H) | [11,33] |
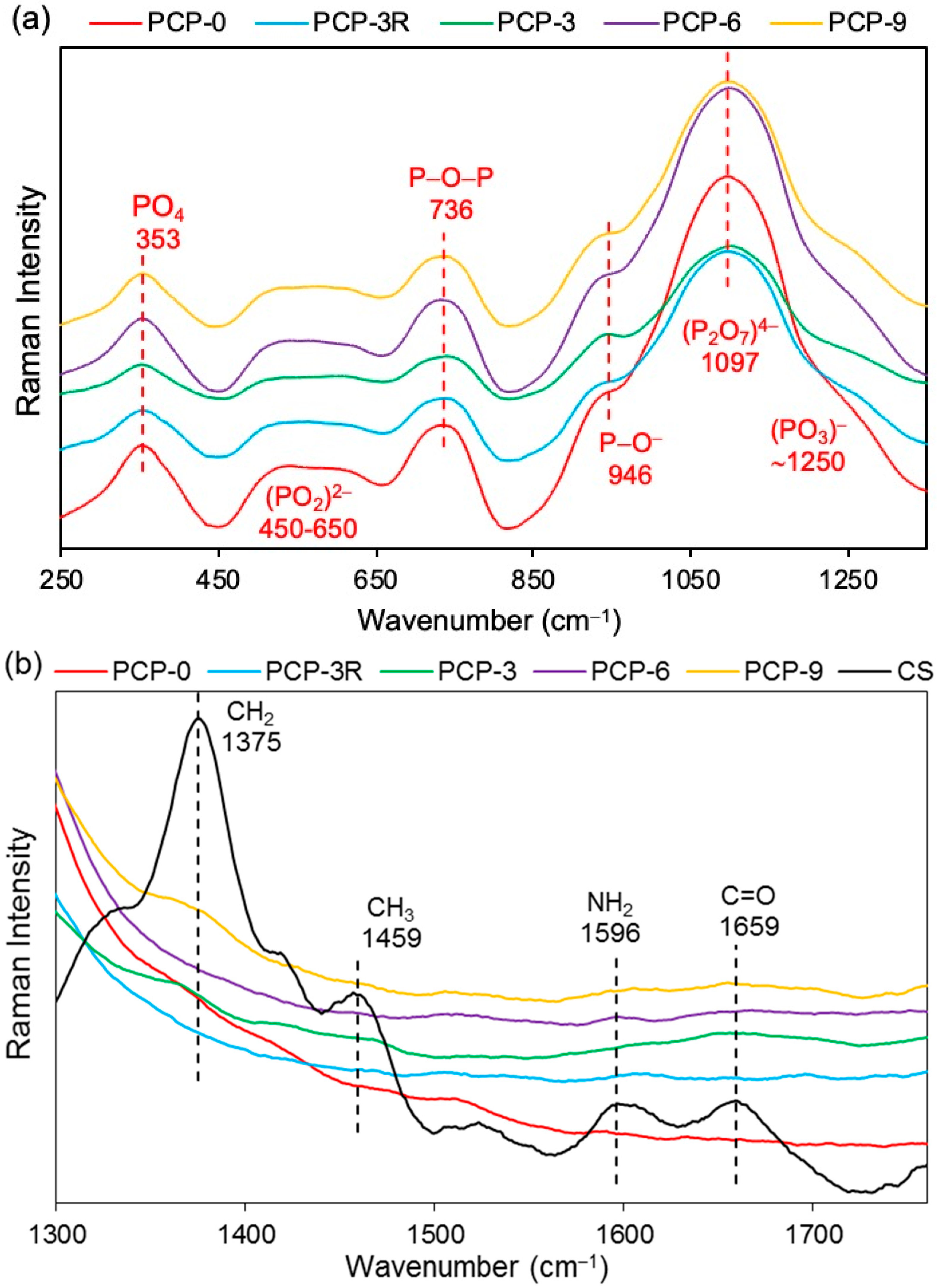
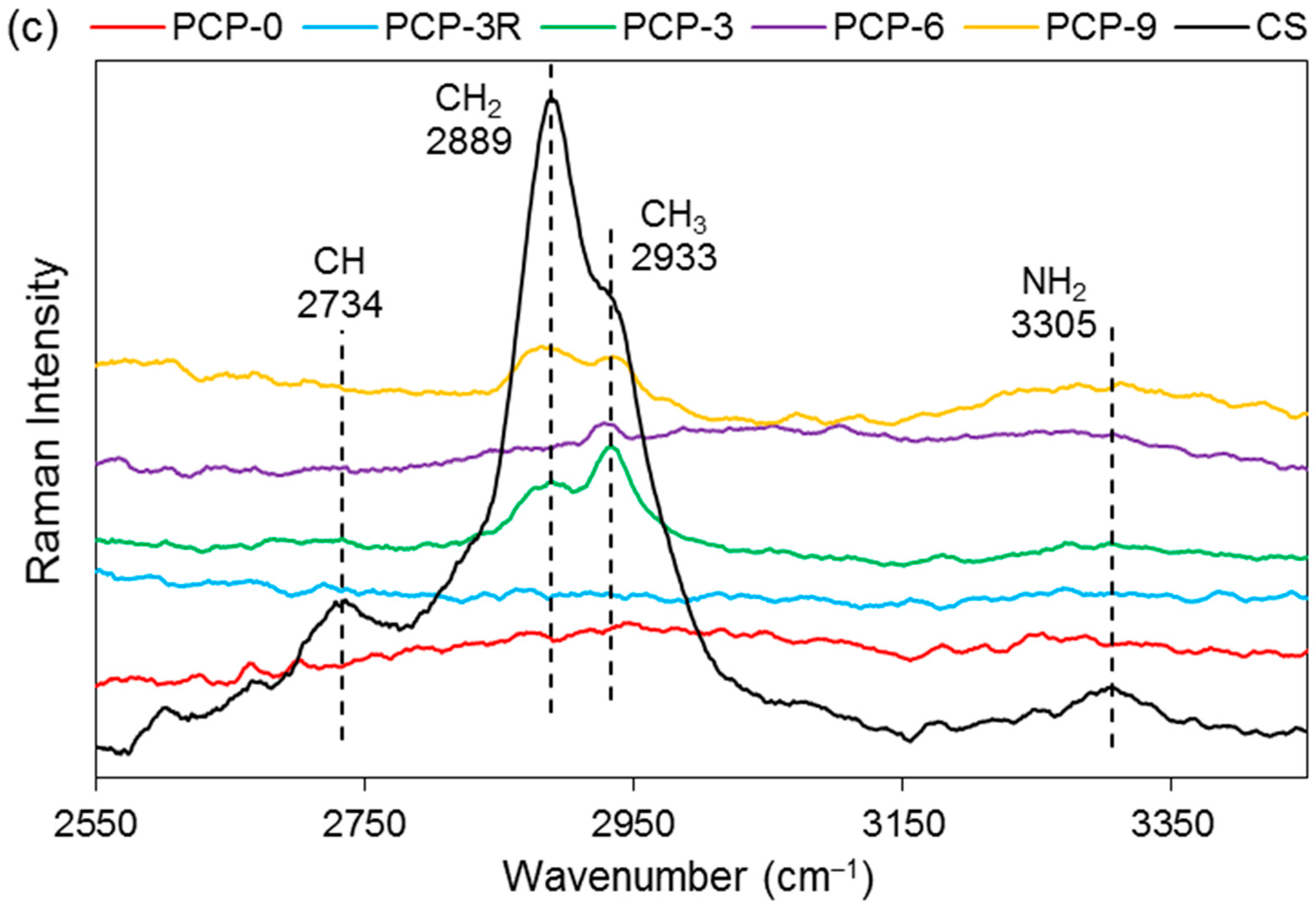
| Raman–PGF | ||
| 353 | δ(PO4) of phosphate polyhedra | [36,37] |
| 450–650 | δs and δas(PO2)2− in Q0 species | [38,39] |
| 736 | υs(P–O–P) of the bridging oxygen in Q1 species | [37,38] |
| 946 | υas(P–O−) of the nonbridging oxygen in Q0 species | [37,39] |
| 1097 | υas(P2O7)4− in Q1 species | [39] |
| ~1250 | υas(PO3)− of the non-bridging oxygen in Q2 species | [36,38] |
| Raman–CS | ||
| 898 | υs(C–O–C) in the pyranose ring | [40,41] |
| 1106 | υ(C–O–C) in the glycosidic linkage and υ(C–C) | [42,43] |
| 1266 | υ(CH) | [41] |
| 1375 | δ(CH2) | [41,44] |
| 1459 | δas(CH3) | [40,41] |
| 1596 | δ(NH2) | [41,44] |
| 1659 | υ(C=O) in the amide group | [44,45] |
| 2734, 2889, 2933 | υ(CH), υ(CH2), and υ(CH3) | [41,44] |
| 3305 | υ(NH2) | [40,41] |
| δs (δas) = symmetric (asymmetric) bending vibration | ||
| υs (υas) = symmetric (asymmetric) stretching vibration | ||
| Sample codes | Binding Energy (eV) | Atomic Ratio |
|---|---|---|
| PCP-0 | - | - |
| PCP-3R | 399.4; 401.5 | 35.2:64.8 |
| PCP-3 | 399.2; 401.3 | 59.8:40.2 |
| PCP-6 | 399.1; 401.1 | 83.2:16.8 |
| PCP-9 | 399.2; 401.4 | 82.9:17.1 |
| CS | 399.1; 400.7 | 94.8:5.2 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tan, C.; Rudd, C.; Parsons, A.; Sharmin, N.; Zhang, J.; Chen, W.; Ahmed, I. Chitosan as a Coupling Agent for Phosphate Glass Fibre/Polycaprolactone Composites. Fibers 2018, 6, 97. https://doi.org/10.3390/fib6040097
Tan C, Rudd C, Parsons A, Sharmin N, Zhang J, Chen W, Ahmed I. Chitosan as a Coupling Agent for Phosphate Glass Fibre/Polycaprolactone Composites. Fibers. 2018; 6(4):97. https://doi.org/10.3390/fib6040097
Chicago/Turabian StyleTan, Chao, Chris Rudd, Andrew Parsons, Nusrat Sharmin, Junxiao Zhang, Wanru Chen, and Ifty Ahmed. 2018. "Chitosan as a Coupling Agent for Phosphate Glass Fibre/Polycaprolactone Composites" Fibers 6, no. 4: 97. https://doi.org/10.3390/fib6040097
APA StyleTan, C., Rudd, C., Parsons, A., Sharmin, N., Zhang, J., Chen, W., & Ahmed, I. (2018). Chitosan as a Coupling Agent for Phosphate Glass Fibre/Polycaprolactone Composites. Fibers, 6(4), 97. https://doi.org/10.3390/fib6040097