
Interactions of the Receptor Binding Domain of SARS-CoV-2 Variants with hACE2: Insights from Molecular Docking Analysis and Molecular Dynamic Simulation

 , , , , ,
, , , , ,  ,
,  and
and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results and Discussion
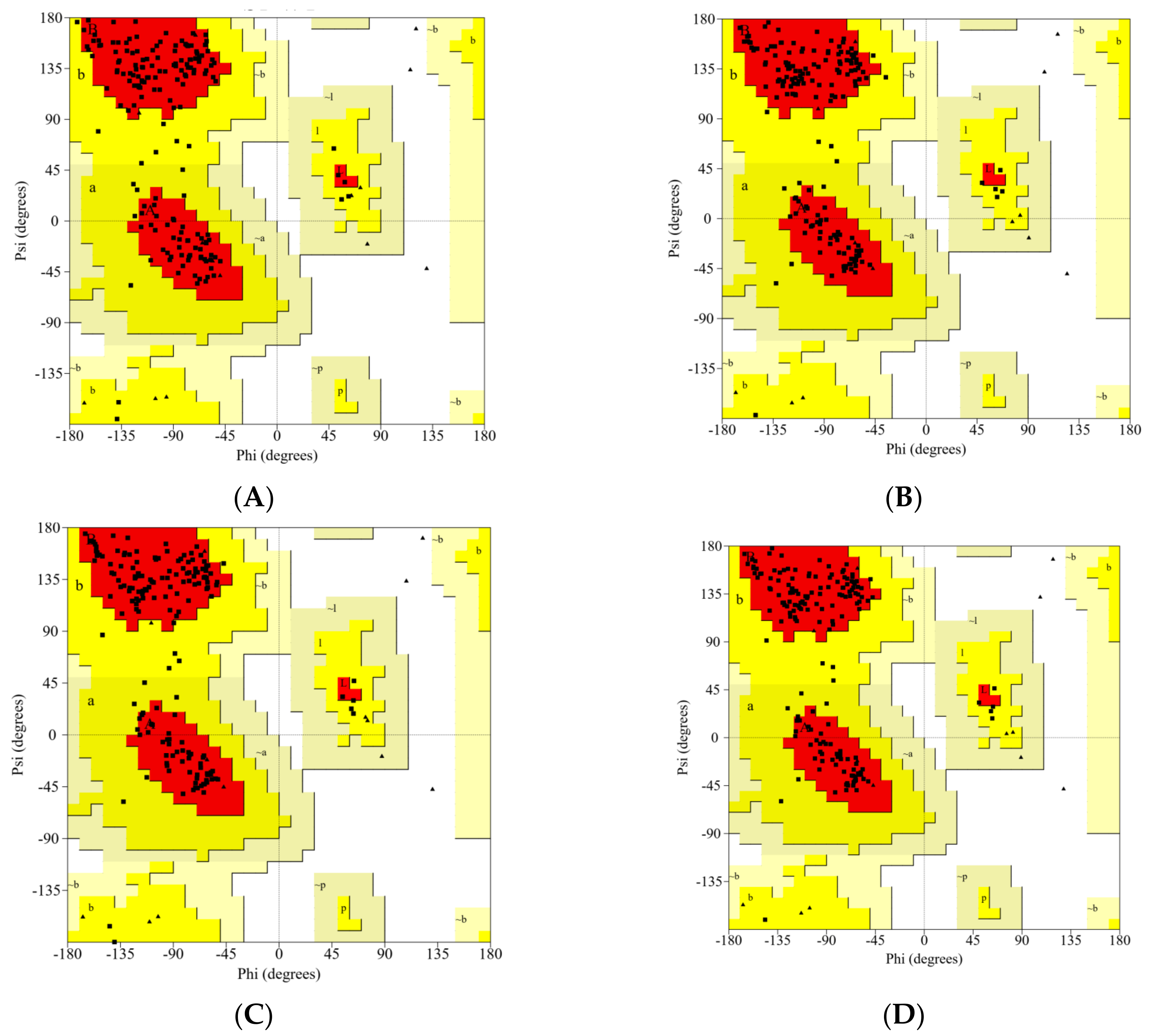
2.1. Analysis of the Modeled RBD Structures
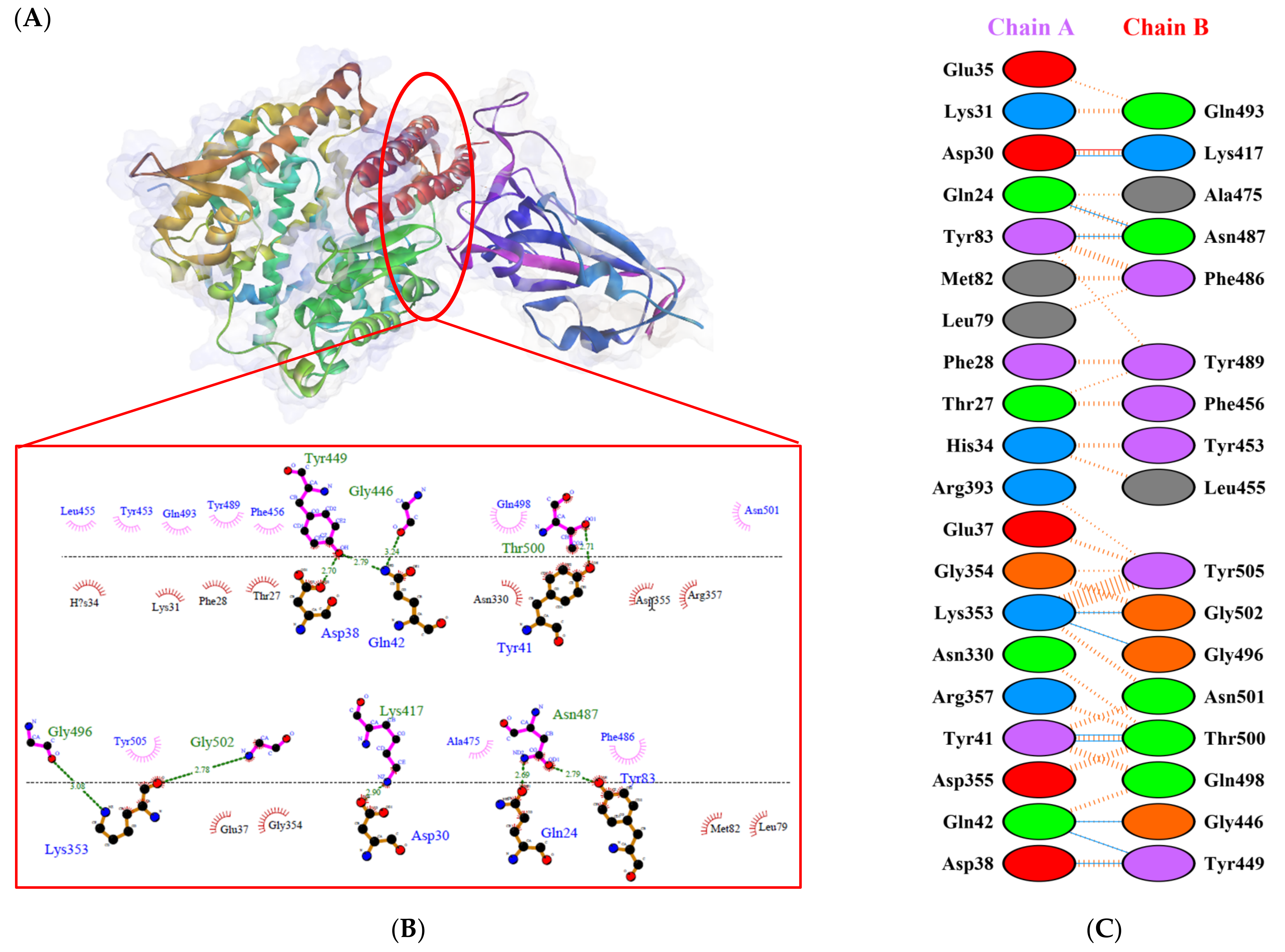
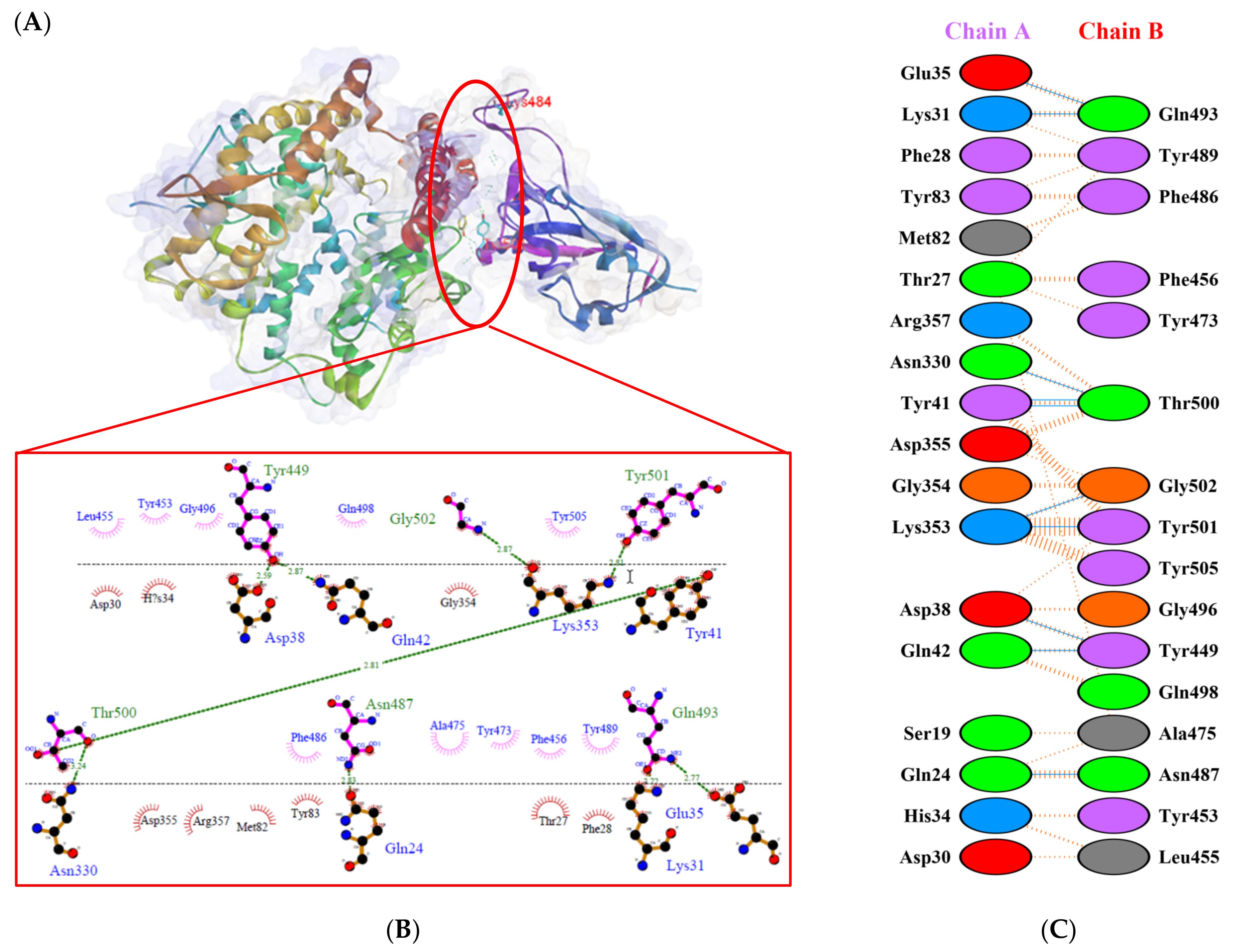
2.2. Molecular Docking Analysis
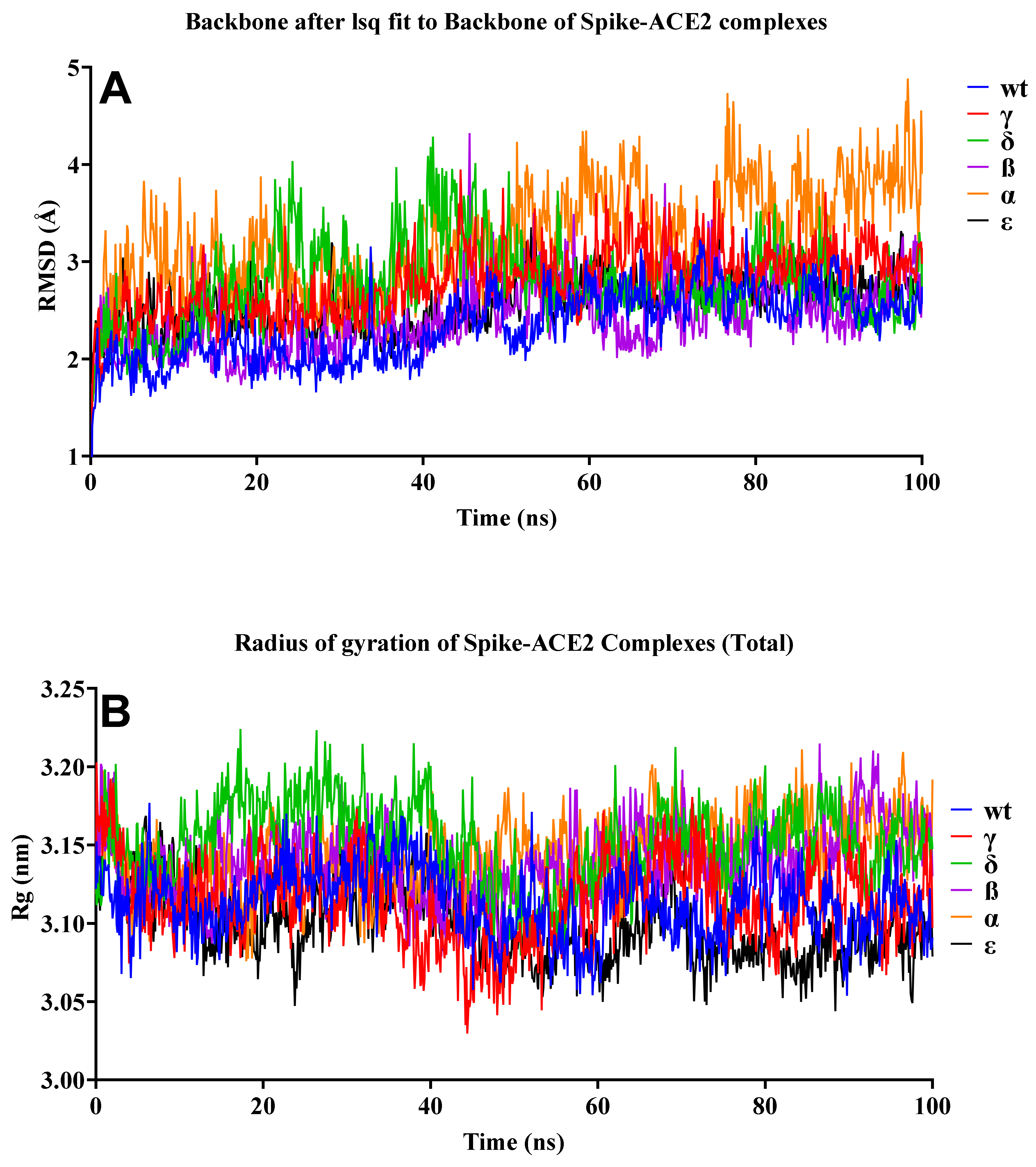
2.3. Molecular Dynamics Simulation of Protein–Protein Complexes
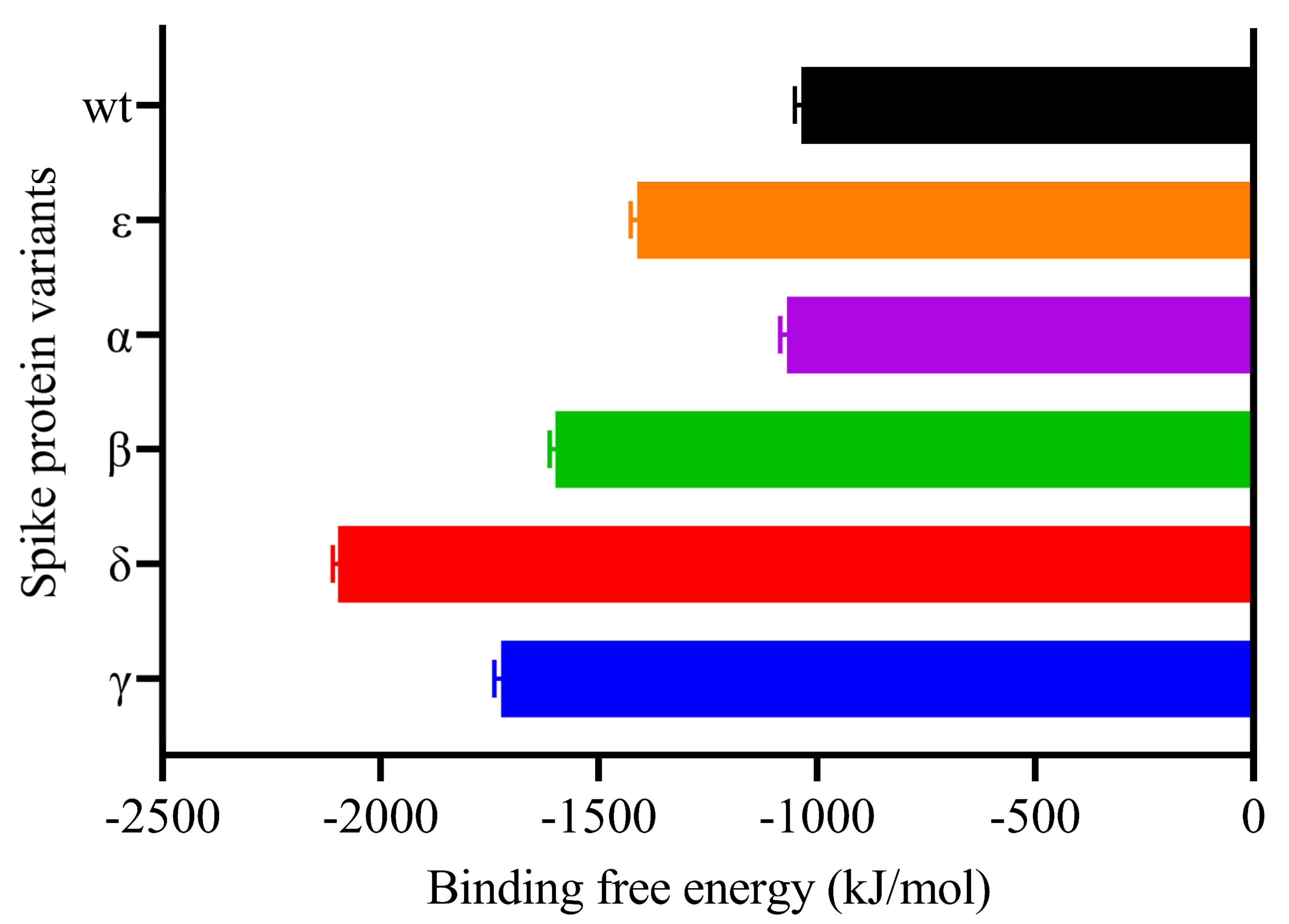
2.4. Binding Free Energy of Protein–Protein Complex Simulation Trajectory
3. Materials and Methods
3.1. Preparation of the Macromolecules
3.2. Molecular Docking Assay
3.3. Molecular Dynamics (MD) Simulations
3.4. Analysis of MD Simulations
3.5. Free-Energy Calculations
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lai, C.C.; Shih, T.P.; Ko, W.C.; Tang, H.J.; Hsueh, P.R. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and coronavirus disease-2019 (COVID-19): The epidemic and the challenges. Int. J. Antimicrob. Agents 2020, 55, 105924. [Google Scholar] [CrossRef]
- Tallei, T.E.; Tumilaar, S.G.; Niode, T.J.; Fatimawali, F.; Kepel, B.J.; Idroes, R.; Effendi, Y.; Sakib, S.A.; Emran, T.B. Potential of plant bioactive compounds as SARS-CoV-2 main protease (Mpro) and spike (S) glycoprotein inhibitors: A molecular docking study. Scientifica 2020, 2020. [Google Scholar] [CrossRef]
- Sharun, K.; Tiwari, R.; Dhama, K.; Emran, T.B.; Rabban, A.A.; Al Mutair, A. Emerging SARS-CoV-2 variants: Impact on vaccine efficacy and neutralizing antibodies. Hum. Vaccines Immunother. 2021, 17, 1–4. [Google Scholar] [CrossRef]
- Romano, M.; Ruggiero, A.; Squeglia, F.; Maga, G.; Berisio, R. A structural view of SARS-CoV-2 RNA replication machinery: RNA synthesis, proofreading and final capping. Cells 2020, 9, 1267. [Google Scholar] [CrossRef]
- Khairan, K.; Idroes, R.; Tallei, T.E.; Nasim, M.J.; Jacob, C. Bioactive compounds from medicinal plants and their possible effect as therapeutics agents against COVID-19: A review. Curr. Nutr. Food Sci. 2021, 17. [Google Scholar] [CrossRef]
- Rakib, A.; Arkajyoti, P.; Uddin, N.C.; Sami, S.A.; Baral, S.K.; Majumder, M.; Tareq, A.M.; Amin, M.N.; Shahriar, A.; Uddin, Z.; et al. Biochemical and computational approach of selected phytocompounds from Tinospora crispa in the management of COVID-19. Molecules 2020, 25, 3936. [Google Scholar] [CrossRef]
- Yadav, R.; Chaudhary, J.K.; Jain, N.; Chaudhary, P.K.; Khanra, S.; Dhamija, P.; Sharma, A.; Kumar, A.; Hadu, S. Role of structural and non-structural proteins and therapeutic targets of SARS-CoV-2 for COVID-19. Cells 2021, 10, 821. [Google Scholar] [CrossRef]
- Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Qiu, Y.; Wang, J.; Liu, Y.; Wei, Y.; et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet 2020, 395, 507–513. [Google Scholar] [CrossRef] [Green Version]
- Ceraolo, C.; Giorgi, F.M. Genomic variance of the 2019-nCoV coronavirus. J. Med. Virol. 2020, 92, 522–528. [Google Scholar] [CrossRef] [Green Version]
- Naqvi, A.A.T.; Fatima, K.; Mohammad, T.; Fatima, U.; Singh, I.K.; Singh, A.; Atif, S.M.; Hariprasad, G.; Hasan, G.M.; Hassan, I. Insights into SARS-CoV-2 genome, structure, evolution, pathogenesis and therapies: Structural genomics approach. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165878. [Google Scholar] [CrossRef]
- Li, W.; Moore, M.J.; Vasilieva, N.; Sui, J.; Wong, S.K.; Berne, M.A.; Somasundaran, M.; Sullivan, J.L.; Luzuriaga, K.; Greenough, T.C.; et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003, 426, 450–454. [Google Scholar] [CrossRef] [Green Version]
- Ou, X.; Liu, Y.; Lei, X.; Li, P.; Mi, D.; Ren, L.; Guo, L.; Guo, R.; Chen, T.; Hu, J.; et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat. Commun. 2020, 11, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Papa, G.; Mallery, D.L.; Albecka, A.; Welch, L.G.; Cattin-Ortola, J.; Luptak, J.; Paul, D.; McMahon, H.T.; Goodfellow, I.G.; Carter, A.; et al. Furin cleavage of SARS-CoV-2 Spike promotes but is not essential for infection and cell-cell fusion. PLoS Pathog. 2021, 17, e1009246. [Google Scholar] [CrossRef]
- Henderson, R.; Edwards, R.J.; Mansouri, K.; Janowska, K.; Stalls, V.; Gobeil, S.; Kopp, M.; Hsu, A.; Borgnia, M.; Parks, R.; et al. Controlling the SARS-CoV-2 Spike glycoprotein conformation. bioRxiv Prepr. Serv. Biol. 2020, 102087. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Yang, C.; Xu, X.F.; Xu, W.; Liu, S.W. Structural and functional properties of SARS-CoV-2 spike protein: Potential antivirus drug development for COVID-19. Acta Pharmacol. Sin. 2020, 41, 1141–1149. [Google Scholar] [CrossRef]
- Faria, N.R.; Mellan, T.A.; Whittaker, C.; Claro, I.M.; da Candido, D.S.; Mishra, S.; Crispim, M.A.E.; Sales, F.C.; Hawryluk, I.; McCrone, J.T.; et al. Genomics and epidemiology of a novel SARS-CoV-2 lineage in Manaus, Brazil. medRxiv Prepr. Serv. Health Sci. 2021, 52554. [Google Scholar] [CrossRef]
- Madhi, S.A.; Baillie, V.; Cutland, C.; Voysey, M.; Phil, D.; Koen, A.L.; Fairlie, L.; Paeds, F.C.; Padayachee, S.D.; Dheda, K.; et al. Efficacy of the ChAdOx1 nCoV-19 Covid-19 vaccine against the B.1.351 variant. N. Engl. J. Med. 2021. [Google Scholar] [CrossRef]
- Noh, J.Y.; Jeong, H.W.; Shin, E.-C. SARS-CoV-2 mutations, vaccines, and immunity: Implication of variants of concern. Signal Transduct. Target. Ther. 2021, 6, 203. [Google Scholar] [CrossRef]
- Wang, Z.; Schmidt, F.; Nussenzweig, M.C. mRNA vaccine-elicited antibodies to SARS-CoV-2 and circulating variants. Nature 2021, 592, 616–622. [Google Scholar] [CrossRef]
- Liu, Z.; VanBlargan, L.A.; Bloyet, L.-M.; Rothlauf, P.W.; Chen, R.E.; Stumpf, S.; Zhao, H.; Errico, J.M.; Theel, E.S.; Liebeskind, M.J.; et al. Identification of SARS-CoV-2 spike mutations that attenuate monoclonal and serum antibody neutralization. Cell Host Microbe 2021, 29, 477–488.e4. [Google Scholar] [CrossRef]
- Wang, P.; Wang, M.; Yu, J.; Cerutti, G.; Nair, M.S.; Huang, Y.; Kwong, P.D.; Shapiro, L.; Ho, D.D. Increased resistance of SARS-CoV-2 variants B.1.351 and B.1.1.7 to Antibody neutralization. bioRxiv Prepr. Serv. Biol. 2021, 428137. [Google Scholar] [CrossRef]
- Buchan, B.W.; Yao, J.D. Severe acute respiratory syndrome Coronavirus 2: The emergence of important genetic variants and testing options for clinical laboratories. Clin. Microbiol. Newsl. 2021, 43, 89–96. [Google Scholar] [CrossRef]
- Ramanathan, M.; Ferguson, I.D.; Miao, W.; Khavari, P.A. SARS-CoV-2 B.1.1.7 and B.1.351 spike variants bind human ACE2 with increased affinity. Lancet Infect. Dis. 2021. [Google Scholar] [CrossRef]
- Wang, P.; Casner, R.G.; Nair, M.S.; Wang, M.; Yu, M.; Cerutti, G.; Liu, L.; Kwong, P.D.; Huang, Y.; Shapiro, L.; et al. Increased resistance of SARS-CoV-2 variant P.1 to antibody neutralization. Cell Host Microbe 2021, 29, 747–751.e4. [Google Scholar] [CrossRef]
- Davies, N.G. Estimated transmissibility and impact of SARS-CoV-2 lineage B.1.1.7 in England. Science 2021, 372, eabg3055. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.W.; Tambyah, P.A.; Hui, D.S. Emergence of a new SARS-CoV-2 variant in the UK. J. Infect. 2021, 82, e27–e28. [Google Scholar] [CrossRef] [PubMed]
- Janik, E.; Niemcewicz, M.; Podogrocki, M.; Majsterek, I.; Bijak, M. The emerging concern and interest SARS-CoV-2 variants. Pathogens 2021, 10, 633. [Google Scholar] [CrossRef]
- Tegally, H.; Wilkinson, E.; Giovanetti, M.; Iranzadeh, A.; Fonseca, V.; Giandhari, J.; Doolabh, D.; Pillay, S.; San, E.J.; Msomi, N.; et al. Emergence and rapid spread of a new severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2) lineage with multiple spike mutations in South Africa. medRxiv 2020, 10, 20248640. [Google Scholar] [CrossRef]
- Yadav, P.; Mohandas, S.; Sarkale, P.; Nyayanit, D.; Shete, A.; Sahay, R.; Potdar, V.; Baradkar, S.; Gupta, N.; Sapkal, G.; et al. Isolation of SARS-CoV-2 B.1.1.28.2 P2 variant and pathogenicity comparison with D614G variant in hamster model. bioRxiv 2021, 445424. [Google Scholar] [CrossRef]
- Callaway, E. Coronavirus variants get Greek names—But will scientists use them? Nature 2021, 162. [Google Scholar] [CrossRef]
- Khan, A.; Zia, T.; Suleman, M.; Khan, T.; Ali, S.S.; Abbasi, A.A.; Mohammad, A.; Wei, D.-Q. Higher infectivity of the SARS-CoV-2 new variants is associated with K417N/T, E484K, and N501Y mutants: An insight from structural data. J. Cell. Physiol. 2021, 236, 7045–7057. [Google Scholar] [CrossRef] [PubMed]
- Leung, K.; Shum, M.H.; Leung, G.M.; Lam, T.T.; Wu, J.T. Early transmissibility assessment of the N501Y mutant strains of SARS-CoV-2 in the United Kingdom, October to November 2020. Eur. Surveill. Bull. Eur. Mal. Transm. Eur. Commun. Dis. Bull. 2021, 26. [Google Scholar] [CrossRef]
- Gobeil, S.M.-C.; Janowska, K.; McDowell, S.; Mansouri, K.; Parks, R.; Stalls, V.; Kopp, M.F.; Manne, K.; Li, D.; Wiehe, K.; et al. Effect of natural mutations of SARS-CoV-2 on spike structure, conformation, and antigenicity. Science 2021, 373, eabi6226. [Google Scholar] [CrossRef]
- Rees-Spear, C.; Muir, L.; Griffith, S.A.; Heaney, J.; Aldon, Y.; Snitselaar, J.L.; Thomas, P.; Graham, C.; Seow, J.; Lee, N.; et al. The effect of spike mutations on SARS-CoV-2 neutralization. Cell Rep. 2021, 34, 108890. [Google Scholar] [CrossRef]
- Kirby, T. New variant of SARS-CoV-2 in UK causes surge of COVID-19. Lancet. Respir. Med. 2021, 9, e20–e21. [Google Scholar] [CrossRef]
- Rakib, A.; Nain, Z.; Islam, M.A.; Sami, S.A.; Mahmud, S.; Islam, A.; Ahmed, S.; Siddiqui, A.B.F.; Babu, S.M.O.F.; Hossain, P.; et al. A molecular modelling approach for identifying antiviral selenium-containing heterocyclic compounds that inhibit the main protease of SARS-CoV-2: An in silico investigation. Brief. Bioinform. 2021, 22, 1476–1498. [Google Scholar] [CrossRef]
- Dutta, M.; Tareq, A.M.; Rakib, A.; Mahmud, S.; Sami, S.A.; Mallick, J.; Islam, M.N.; Majumder, M.; Uddin, M.Z.; Alsubaie, A.; et al. Phytochemicals from Leucas zeylanica targeting main protease of SARS–CoV–2: Chemical profiles, molecular docking, and molecular dynamics simulations. Biology 2021, 10, 789. [Google Scholar] [CrossRef]
- Chowdhury, K.H.; Chowdhury, R.; Mahmud, S.; Tareq, A.M.; Hanif, N.B.; Banu, N.; Reza, A.S.M.; Emran, T.B.; Simal-Gandara, J. Drug repurposing approach against novel coronavirus disease (COVID-19) through virtual screening targeting SARS-CoV-2 main protease. Biology 2021, 10, 2. [Google Scholar] [CrossRef]
- Li, Q.; Wu, J.; Nie, J.; Zhang, L.; Hao, H.; Liu, S.; Zhao, C.; Zhang, Q.; Liu, H.; Nie, L.; et al. The impact of mutations in SARS-CoV-2 spike on viral infectivity and antigenicity. Cell 2020, 182, 1284–1294.e9. [Google Scholar] [CrossRef]
- Weisblum, Y.; Schmidt, F.; Zhang, F.; DaSilva, J.; Poston, D.; Lorenzi, J.C.C.; Muecksch, F.; Rutkowska, M.; Hoffmann, H.-H. Escape from neutralizing antibodies by SARS-CoV-2 spike protein variants. Elife 2020, 9. [Google Scholar] [CrossRef]
- Singh, A.; Steinkellner, G.; Köchl, K.; Gruber, K.; Gruber, C.C. Serine 477 plays a crucial role in the interaction of the SARS-CoV-2 spike protein with the human receptor ACE2. Sci. Rep. 2021, 11. [Google Scholar] [CrossRef]
- Kleywegt, G.J.; Jones, T.A. Phi/Psichology: Ramachandran revisited. Structure 1996, 4, 1395–1400. [Google Scholar] [CrossRef] [Green Version]
- Lovell, S.C.; Ian, W.; Davis, W.; Arendall, B., III; de Bakker, P.I.W.; Word, M.; Prisant, M.G.; Richardson, J.S.; Richardson, D.C. Structure validation by Cα geometry: φ,ψ and Cβ deviation. Proteins Struct. Funct. Genet. 2003, 50, 437–450. [Google Scholar] [CrossRef]
- Gowder, S.M.; Chatterjee, J.; Chaudhuri, T.; Paul, K. Prediction and analysis of surface hydrophobic residues in tertiary structure of proteins. Sci. World J. 2014, 2014, 971258. [Google Scholar] [CrossRef]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [Green Version]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2021, 581, 215–220. Available online: https://www.nature.com/articles/s41586-020-2180-5 (accessed on 11 May 2021). [CrossRef] [PubMed] [Green Version]
- Koley, T.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structural analysis of COVID-19 spike protein in recognizing the ACE2 receptor of different mammalian species and its susceptibility to viral infection. 3 Biotech. 2021, 11. [Google Scholar] [CrossRef]
- Harapan, H.; Ryan, M.; Yohan, B.; Abidin, R.S.; Nainu, F.; Rakib, A.; Jahan, I.; Emran, T.B.; Ullah, I.; Panta, K.; et al. COVID-19 and dengue: Double punches for dengue-endemic countries in Asia. Rev. Med. Virol. 2021, 31, e2161. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Wang, J.; Peng, W.; Wu, F.-X.; Pan, Y. Protein–protein interactions: Detection, reliability assessment and applications. Brief. Bioinform. 2017, 18, 798–819. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.; Chen, Q.; Zhou, Y.; Fan, H.; Deng, Y.-Q.; Wang, Y.; Teng, Y.; Zhao, Z.; Cui, Y.; Li, Y.; et al. Adaptation of SARS-CoV-2 in BALB/c mice for testing vaccine efficacy. Science 2020, 369, 1603–1607. [Google Scholar] [CrossRef]
- Luan, B.; Wang, H.; Huynh, T. Enhanced binding of the N501Y-mutated SARS-CoV-2 spike protein to the human ACE2 receptor: Insights from molecular dynamics simulations. FEBS Lett. 2021, 595, 1454–1461. [Google Scholar] [CrossRef] [PubMed]
- Gan, H.H.; Twaddle, A.; Marchand, B.; Gunsalus, K.C. Structural modeling of the SARS-CoV-2 spike/human ACE2 complex interface can identify high-affinity variants associated with increased transmissibility. J. Mol. Biol. 2021, 433, 167051. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Hofmann-Winkler, H.; Kruger, N.; Kempf, A.; Nehlmeier, I.; Graiche, L.; Arora, P.; Sidarovich, A.; Moldenhauer, A.-S.; Winkler, M.S.; et al. SARS-CoV-2 variant B.1.617 is resistant to Bamlanivimab and evades antibodies induced by infection and vaccination. Cell Rep. 2021, 109415. [Google Scholar] [CrossRef]
- Ashwaq, O.; Manickavasagam, P.; Haque, S.M. V483A: An emerging mutation hotspot of SARS-CoV-2. Future Virol. 2021, 16. [Google Scholar] [CrossRef] [PubMed]
- Yadav, P.D.; Mohandas, S.; Shete, A.M.; Nyayanit, D.A.; Gupta, N.; Patil, D.Y.; Sapkal, G.N.; Potdar, V.; Kadam, M.; Kumar, A.; et al. SARS CoV-2 variant B.1.617.1 is highly pathogenic in hamsters than B.1 variant. bioRxiv 2021, 442760. [Google Scholar] [CrossRef]
- Kim, S.; Lei, Z.; Dicker, J.; Cao, Y.; Zhang, X.F.; Im, W. Differential interactions between human ACE2 and spike RBD of SARS-CoV-2 variants of concern. bioRxiv Prepr. Server Biol. 2021, 453598. [Google Scholar] [CrossRef]
- Laffeber, C.; de Koning, K.; Kanaar, R.; Lebbink, J.H.G. Experimental evidence for enhanced receptor binding by rapidly spreading SARS-CoV-2 variants. J. Mol. Biol. 2021, 433, 167058. [Google Scholar] [CrossRef]
- Shahhosseini, N.G.; Babuadze, G.; Wong, G.; Kobinger, G.P. Mutation signatures and in silico docking of novel SARS-CoV-2 variants of concern. Microorganisms 2021, 9, 926. [Google Scholar] [CrossRef]
- Liu, H.; Wei, P.; Zhang, Q.; Chen, Z.; Aviszus, K.; Downing, W.; Peterson, S.; Reynoso, L.; Downey, G.P.; Frankel, S.K.; et al. 501Y.V2 and 501Y.V3 variants of SARS-CoV-2 lose binding to Bamlanivimab in vitro. bioRxiv Prepr. Server Biol. 2021, 431305. [Google Scholar] [CrossRef]
- Chakraborty, S. Evolutionary and structural analysis elucidates mutations on SARS-CoV2 spike protein with altered human ACE2 binding affinity. Biochem. Biophys. Res. Commun. 2021, 538, 97–103. [Google Scholar] [CrossRef]
- Bastolla, U.; Demetrius, L. Stability constraints and protein evolution: The role of chain length, composition and disulfide bonds. Protein Eng. Des. Sel. 2005, 18, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Schwede, T.; Kopp, J.; Guex, N.; Peitsch, M.C. SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res. 2003, 31, 3381–3385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Yan, Y.; Zhang, D.; Zhou, P.; Li, B.; Huang, S.Y. HDOCK: A web server for protein-protein and protein-DNA/RNA docking based on a hybrid strategy. Nucleic Acids Res. 2017, 45, W365–W373. [Google Scholar] [CrossRef]
- Yan, Y.; Tao, H.; He, J.; Huang, S.Y. The HDOCK server for integrated protein–protein docking. Nat. Protoc. 2020, 15, 1829–1852. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Jabłońska, J.; Pravda, L.; Vařeková, R.S.; Thornton, J.M. PDBsum: Structural summaries of PDB entries. Protein Sci. 2018, 27, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Pall, S.; Smith, J.C.; Hess, B.; Lindahl, E. Gromacs: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; O’Dror, R.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins Struct. Funct. Bioinform. 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [Green Version]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T. End-point binding free energy calculation with MM/PBSA and MM/GBSA: Strategies and applications in drug design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef] [PubMed]
- Kumari, R.; Kumar, R.; Lynn, A. G-mmpbsa-A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]



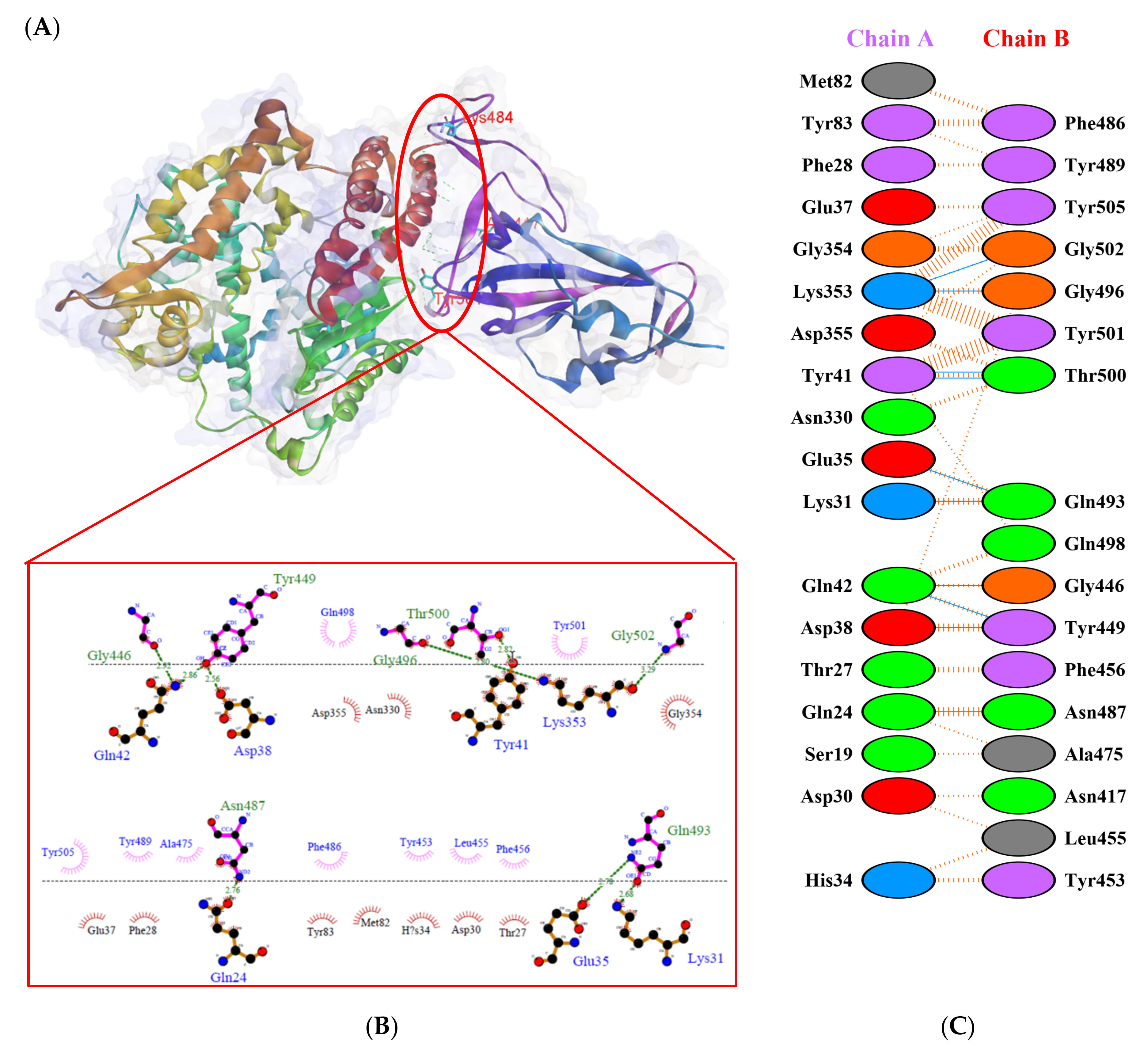
 ), salt bridges (
), salt bridges (  ), and non-bonded (
), and non-bonded (  ) interactions. Chain A represents hACE2 and chain B represents RBD WT. Residue colors: positive (blue): H,K,R; negative (red): D,E; neutral (green): S,T,N,Q; aliphatic (grey): A,V,L,I,M; aromatic (purple): F,Y,W; Pro & Gly (orange): P,G.
), salt bridges ( ), and non-bonded ( ) interactions. Chain A represents hACE2 and chain B represents RBD WT. Residue colors: positive (blue): H,K,R; negative (red): D,E; neutral (green): S,T,N,Q; aliphatic (grey): A,V,L,I,M; aromatic (purple): F,Y,W; Pro & Gly (orange): P,G.
) interactions. Chain A represents hACE2 and chain B represents RBD WT. Residue colors: positive (blue): H,K,R; negative (red): D,E; neutral (green): S,T,N,Q; aliphatic (grey): A,V,L,I,M; aromatic (purple): F,Y,W; Pro & Gly (orange): P,G.
), salt bridges ( ), and non-bonded ( ) interactions. Chain A represents hACE2 and chain B represents RBD WT. Residue colors: positive (blue): H,K,R; negative (red): D,E; neutral (green): S,T,N,Q; aliphatic (grey): A,V,L,I,M; aromatic (purple): F,Y,W; Pro & Gly (orange): P,G. ), salt bridges ( ), and non-bonded ( ) interactions. Chain A represents hACE2 and chain B represents RBD Gamma. Residue colors: positive (blue): H,K,R; negative (red): D,E; neutral (green): S,T,N,Q; aliphatic (grey): A,V,L,I,M; aromatic (purple): F,Y,W; Pro & Gly (orange): P,G.
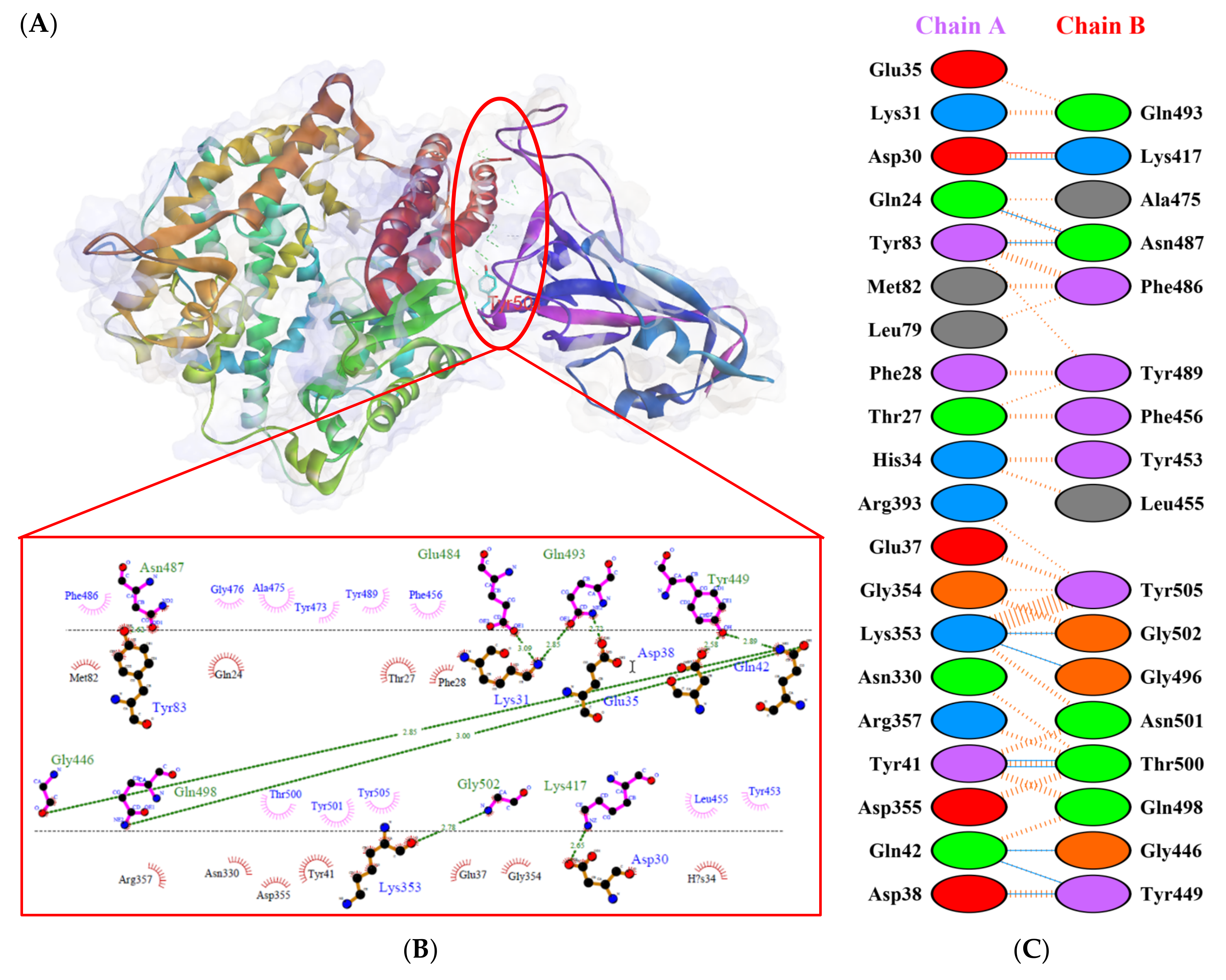
), salt bridges ( ), and non-bonded ( ) interactions. Chain A represents hACE2 and chain B represents RBD Gamma. Residue colors: positive (blue): H,K,R; negative (red): D,E; neutral (green): S,T,N,Q; aliphatic (grey): A,V,L,I,M; aromatic (purple): F,Y,W; Pro & Gly (orange): P,G.
), salt bridges ( ), and non-bonded ( ) interactions. Chain A represents hACE2 and chain B represents RBD Gamma. Residue colors: positive (blue): H,K,R; negative (red): D,E; neutral (green): S,T,N,Q; aliphatic (grey): A,V,L,I,M; aromatic (purple): F,Y,W; Pro & Gly (orange): P,G.
), salt bridges ( ), and non-bonded ( ) interactions. Chain A represents hACE2 and chain B represents RBD Gamma. Residue colors: positive (blue): H,K,R; negative (red): D,E; neutral (green): S,T,N,Q; aliphatic (grey): A,V,L,I,M; aromatic (purple): F,Y,W; Pro & Gly (orange): P,G. ), salt bridges ( ), and non-bonded ( ) interactions. Chain A represents hACE2 and chain B represents RBD Delta. Residue colors: positive (blue): H,K,R; negative (red): D,E; neutral (green): S,T,N,Q; aliphatic (grey): A,V,L,I,M; aromatic (purple): F,Y,W; Pro & Gly (orange): P,G.
), salt bridges ( ), and non-bonded ( ) interactions. Chain A represents hACE2 and chain B represents RBD Delta. Residue colors: positive (blue): H,K,R; negative (red): D,E; neutral (green): S,T,N,Q; aliphatic (grey): A,V,L,I,M; aromatic (purple): F,Y,W; Pro & Gly (orange): P,G.
), salt bridges ( ), and non-bonded ( ) interactions. Chain A represents hACE2 and chain B represents RBD Delta. Residue colors: positive (blue): H,K,R; negative (red): D,E; neutral (green): S,T,N,Q; aliphatic (grey): A,V,L,I,M; aromatic (purple): F,Y,W; Pro & Gly (orange): P,G.
), salt bridges ( ), and non-bonded ( ) interactions. Chain A represents hACE2 and chain B represents RBD Delta. Residue colors: positive (blue): H,K,R; negative (red): D,E; neutral (green): S,T,N,Q; aliphatic (grey): A,V,L,I,M; aromatic (purple): F,Y,W; Pro & Gly (orange): P,G. ), salt bridges ( ), and non-bonded ( ) interactions. Chain A represents hACE2 and chain B represents RBD Beta. Residue colors: positive (blue): H,K,R; negative (red): D,E; neutral (green): S,T,N,Q; aliphatic (grey): A,V,L,I,M; aromatic (purple): F,Y,W; Pro & Gly (orange): P,G.
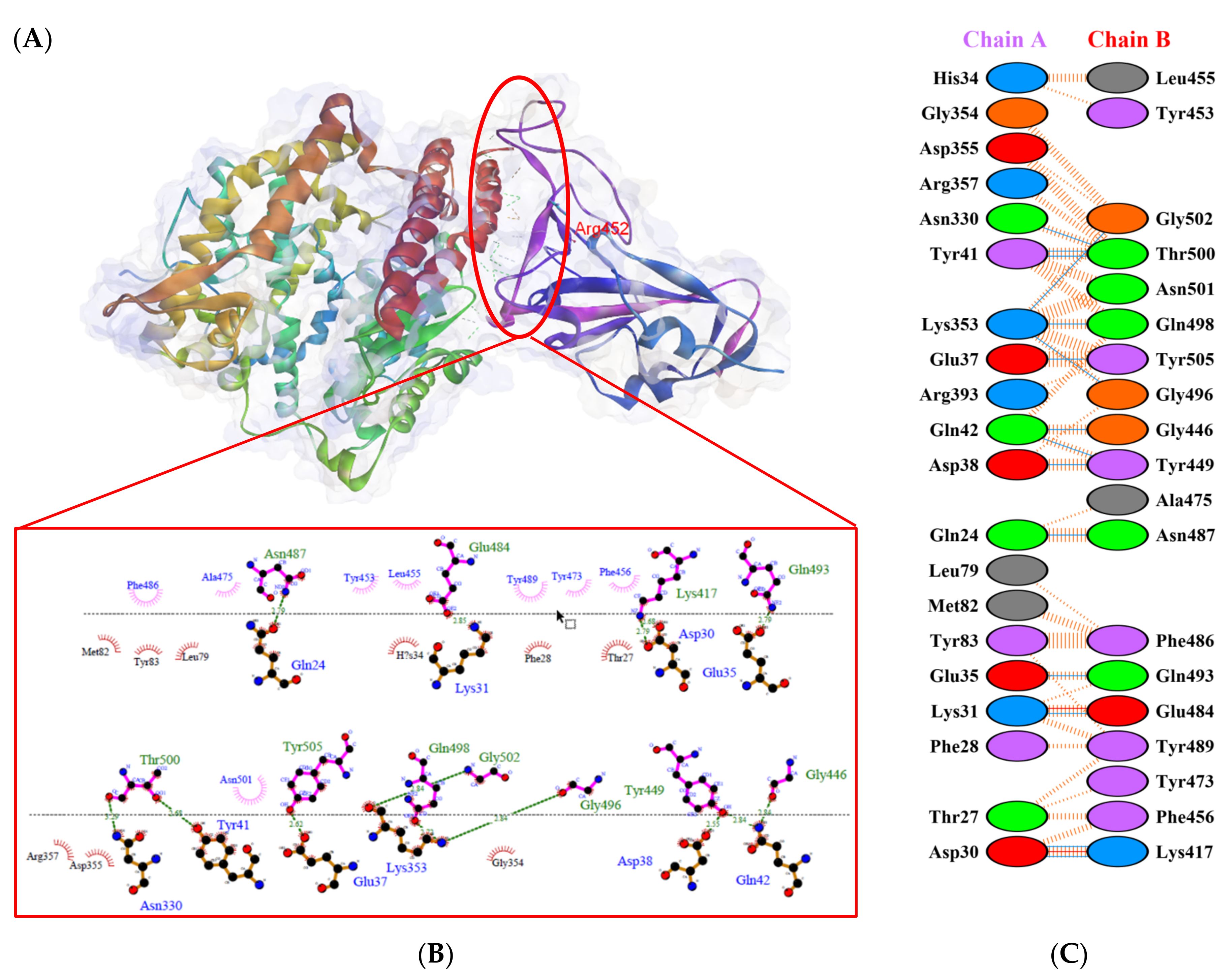
), salt bridges ( ), and non-bonded ( ) interactions. Chain A represents hACE2 and chain B represents RBD Beta. Residue colors: positive (blue): H,K,R; negative (red): D,E; neutral (green): S,T,N,Q; aliphatic (grey): A,V,L,I,M; aromatic (purple): F,Y,W; Pro & Gly (orange): P,G.
), salt bridges ( ), and non-bonded ( ) interactions. Chain A represents hACE2 and chain B represents RBD Beta. Residue colors: positive (blue): H,K,R; negative (red): D,E; neutral (green): S,T,N,Q; aliphatic (grey): A,V,L,I,M; aromatic (purple): F,Y,W; Pro & Gly (orange): P,G.
), salt bridges ( ), and non-bonded ( ) interactions. Chain A represents hACE2 and chain B represents RBD Beta. Residue colors: positive (blue): H,K,R; negative (red): D,E; neutral (green): S,T,N,Q; aliphatic (grey): A,V,L,I,M; aromatic (purple): F,Y,W; Pro & Gly (orange): P,G. ), salt bridges ( ), and non-bonded ( ) interactions. Chain A represents hACE2 and chain B represents RBD Alpha. Residue colors: positive (blue): H,K,R; negative (red): D,E; neutral (green): S,T,N,Q; aliphatic (grey): A,V,L,I,M; aromatic (purple): F,Y,W; Pro & Gly (orange): P,G.
), salt bridges ( ), and non-bonded ( ) interactions. Chain A represents hACE2 and chain B represents RBD Alpha. Residue colors: positive (blue): H,K,R; negative (red): D,E; neutral (green): S,T,N,Q; aliphatic (grey): A,V,L,I,M; aromatic (purple): F,Y,W; Pro & Gly (orange): P,G.
), salt bridges ( ), and non-bonded ( ) interactions. Chain A represents hACE2 and chain B represents RBD Alpha. Residue colors: positive (blue): H,K,R; negative (red): D,E; neutral (green): S,T,N,Q; aliphatic (grey): A,V,L,I,M; aromatic (purple): F,Y,W; Pro & Gly (orange): P,G.
), salt bridges ( ), and non-bonded ( ) interactions. Chain A represents hACE2 and chain B represents RBD Alpha. Residue colors: positive (blue): H,K,R; negative (red): D,E; neutral (green): S,T,N,Q; aliphatic (grey): A,V,L,I,M; aromatic (purple): F,Y,W; Pro & Gly (orange): P,G. ), salt bridges ( ), and non-bonded ( ) interactions. Chain A represents hACE2 and chain B represents RBD Epsilon. Residue colors: positive (blue): H,K,R; negative (red): D,E; neutral (green): S,T,N,Q; aliphatic (grey): A,V,L,I,M; aromatic (purple): F,Y,W; Pro & Gly (orange): P,G.
), salt bridges ( ), and non-bonded ( ) interactions. Chain A represents hACE2 and chain B represents RBD Epsilon. Residue colors: positive (blue): H,K,R; negative (red): D,E; neutral (green): S,T,N,Q; aliphatic (grey): A,V,L,I,M; aromatic (purple): F,Y,W; Pro & Gly (orange): P,G.
), salt bridges ( ), and non-bonded ( ) interactions. Chain A represents hACE2 and chain B represents RBD Epsilon. Residue colors: positive (blue): H,K,R; negative (red): D,E; neutral (green): S,T,N,Q; aliphatic (grey): A,V,L,I,M; aromatic (purple): F,Y,W; Pro & Gly (orange): P,G.
), salt bridges ( ), and non-bonded ( ) interactions. Chain A represents hACE2 and chain B represents RBD Epsilon. Residue colors: positive (blue): H,K,R; negative (red): D,E; neutral (green): S,T,N,Q; aliphatic (grey): A,V,L,I,M; aromatic (purple): F,Y,W; Pro & Gly (orange): P,G.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variants | Most Favored Region (%) | Additional Allowed Region (%) | Generously Allowed Region (%) | Disallowed Region (%) |
|---|---|---|---|---|
| Wild-type * | 89.3 | 10.7 | 0.0 | 0.0 |
| Gamma | 90.5 | 9.5 | 0.0 | 0.0 |
| Delta | 90.5 | 9.5 | 0.0 | 0.0 |
| Beta | 92.3 | 7.7 | 0.0 | 0.0 |
| Alpha | 91.7 | 8.3 | 0.0 | 0.0 |
| Epsilon | 90.5 | 9.5 | 0.0 | 0.0 |
| hACE2 Residues | RBD Residues | |||||
|---|---|---|---|---|---|---|
| WT | Gamma | Delta | Beta | Alpha | Epsilon | |
| Ser19 | - | - | Ala475 | - | - | - |
| Gln24 | Asn487 | Asn487 | Asn487 | Asn487 | - | Asn487 |
| Asp30 | Lys417 | - | Lys417 | - | Lys417 | Lys417 |
| Lys31 | - | - | - | - | Glu484 | Glu484 |
| - | Gln493 | Gln493 | Gln493 | Gln493 | - | |
| Glu35 | - | Gln493 | Gln493 | Gln493 | Gln493 | Gln493 |
| Glu37 | - | - | - | - | - | Tyr505 |
| Asp38 | Tyr449 | Tyr449 | Tyr449 | Tyr449 | Tyr449 | Tyr449 |
| Tyr41 | Thr500 | Thr500 | Thr500 | Thr500 | - | Thr500 |
| Gln42 | Gly446 | - | Gly446 | Gly446 | Gly446 | Gly446 |
| Tyr449 | Tyr449 | Tyr449 | Tyr449 | Tyr449 | Tyr449 | |
| - | - | - | - | Gln498 | - | |
| Tyr83 | Asn487 | - | - | - | Asn487 | - |
| - | - | Tyr489 | - | - | - | |
| Asn330 | - | Thr500 | - | - | Thr500 | |
| Lys353 | Gly496 | - | Gly496 | Gly496 | - | Gly496 |
| - | - | Gln498 | - | - | Gln498 | |
| - | Tyr501 | - | - | - | - | |
| Gly502 | Gly502 | Gly502 | Gly502 | Gly502 | Gly502 | |
| Hydrogen Bonds | WT | Gamma | Delta | Beta | Alpha | Epsilon | |
|---|---|---|---|---|---|---|---|
| Donor | Acceptor | Occupancy | |||||
| Tyr83-side | Asn87-side | 49.75% | 47.95% | 46.95% | 48.85% | - | 47.65% |
| Gln493-side | Glu35-side | 47.05% | 47.35% | 47.65% | 43.16% | - | 42.96% |
| Tyr505-side | Glu37-side | 45.85% | 9.39% | 2.20% | 7.49% | - | 45.55% |
| Thr500-side | Asp355-side | 33.47% | 53.35% | 37.16% | 46.05% | - | 26.77% |
| Gly502-main | Lys353-main | 30.77% | 44.66% | 28.47% | 48.55% | - | 40.56% |
| Lys417-side | Asp30-side | 27.97% | - | 10.29% | - | 30.67% | 27.27% |
| Ser19-side | Ala475-main | 26.67% | 33.87% | 36.26% | 1.90% | 2.50% | 4.20% |
| Lys31-side | Gln493-side | 20.88% | 7.99% | 12.69% | 8.39% | - | 9.89% |
| Thr500-side | Tyr41-side | 18.98% | 4.60% | 13.09% | 5.79% | - | 26.17% |
| Tyr489-side | Tyr83-side | 18.88% | - | - | - | - | - |
| Lys353-side | Tyr495-main | 13.19% | - | 4.90% | 0.20% | - | 16.48% |
| Tyr449-side | Asp38-side | 3.00% | 35.76% | 48.95% | 43.16% | 27.27% | 60.04% |
| Tyr501-side | Asp38-side | - | 14.89% | - | - | - | - |
| Ser477-side | Glu23-side | - | 10.99% | - | 1.70% | - | - |
| Tyr505-side | Ala386-main | - | - | 19.68% | 1.70% | - | - |
| Gln498-side | Asp355-side | - | - | 15.88% | - | - | - |
| Tyr453-side | His34-side | - | 0.30% | 0.10% | 32.97% | 3.50% | 0.20% |
| Van der Waal Energy kJ/mol | Electrostatic Energy kJ/mol | Polar Solvation Energy kJ/mol | SASA Energy kJ/mol | Binding Energy kJ/mol | |
|---|---|---|---|---|---|
| WT | −383,845 (2.17) | −1,427,047 (7.577) | 820,013 (18.49) | −45,734 (0.374) | −1,036,072 (14.875) |
| Gamma | −395,679 (2.335) | −1,757,205 (5.821) | 473,056 (14.625) | −4463 (0.335) | −1,723.82 (15.445) |
| Delta | −371,792 (2.857) | −2,384,008 (6.377) | 705,859 (12.583) | −46,789 (0.346) | −2,097,241 (11.978) |
| Beta | −379,902 (2.127) | −1,718,065 (7.052) | 542,671 (12.887) | −44,012 (0.403) | −1,599,527 (12.756) |
| Alpha | −366.51 (2.228) | −1,358,149 (9.501) | 701,388 (16.644) | −4414 (0.371) | −1,069.033 (15.474) |
| Epsilon | −364,326 (2.824) | −1972,844 (7.259) | 970,525 (13.802) | −4525 (0.394) | −1412,592 (14.606) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Celik, I.; Yadav, R.; Duzgun, Z.; Albogami, S.; El-Shehawi, A.M.; Fatimawali; Idroes, R.; Tallei, T.E.; Emran, T.B. Interactions of the Receptor Binding Domain of SARS-CoV-2 Variants with hACE2: Insights from Molecular Docking Analysis and Molecular Dynamic Simulation. Biology 2021, 10, 880. https://doi.org/10.3390/biology10090880
Celik I, Yadav R, Duzgun Z, Albogami S, El-Shehawi AM, Fatimawali, Idroes R, Tallei TE, Emran TB. Interactions of the Receptor Binding Domain of SARS-CoV-2 Variants with hACE2: Insights from Molecular Docking Analysis and Molecular Dynamic Simulation. Biology. 2021; 10(9):880. https://doi.org/10.3390/biology10090880
Chicago/Turabian StyleCelik, Ismail, Rohitash Yadav, Zekeriya Duzgun, Sarah Albogami, Ahmed M. El-Shehawi, Fatimawali, Rinaldi Idroes, Trina Ekawati Tallei, and Talha Bin Emran. 2021. "Interactions of the Receptor Binding Domain of SARS-CoV-2 Variants with hACE2: Insights from Molecular Docking Analysis and Molecular Dynamic Simulation" Biology 10, no. 9: 880. https://doi.org/10.3390/biology10090880
APA StyleCelik, I., Yadav, R., Duzgun, Z., Albogami, S., El-Shehawi, A. M., Fatimawali, Idroes, R., Tallei, T. E., & Emran, T. B. (2021). Interactions of the Receptor Binding Domain of SARS-CoV-2 Variants with hACE2: Insights from Molecular Docking Analysis and Molecular Dynamic Simulation. Biology, 10(9), 880. https://doi.org/10.3390/biology10090880