1. Introduction
Single-cell RNA sequencing (scRNA-seq) technology is very popular in biomedical research. This technology examines the gene expressions among different cells in a tissue and provides the gene expression profile on a cell level [
1]. Compared with the traditional RNA-seq technology, which examines the average gene expressions in a tissue, scRNA-seq is a recent development and a very advanced technology.
scRNA-seq technology is also useful for identifying cellular heterogeneity. On the other hand, scRNA-seq has low capture efficiency and high dropouts compared with regular RNA-seq technologies. Yet, quality control is necessary for removing technical noise from scRNA-seq data [
2]. scRNA-seq protocols can be classified into two categories, full-length transcript sequencing approaches and 3
-end or 5
-end transcript sequencing [
2]. In addition, 3
-end or 5
-end transcript sequencing are known as unique molecular identifier (UMI) tag-based protocols, and full-length transcript sequencing is known as a non-UMI-based protocol [
3]. UMI tag-based scRNA-seq protocols uses UMI tags for different transcript molecules. During the scRNA-seq process, the transcript molecules get attached with their respective UMI tags, and then these UMIs are counted to obtain the number of transcripts for each gene [
4]. UMI tag-based protocols include Drop-seq and 10× Genomics Chromium. Compared with UMI tag-based sequencing, the Non-UMI-based scRNA-seq protocols sequence whole transcripts [
2]. Non-UMI-based protocols include Smart-seq2, MATQ-seq, and Fluidigm C1 [
3].
One of the many applications of scRNA-seq technology is differentiating cell groups by comparing their molecular signatures, for instance, identifying highly differentiated genes to cluster knockout and wild type cells. Similarly, by comparing the gene expression profile of cancer cells with that of healthy cells, one can identify genes with altered expression that might be responsible for cancer. The applications of scRNA-seq technology are numerous and of high impact in genomic research. However, the scRNA-seq data come with certain challenges as well [
5], especially requiring high computation time and resources. Methods dealing with these challenges typically include shifting and scaling, batch effect correction, dimensionality reduction, missing data imputation, and selection of important features. High dimensionality is a major computational challenge in analyzing scRNA-seq data. Therefore, dimensionality reduction methods such as principal component analysis (PCA), T-distributed stochastic neighbor embedding (t-SNE), and feature selection are often performed in scRNA-seq data analysis [
2].
scRNA-seq data usually contain a larger number of features (genes) than the number of samples (cells). People typically represent
p as the dimension of features and
n as the dimension of samples. When
, we call the data having the “large-p-small-n” problem. Statistical models could result in poor prediction performance due to over-fitting when training data contain fewer samples compared to the number of features [
6]. There are several methods to deal with the “large-p-small-n” problem in Machine Learning (ML), of which feature selection is the most useful.
Feature selection is the method of selecting variables that contain better signal than the noise variables for the target variable of interest Random forests, Recursive Feature Elimination (RFE), and penalized regression are often used for feature selection in ML.
In this research, we explored the penalized regression methods to select feature variables over RFE and random forest. The rationale behind this choice is that RFE is computationally expensive when applied to high-dimensional data [
7]. Therefore, RFE is not an ideal choice for feature selection in scRNA-seq data. Random forest and its application on scRNA-seq data have been examined thoroughly in other studies [
8,
9,
10,
11]. Moreover, some penalized regression methods are worth exploring to compare their performances for feature selection in scRNA-seq data because these methods were developed primarily for tackling the challenges of “large-p-small-n” problems [
12].
Some popular variants of penalized regression in machine learning are ridge regression, least absolute shrinkage and selection operator (lasso) [
13], and elastic net regression [
14], where the latter is a combination of ridge and lasso. The applicability of these methods depends on the problem one is addressing in modeling scRNA-seq data. Ridge regression reduces the dimension of feature variables by making their coefficients closer to 0. The features for which the coefficients are close to zero are forced to be excluded while the remaining features are retained in the model. This notion of feature selection using ridge regression is known as hard thresholding. Lasso regression [
13] implements soft thresholding for feature selection. In soft thresholding, the regression coefficients are made exactly equal to zero. The limitation of penalized regression such as ridge, lasso, and elastic net regression is that these algorithms do not consider any previously known relationships between the features, such as the grouping information of the genes in the scRNA-seq data, while selecting them [
15,
16]. There are improved lasso methods such as group lasso [
17] and sparse group lasso [
18] that allow us to use grouping information of features, which could account for the interrelationship of variables. Drop lasso [
12] and big lasso [
19] are two other interesting variants of penalized regression. Our primary objective in this article is to explore scRNA-seq data specific to different species using the collection of penalized regression methods, and determine which method is more suitable for the problem under study.
There are some other variants of lasso regression such as nuisance penalized regression [
20], fused lasso [
21], adaptive lasso [
22], and prior lasso [
23]. Nuisance penalized regression is suitable when some of the feature variables are of particular interest and others are considered noise (nuisance). However, this distinction between genes is not always known in advance. In this study, we are comparing methods that do not require prior knowledge of genes for feature selection. Therefore, nuisance penalized regression is not selected for analysis in our research. While prior lasso can be applied to biological data, it requires prior information to be incorporated [
23]. Fused lasso was proposed for processing image-based time series data, which is very different from scRNA-seq application. Meanwhile, adaptive lasso incorporates penalty to proportional hazards regression. These methods may not be suitable for our applications. Therefore, they are excluded from our study.
There exist studies in penalized regression methods for high-dimensional data [
24,
25]. However, these studies did not include a performance comparison of some of the selected methods for our research. The performance of all the methods are thoroughly investigated in our research, which may not have been done in scRNA-seq data before. The compelling rationale for our research is to fill this gap in knowledge and form a general recommendation on the performance of these methods. Furthermore, we propose a new method (an algorithm for feature selection using penalized regression methods) that uses fewer genes to execute the best of the selected methods and improve its prediction performance measured via AUC. This study is an extension of the research presented at the Comparative Genomics: 19th International Conference, RECOMB-CG 2022, La Jolla, CA, USA, 20–21 May 2022, Proceedings [
26]. The R codes for the analysis are provided in
Appendix A.
The rest of this article is organized as follows.
Section 2 explains different methods and metrics used in this study.
Section 3 introduces the scRNA-seq data sets and the research design.
Section 4 and
Section 5 showcase the results, discuss the findings and their biological interpretations, and briefly introduce potential future directions for this research.
4. Results
4.1. Benchmarking Penalized Regression
The first objective of this study is to compare the performance of the selected penalized regression methods. The results in terms of cross-validated AUC (CV-AUC) and computational time are shown in
Table 2 and
Table 3, respectively.
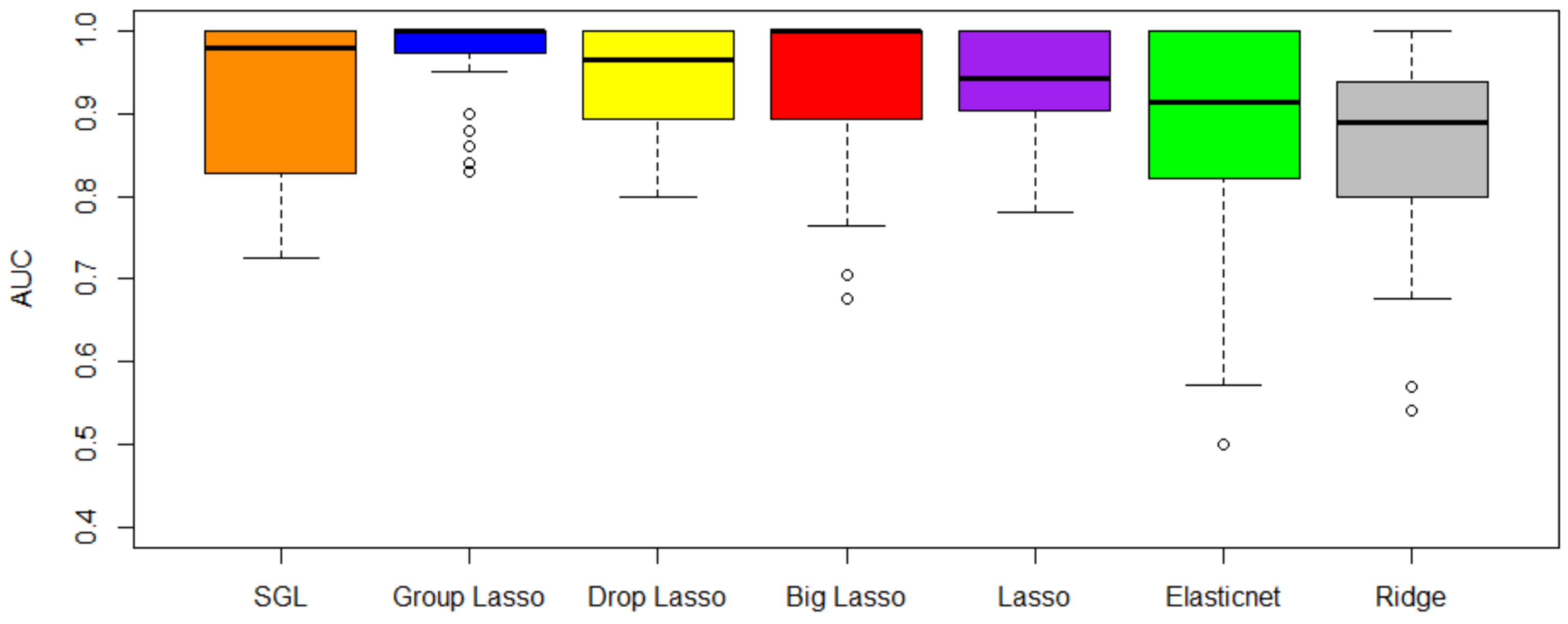
Figure 5 shows the CV-AUC by method for the four data sets. From
Table 2 and
Figure 5, we observe that the top 5 methods in order of importance are SGL, grplasso, droplasso, biglasso, and lasso. As evident from
Table 4, the variance of CV-AUC is close to 0 for all methods when rounded to 2 decimal points. Notice that SGL and grplasso outperform the other methods in terms of CV-AUC, whereas ridge regression method has the least CV-AUC. This could be because grplasso and SGL incorporated grouping of genes into the model, whereas ridge regression treats all the genes equally. After performing Friedman test (a non-parametric statistical test) with the CV-AUC results of the 7 methods, we found statistically significant differences between their performance at
p-value = 15%. An Analysis of Variance (ANOVA) test such as the Friedman test alone cannot reveal which of these methods has a significant difference in performance [
47]. Therefore, we use a post-hoc test in the next step. Post-hoc tests are statistical tests carried out on the subgroups in data already tested in other analyses such as an ANOVA test [
47]. A Nemenyi test is one such post-hoc test. A Nemenyi test on our results revealed that the difference in performance is between SGL and ridge regression. However, more data sets may need to be analyzed to further verify these results.
After comparing the computation time of the methods given in
Table 3, it is evident that the SGL method has the least average computation time across the data sets. Ridge regression requires the highest computation time on average across the four data sets. This is because of its computational complexity arising from using all of the features (genes) in the data. SGL can deselect an entire group of genes or some of the genes within a group, which reduces the computational complexity. Data set GSE60749 has lesser computation time than GSE81861 across all methods because it has fewer non-zero coefficients. Average computation time for lasso, elastic net, and big lasso increased with an increase in the number of samples in the data.
4.2. Performance of the Proposed Algorithm for Feature Selection
The second objective of this study was to combine the top-performing penalized regression methods to improve predictive AUC and perform gene selection. From the discussion of the results of the first objective, we observed that SGL and grplasso were better candidates to form a new method. In terms of gene selection, SGL outperforms grplasso. SGL could identify the top important genes in one fold of 10-fold CV, whereas grplasso takes multiple folds to obtain the same result. In other words, the results of top important genes changed from fold to fold of the 10-fold CV for group lasso. Therefore, SGL was chosen over grplasso for the proposed algorithm. SGL achieves better AUC than biglasso in comparable time for data sets of size 63 MB to 252 MB when tested on a computer with a 32 GB processor. Since biglasso did not show a significant improvement in computation time relative to SGL, biglasso is not included in the proposed method.
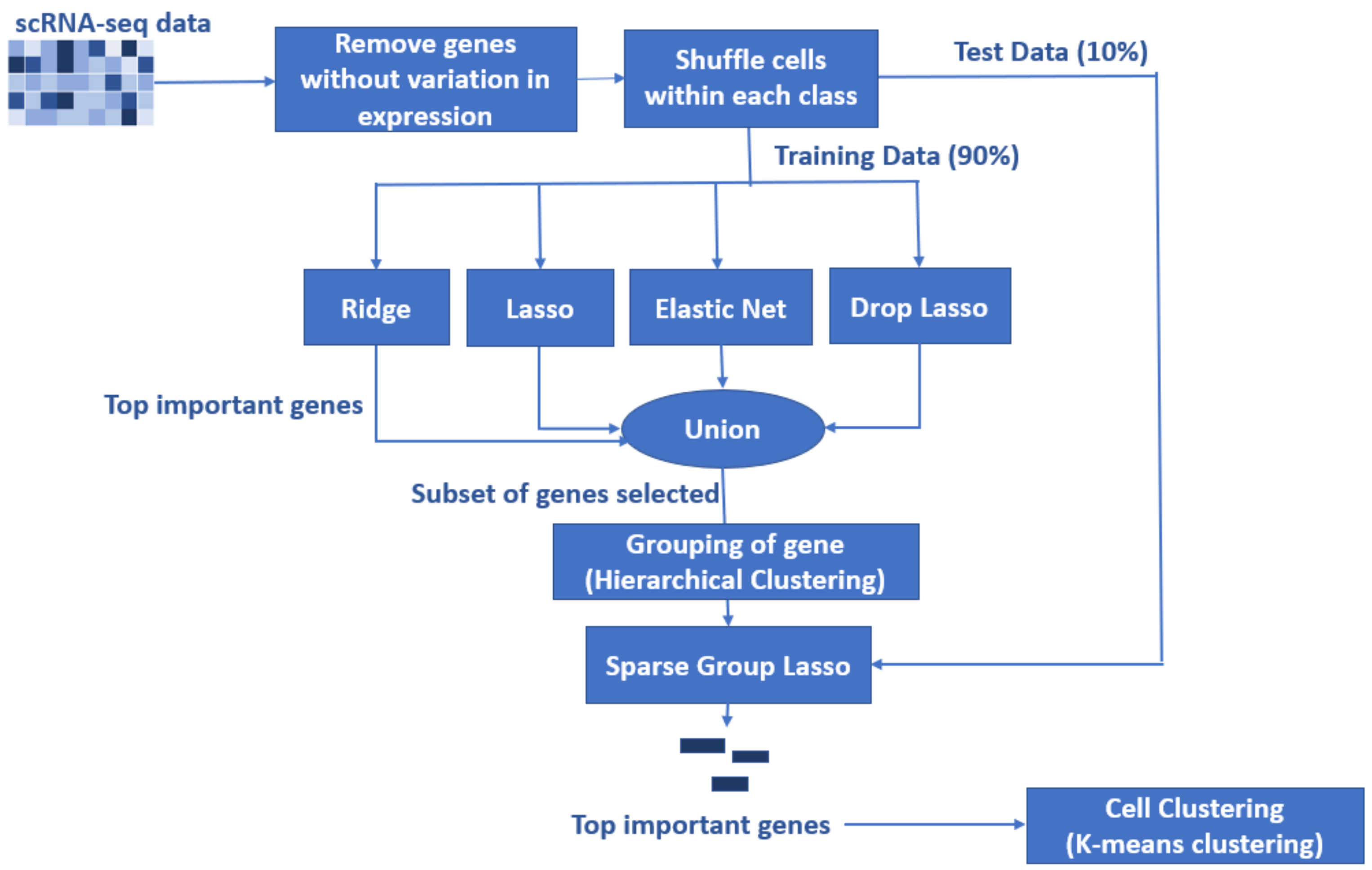
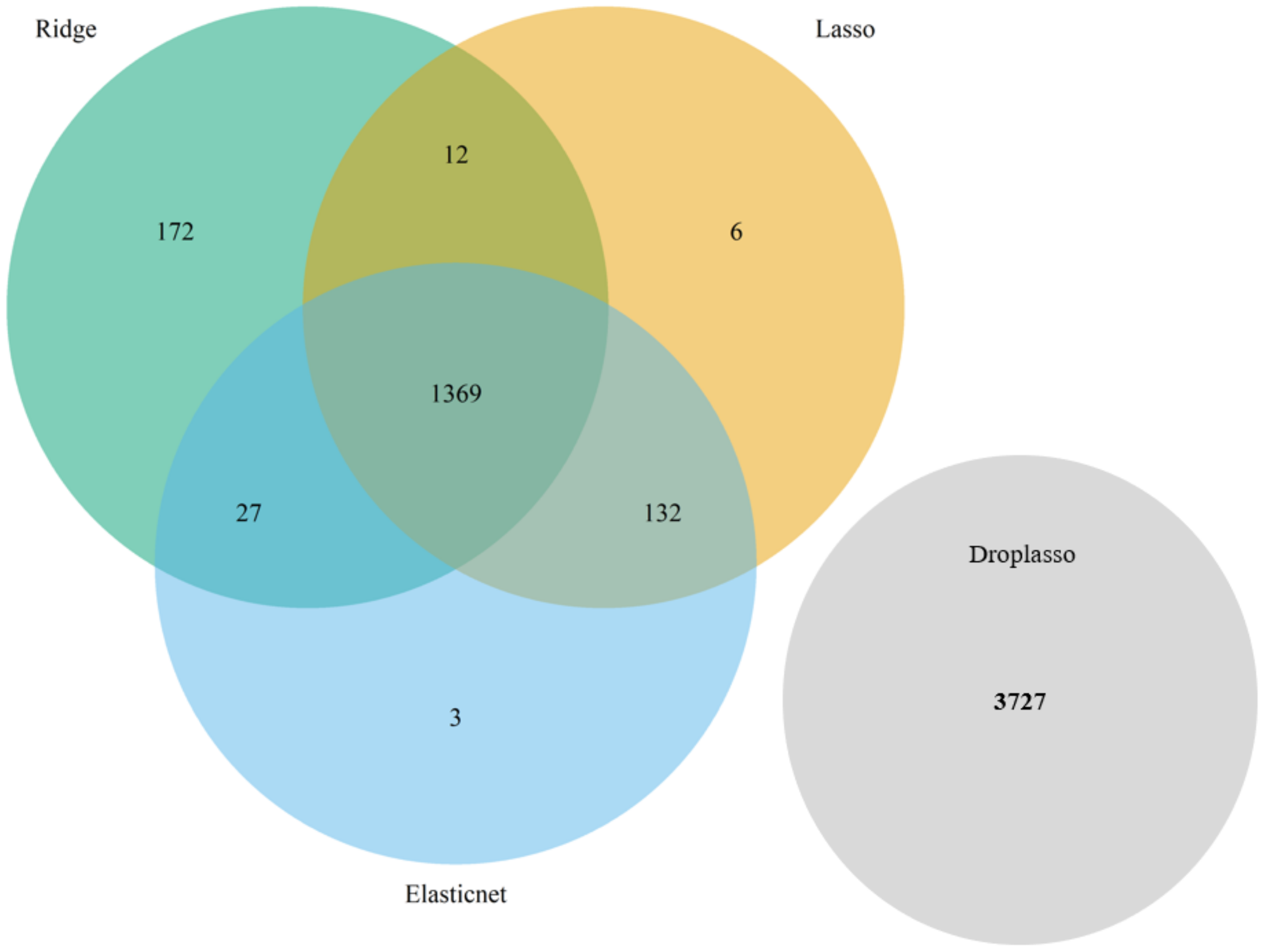
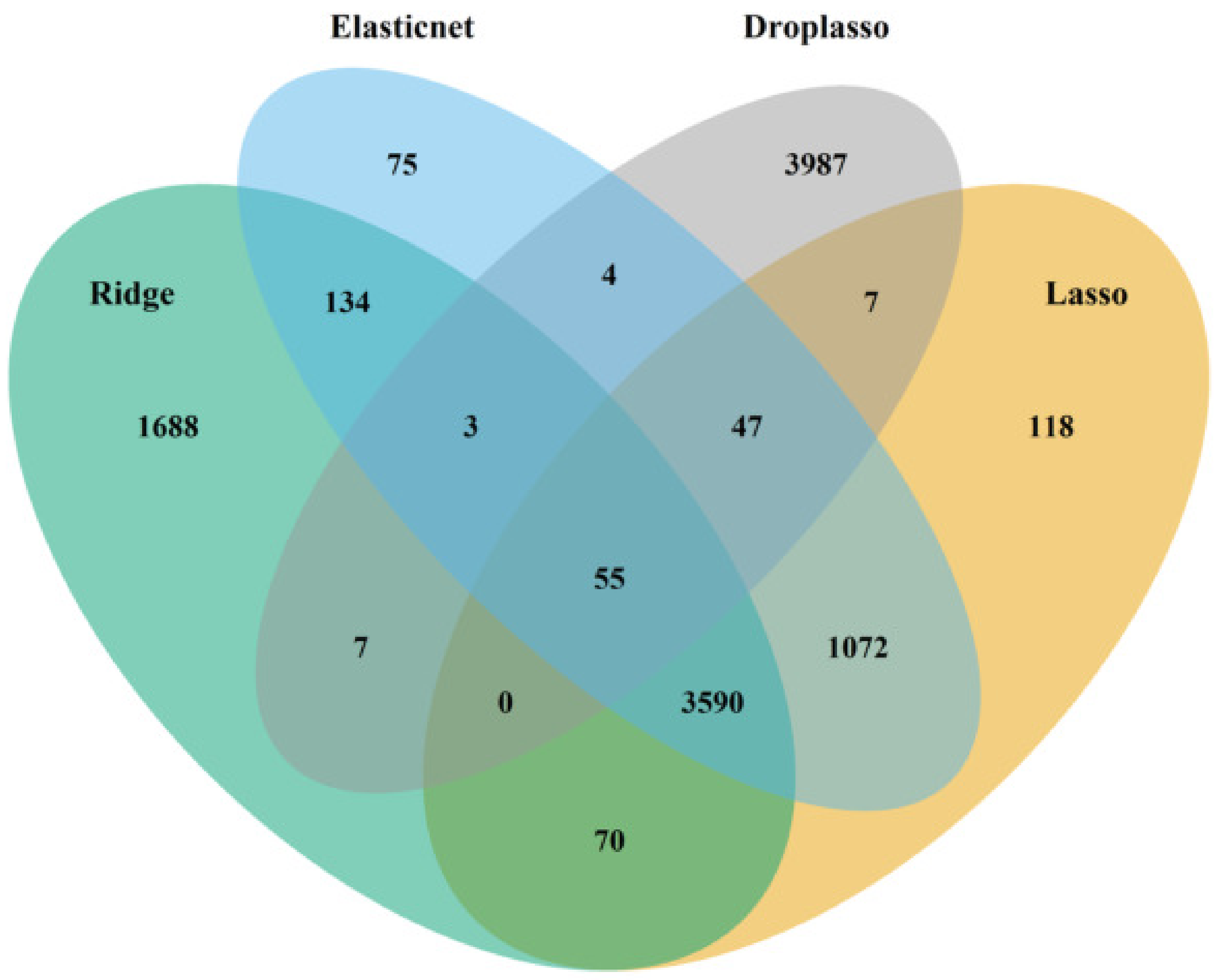
To develop the proposed algorithm using penalized regressions, we selected the ridge, lasso, elastic net, and droplasso to form a filter that produces a gene pool with the number of genes reduced considerably relative to all the available genes. The gene pool was formed by taking a union of the top important genes selected from the four methods as the top important genes have some variations between methods. A union of top important genes is, therefore, more likely to capture the important and differentially expressed genes. The gene pool thus formed is grouped and sent down to SGL to calculate predictive AUC.
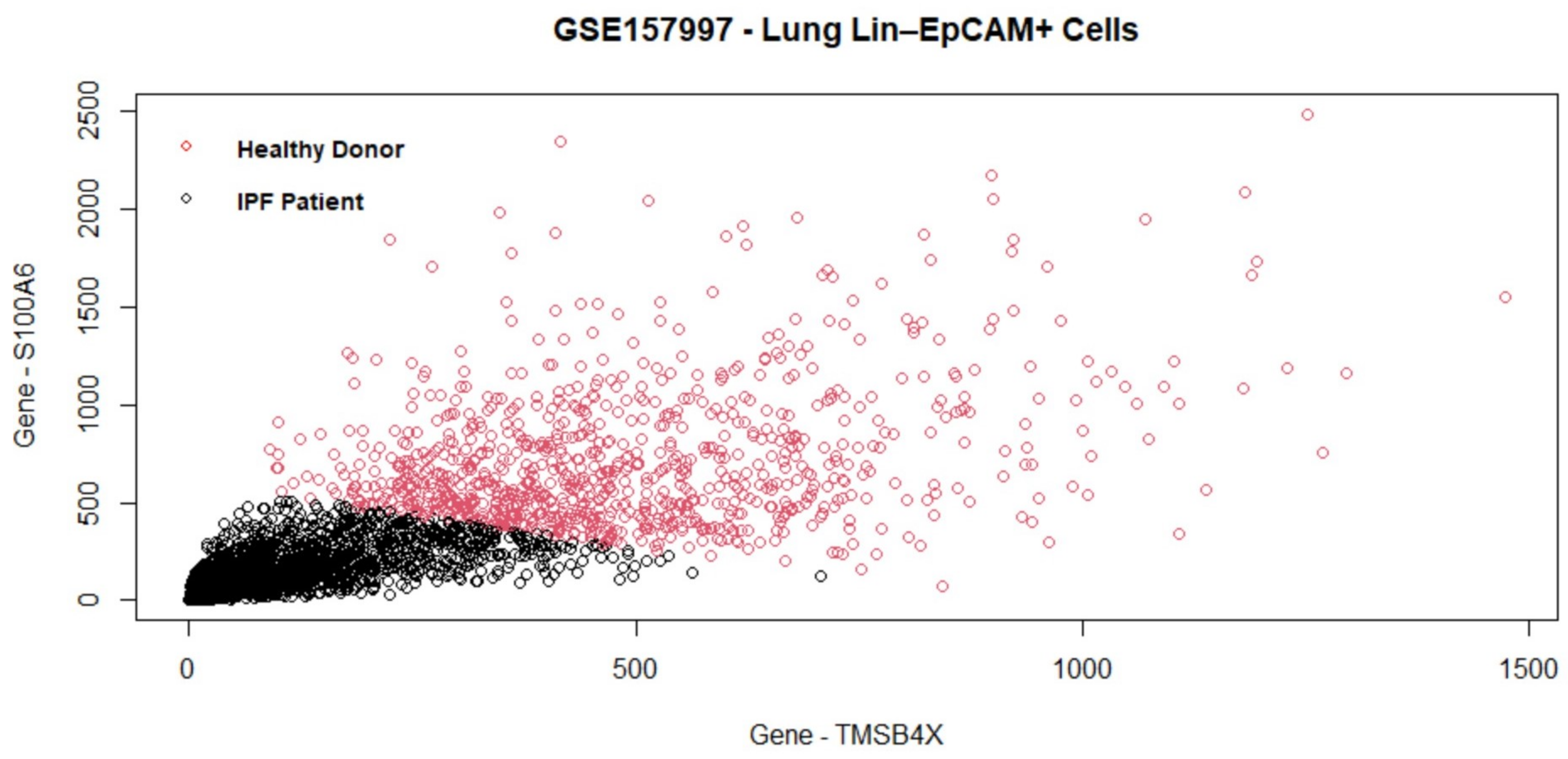
The experimental data used to test the performance of the proposed method include all four data sets in the benchmarking, and also a new independent data set (GSE157997). This data set has the largest number of cells among all five data sets, and was not used to develop the proposed algorithm. It is therefore expected to generate a fair and independent comparison between the proposed method and others.
In the proposed algorithm for feature selection using penalized regression methods, feature selection is performed in two stages. The first stage creates a gene pool that has a significantly reduced number of genes (less than 50%) compared to the original data as shown in
Table 5. Then, the gene pool is grouped using hierarchical clustering and the grouping information is used as input to SGL.
From our experiments, the hierarchical clustering and SGL originally required a 32 GB RAM processor for the computation. With the contribution of the proposed method, large numbers of genes are reduced in the gene pool. We notice that the proposed method can be executed on an 8 GB RAM processor for the following data sets: GSEGSE60749 and GSE71585. The proposed algorithm can be used for other high-dimensional data sets and feature selection as well. As shown in
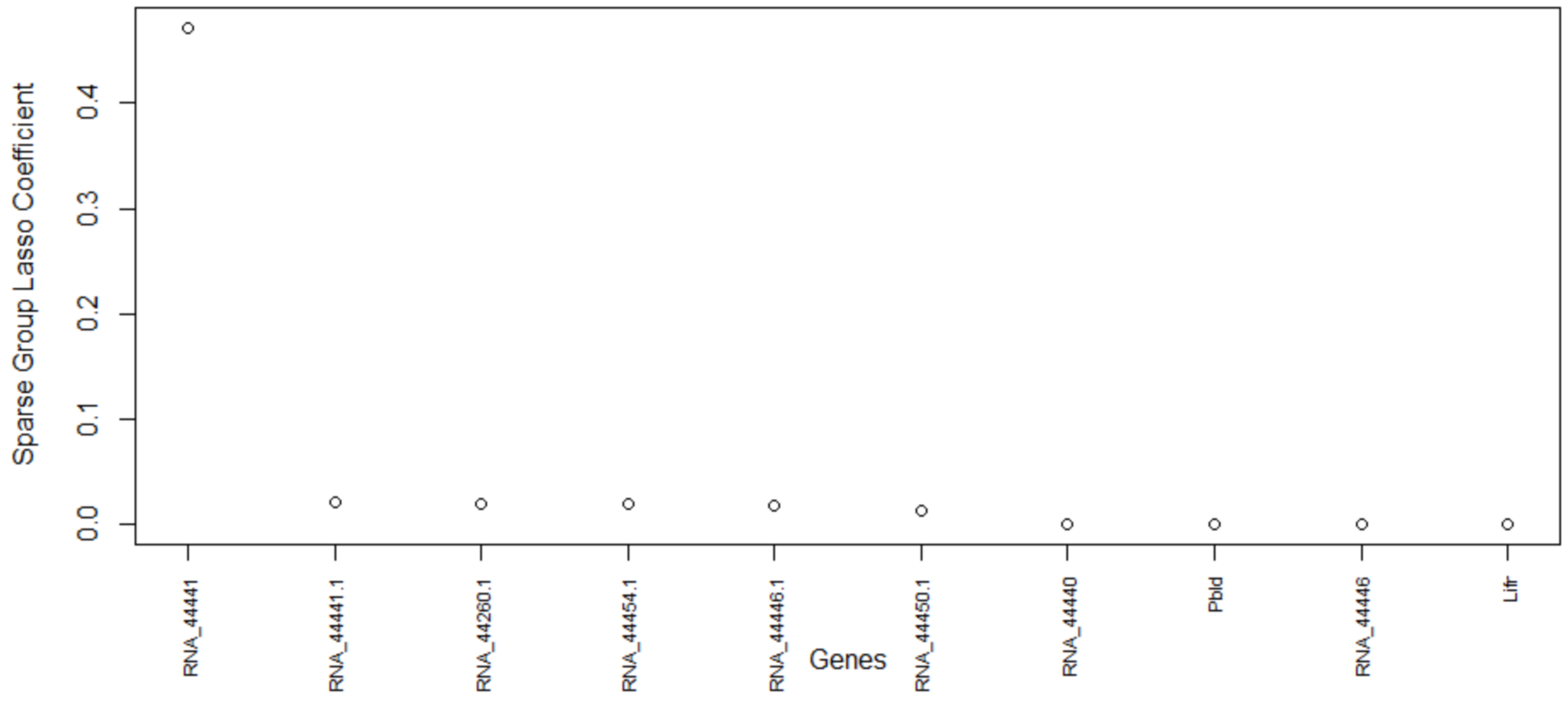
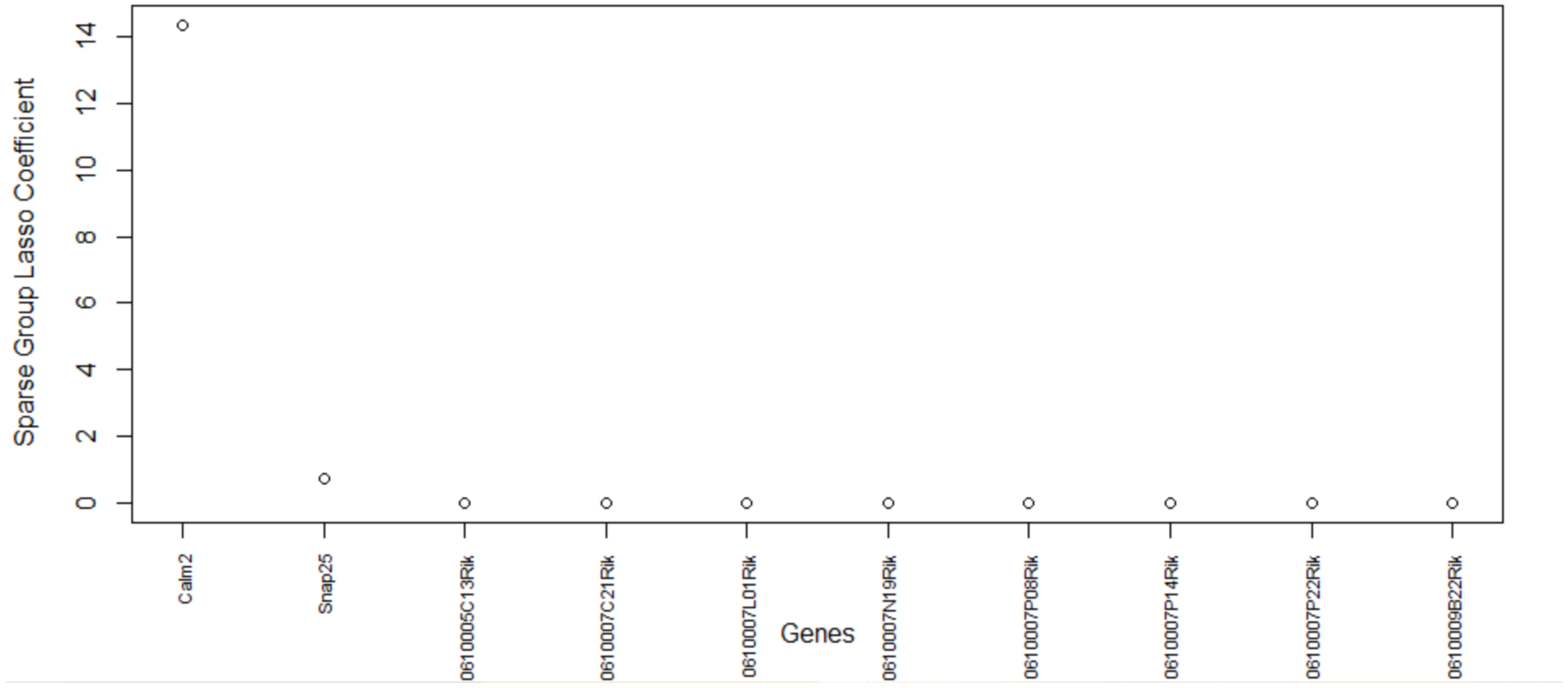
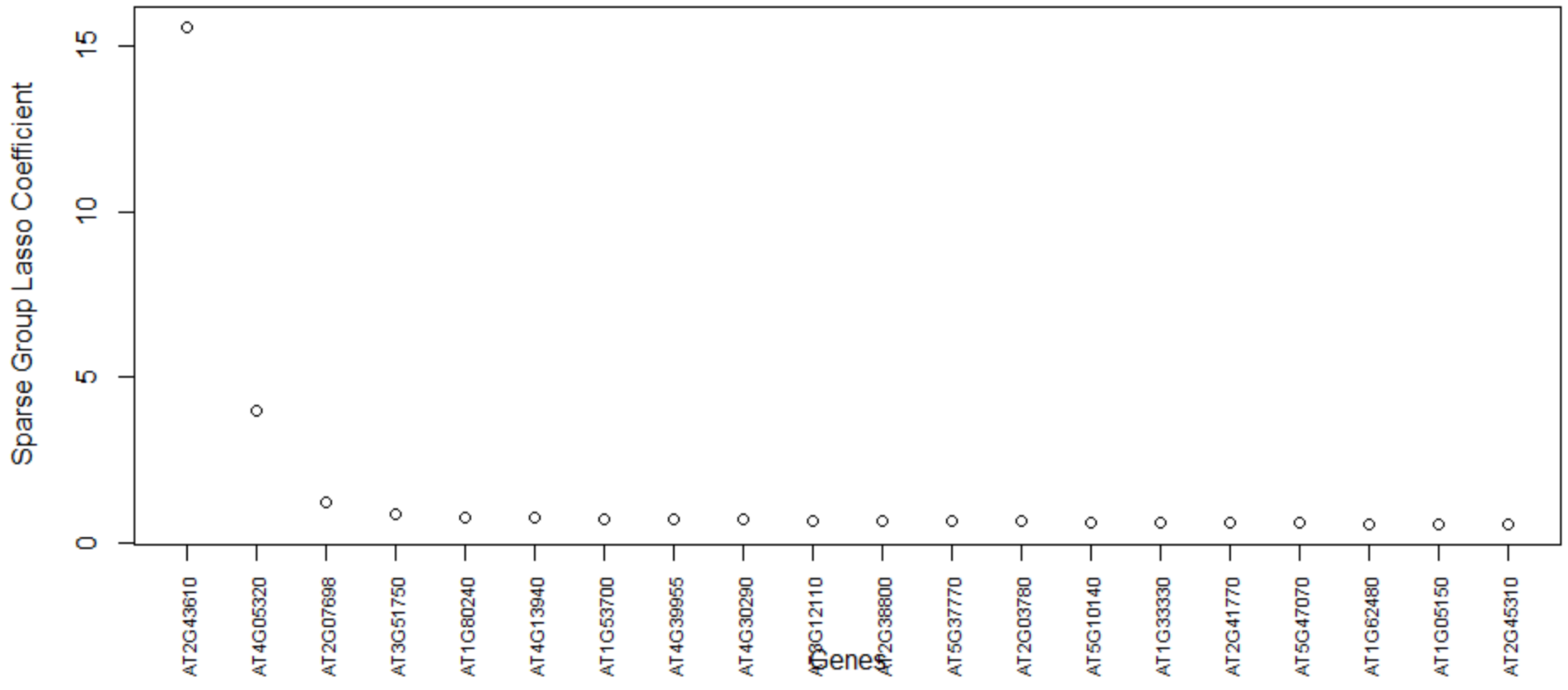
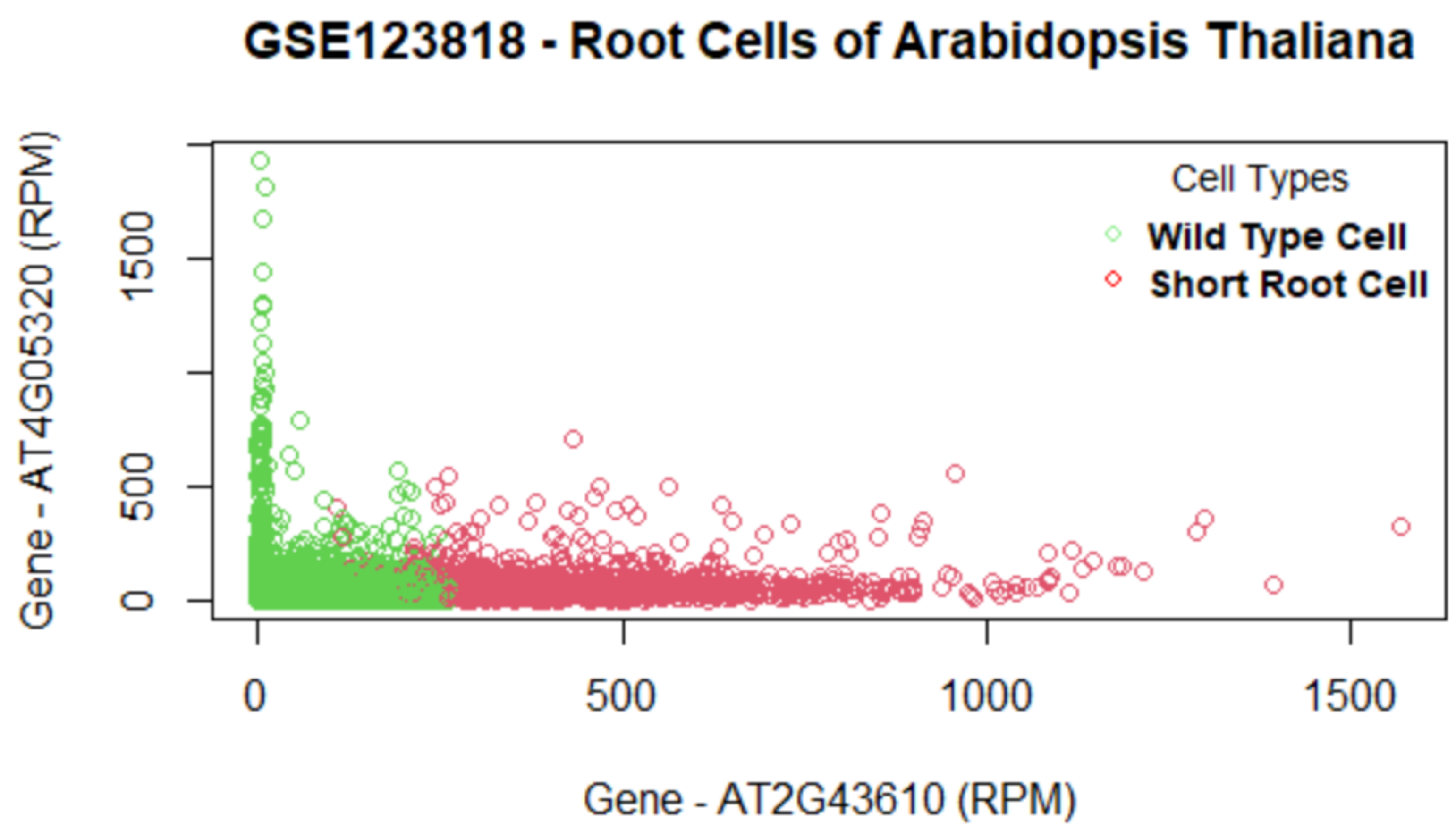
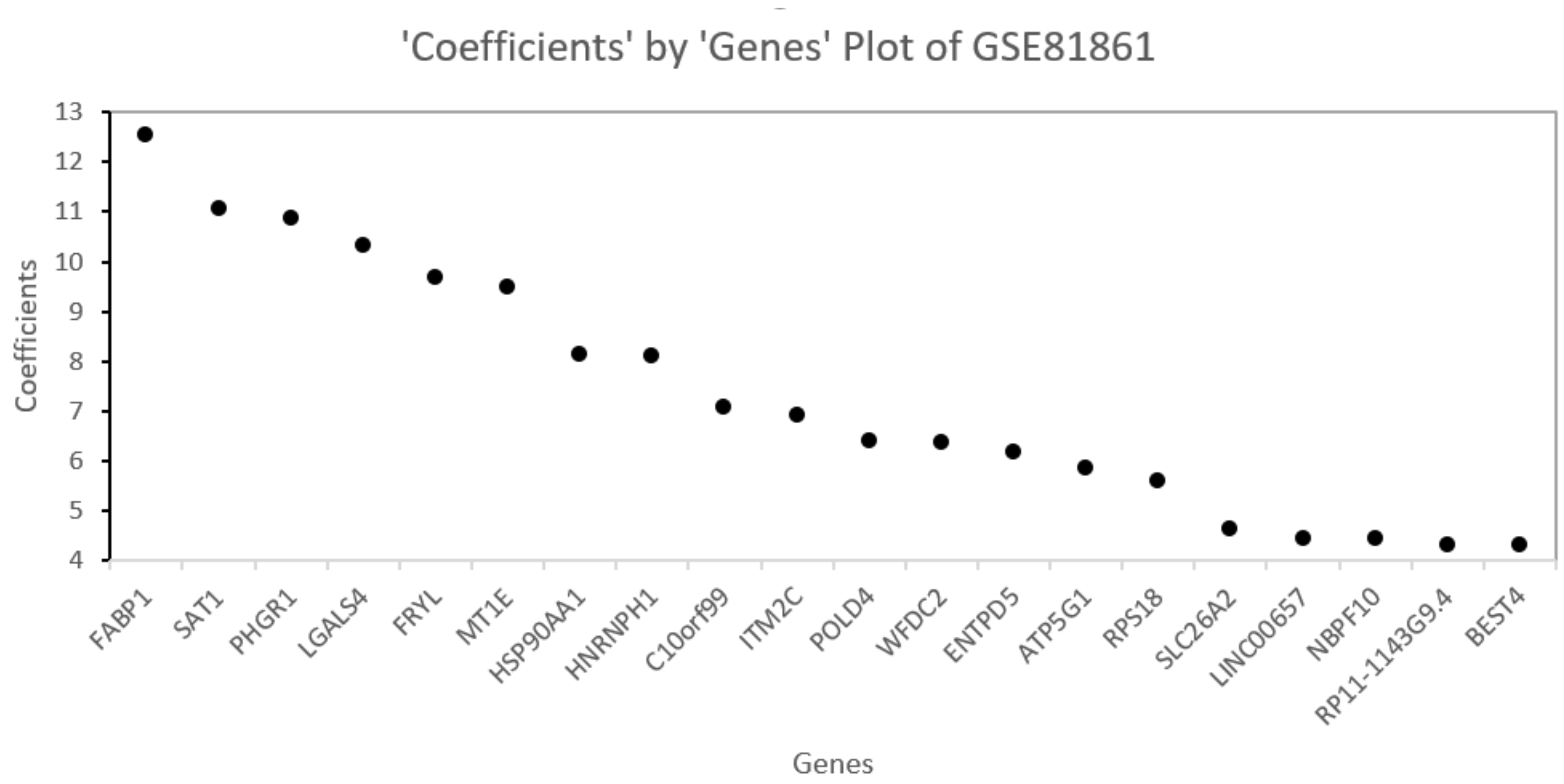
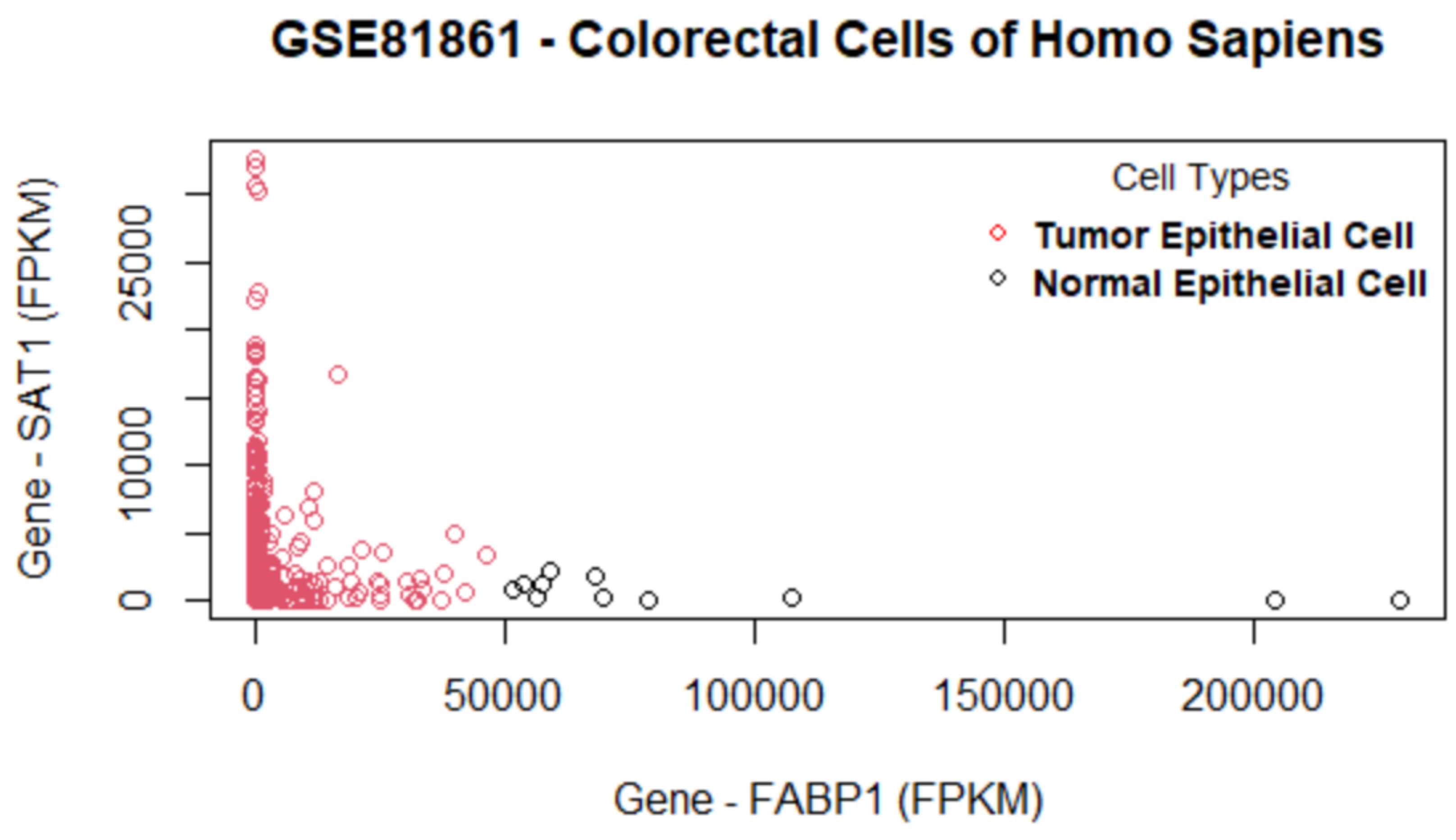
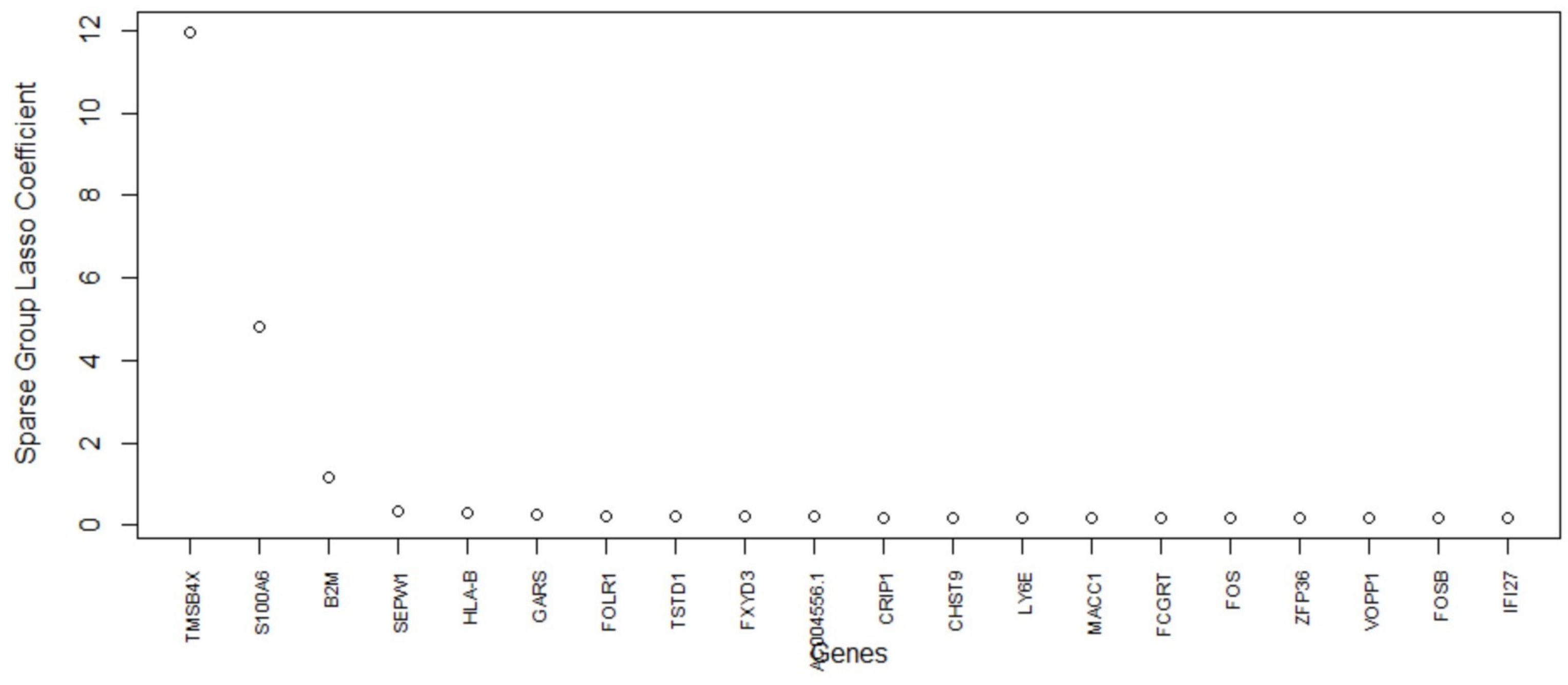
Table 5, this method consistently improves the predictive AUC of SGL. The genes in the SGL model are visualized in a gene versus coefficients plot to select the final subset of genes. Then, this subset of genes is used for cell clustering of each data set using
K-Means clustering.
Essentially, the proposed algorithm tackles the “large-p-small-n” problem by applying feature selection, on the high-dimensional scRNA-seq data, in a sequential fashion. At each stage, different penalized regression methods are used for feature selection. The first stage (ridge, lasso, elastic net, and drop lasso) reduces the number of genes to less than 50% of the original size, and the second stage (SGL) reduces it further to make the number of genes comparable to the number of cells.
6. Conclusions
This study benchmarks several state-of-the-art penalized regression methods on their performance to scRNA-seq classification. Based on the findings of the benchmarking, we proposed an algorithm of penalized regressions, which improved the prediction performance.
The results and findings show that the SGL method outperforms other methods in terms of predictive AUC and computation time. We note that the penalized regression methods can have many hyperparameters, and changes in these hyperparameters may affect the results. For instance, the number of groups of genes and the methods used for grouping the genes can cause differences in predictive AUC, computation time, and final gene selection by group lasso and SGL. The proposed algorithm for feature selection shows a better prediction compared with SGL.
The advantages of our proposed method are two-fold in the sense that it uses hierarchical clustering to find the grouping information of genes that bypasses the need to have much knowledge of genes in scRNA-seq data prior to the execution of SGL, and yet the differentially expressed genes selected by the proposed method and the cell clusters in the data have a strong association. It is because the proposed algorithm for feature selection carries out the feature selection by creating a gene pool based on the union of the top genes from four different methods. This step ensures that any relevant genes that may have been missed by any one of the penalized regression methods are highly likely to be captured by other methods and subsequently included in the gene pool. Due to the sequential nature of analysis and two stages of feature selection, the proposed algorithm may require more time to execute than SGL alone. However, the proposed method uses a smaller subset of genes as input for SGL, thereby reducing computational memory requirements from 32 GB RAM to 8 GB RAM.
This research has the potential to be extended to include more methods and relevant R packages, such as the Seagull [
69], which also implements lasso, group lasso, and sparse group lasso. The proposed method sometimes can be time consuming as it is implemented in a sequential manner. Therefore, further reduction in computational time is possible via parallel computing. The use of other possible machine learning methods such as XGBoost [
70] on top of SGL is a possible research direction worth exploring. In this study, we have explored two groups of cells. Any subpopulations inside the two groups are not investigated. Yet, scRNA-seq data sets may have more than two groups of cells. Therefore, the use of other R packages such as msgl [
71], which can implement the multinomial classification method, is a potential future work to be explored.




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}