
The First Mitochondrial Genomes of the Family Haplodiplatyidae (Insecta: Dermaptera) Reveal Intraspecific Variation and Extensive Gene Rearrangement


 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Insect Collection and DNA Extraction
2.2. Mitogenome Sequencing and Assembly
2.3. Mitogenome Annotation and Analysis
2.4. Phylogenetic Analysis
3. Results
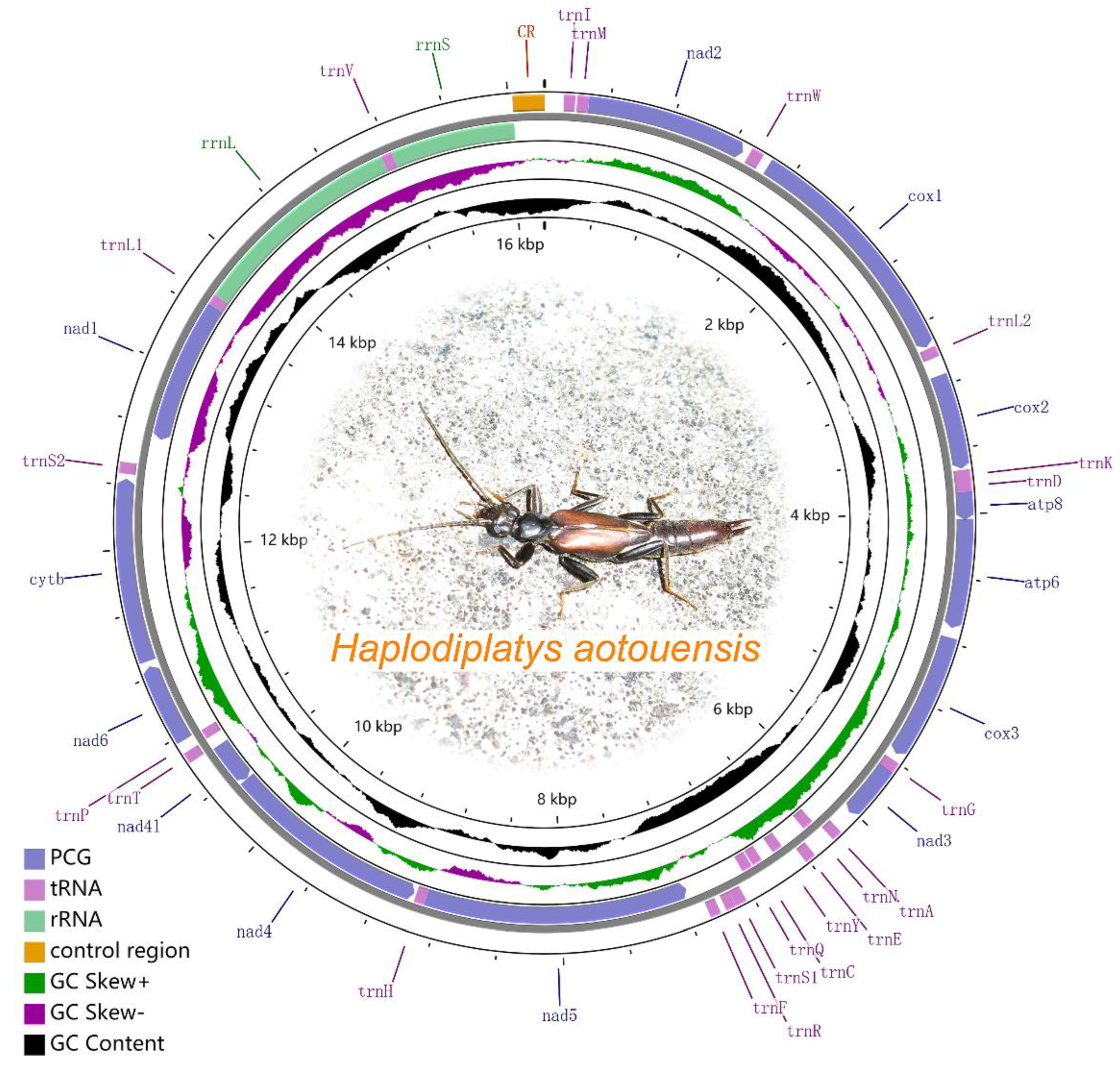
3.1. Mitogenome Structure and Nucleotide Composition
3.2. Gene Rearrangement
3.3. PCGs
3.4. tRNAs, rRNAs and the Control Region
3.5. Phylogenetic Analyses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Haas, F. Biodiversity of Dermaptera. In Insect Biodiversity: Science and Society, 1st ed.; Foottit, G.R., Adler, P.H., Eds.; John Wiley & Sons Ltd.: Hoboken, NJ, USA, 2018; Volume II, pp. 315–334. [Google Scholar]
- Hopkins, H.; Michael, D.M.; Haas, F.; Lesley, S.D. Dermaptera Species File Online. Available online: http://dermaptera.speciesfile.org/HomePage/Dermaptera/HomePage.aspx (accessed on 25 June 2021).
- Haas, F.; Gorb, S.N.; Wootton, R.J. Elastic joints in dermapteran hind wings: Materials and wing folding. Arthropod Struct. Dev. 2000, 29, 137–146. [Google Scholar] [CrossRef]
- Haas, F.; Kukalova-Peck, J. Dermaptera hindwing structure and folding: New evidence for familial, ordinal and superordinal relationships within Neoptera (Insecta). Eur. J. Entomol. 2001, 98, 445–509. [Google Scholar] [CrossRef] [Green Version]
- Haas, F.; Gorb, S.N. Evolution of locomotory attachment pads in the Dermaptera (Insecta). Arthropod Struct. Dev. 2004, 33, 45–66. [Google Scholar] [CrossRef]
- Suzuki, S.; Kitamura, M.; Matsubayashi, K. Matriphagy in the hump earwig, Anechura harmandi (Dermaptera: Forficulidae), increases the survival rates of the offspring. J. Ethol. 2005, 23, 211–213. [Google Scholar] [CrossRef]
- Staerkle, M.; Koelliker, M. Maternal food regurgitation to nymphs in earwigs (Forficula auricularia). Ethology 2008, 114, 844–850. [Google Scholar] [CrossRef]
- Hincks, W.D. (Ed.) A Systematic Monograph of the Dermaptera of the World Based on Material in the British Museum (Natural History). Part One, Pygidicranidae Subfamily Diplatyinae; Natural History Museum: London, UK, 1955; pp. 1–132. [Google Scholar]
- Engel, M.S.; Huang, D.; Thomas, J.C.; Cai, C. A new genus and species of pygidicranid earwigs from the Upper Cretaceous of southern Asia (Dermaptera: Pygidicranidae). Cretac. Res. 2017, 69, 178–183. [Google Scholar] [CrossRef]
- Haas, F. The phylogeny of the Forficulina, a suborder of the Dermaptera. Syst. Entomol. 1995, 20, 85–98. [Google Scholar] [CrossRef]
- Haas, F.; Klass, K.D. The basal phylogenetic relationships in the Dermaptera. In Proceedings of the 1st Dresden Meeting on Insect Phylogeny: Phylogenetic Relationships within the Insect Orders, Dresden, Germany, 19–21 September 2003; Klass, K.D., Ed.; Entomologische Abhandlungen: Dresden, Germany, 2003; pp. 138–142. [Google Scholar]
- Klass, K.D. The female genitalic region in basal earwigs (Insecta: Dermaptera: Pygidicranidae s.l.). Entomol. Abh. 2003, 61, 173–225. [Google Scholar]
- Ross, A.J.; Engel, M.S. The first diplatyid earwig in Tertiary amber (Dermaptera: Diplatyidae): A new species from Miocene Mexican amber. Insect. Syst. Evol. 2013, 44, 157–166. [Google Scholar] [CrossRef]
- Srivastava, G.K. Studies on Oriental Dermaptera preseved in the B. P. Museum, Hawaii, U.S.A. Rec. Zool. Surv. India Occas. Pap. 2003, 210, 1–72. [Google Scholar]
- Ma, W.Z.; Chen, Y.X. The Diplatyidae of China (Dermaptera). Sinozoologoa 1991, 8, 197–202. [Google Scholar]
- Chen, Y.X.; Ma, W.Z. (Eds.) Fauna Sinica, Insecta; Science Press: Beijing, UK, 2004; Volume 35, pp. 1–420. [Google Scholar]
- Wan, X.; Kim, M.I.; Kim, M.J.; Kim, I. Complete mitochondrial genome of the free-living earwig, Challia fletcheri (Dermaptera: Pygidicranidae) and phylogeny of Polyneoptera. PLoS ONE 2012, 7, e42056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beutel, R.; Wipfler, B.; Gottardo, M.; Dallai, R. Polyneoptera or “Lower Neoptera”-New light on old and difficult phylogenetic problems. Atti Accad. Naz. Ital. Entomol. 2013, 61, 113–142. [Google Scholar]
- Naegle, M.A.; Mugleston, J.D.; Bybee, S.M.; Whiting, M.F. Reassessing the phylogenetic position of the epizoic earwigs (Insecta: Dermaptera). Mol. Phylogenet. Evol. 2016, 100, 382–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.T. Comparative mitogenomic analysis of two earwigs (Insecta, Dermaptera) and the preliminary phylogenetic implications. ZooKeys 2022, 1087, 105–122. [Google Scholar] [CrossRef]
- Cameron, S.L. Insect mitochondrial genomics: Implications for evolution and phylogeny. Annu. Rev. Entomol. 2014, 59, 95–117. [Google Scholar] [CrossRef] [Green Version]
- Song, N.; Li, H.; Song, F.; Cai, W. Molecular phylogeny of Polyneoptera (Insecta) inferred from expanded mitogenomic data. Sci. Rep. 2016, 6, 36175. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Ding, S.; Yang, D. The complete mitochondrial genome of a stonefly species, Kamimuria chungnanshana Wu, 1948 (Plecoptera: Perlidae). Mitochondrial DNA Part A 2016, 27, 3810–3811. [Google Scholar] [CrossRef]
- Jin, J.J.; Yu, W.B.; Yang, J.B.; Song, Y.; Depamphilis, C.W.; Yi, T.S.; Li, D.Z. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef]
- Grant, J.R.; Stothard, P. The CGView server: A comparative genomics tool for circular genomes. Nucleic Acids Res. 2008, 36, 181–184. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Bernt, M. CREx: Inferring genomic rearrangements using common intervals. Bioinformatics 2007, 23, 2957–2958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burla, H. Zur Kenntnis der Drosophiliden der Elfenbeinkuste (Franzosisch West-Afrika). Rev. Suisse Zool. 1954, 61, 1–218. [Google Scholar] [CrossRef]
- Clary, D.O.; Wolstenholme, D.R. The ribosomal RNA genes of Drosophila mitochondrial DNA. Nucleic Acids Res. 1985, 13, 4029–4045. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Zhang, Y.; Zhang, Z.; Zhu, J.; Yu, J. KaKs_Calculator 2.0: A toolkit incorporating gamma-series methods and sliding window strategies. Genom. Broteom. Bioinform. 2010, 8, 77–80. [Google Scholar] [CrossRef] [Green Version]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Xia, X. DAMBE5: A comprehensive software package for data analysis in molecular biology and evolution. Mol. Biol. Evol. 2013, 35, 1550–1552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaidya, G.; Lohman, D.J.; Meier, R. SequenceMatrix: Concatenation software for the fast assembly of multi-gene datasets with character set and codon information. Cladistics 2011, 27, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 2016, 34, 772–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lartillot, N.; Lepage, T.; Blanquart, S. PhyloBayes 3: A Bayesian software package for phylogenetic reconstruction and molecular dating. Bioinformatics 2009, 25, 2286–2288. [Google Scholar] [CrossRef] [Green Version]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and postanalysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Rand, D.M.; Kann, L.M. Mutation and selection at silent and replacement sites in the evolution of animal mitochondrial DNA. Genetica 1998, 102/103, 393–407. [Google Scholar] [CrossRef]
- Wei, L.; He, J.; Jia, X.; Qi, Q.; Liang, Z.; Zheng, H.; Ping, Y.; Liu, S.; Sun, J. Analysis of codon usage bias of mitochondrial genome in Bombyx moriand its relation to evolution. BMC Evol. Biol. 2014, 14, 262. [Google Scholar] [CrossRef] [Green Version]
- Fauron, C.M.R.; Wolstenholme, D.R. Extensive diversity among Drosophila species with respect to nucleotide sequences within the adenine+thymine-rich region of mitochondrial DNA molecules. Nucleic Acids Res. 1980, 8, 2439–2452. [Google Scholar] [CrossRef] [Green Version]
- Inohira, K.; Hara, T.; Matsuura, E.T. Nucleotide sequence divergence in the A+T-rich region of mitochondrial DNA in Drosophila simulans and Drosophila mauritiana. Mol. Biol. Evol. 1997, 14, 814–822. [Google Scholar] [CrossRef]
- Burr, M. Observations on the Dermatoptera, including revisions of several genera, and descriptions of new genera and species. Trans. Am. Entomol. Soc. 1904, 5, 277–322. [Google Scholar] [CrossRef]
- Bormans, A.D.E. Viaggio di Leonardo Fea in Birmaniae regioni vicne. LXI. Dermapters (2nd Partie). Anna. Mus. Civ. Stor. Nat. Giacomo Doria 1894, 14, 371–409. [Google Scholar]
- Garey, J.R.; Wolstenholme, D.R. Platyhelminth mitochondrial DNA: Evidence for early evolutionary origin of a tRNAserAGN that contains a dihydrouridine arm replacement loop, and of serine-specifying AGA and AGG codons. J. Mol. Evol. 1989, 28, 374–387. [Google Scholar] [CrossRef]
- Aboul-Ela, F.; Koh, D.; Tinoco, I., Jr.; Martin, F.H. Base-base mismatches. Thermodynamics of double helix formation for dCA3XA3G+ dCT3YT3G (X, Y = A, C, G, D. Nucleic Acids Res. 1985, 13, 4811–4824. [Google Scholar] [CrossRef] [PubMed]
- Erdmann, V.A.; Wolters, J.; Huysmans, E.; De Wachter, R. Collection of published 5S, 5.8s and 4.5s ribosomal RNA sequences. Nucleic Acids Res. 1985, 13, 105–153. [Google Scholar] [CrossRef] [Green Version]
- Simmons, M.P.; Ochoterena, H.; Freudenstein, J.V. Amino acid vs. nucleotide characters: Challenging preconceived notions. Mol. Phylogenet. Evol. 2002, 24, 78–90. [Google Scholar] [CrossRef]
- Simmons, M.P.; Carr, T.G.; O’Neill, K. Relative character-state space, amount of potential phylogenetic information, and heterogeneity of nucleotide and amino acid characters. Mol. Phylogenet. Evol. 2004, 32, 913–926. [Google Scholar] [CrossRef]
- Regier, J.C.; Shultz, J.W. Elongation factor-2: A useful gene for arthropod phylogenetics. Mol. Phylogenet. Evol. 2001, 20, 136–148. [Google Scholar] [CrossRef] [Green Version]
- Wipfler, B.; Koehler, W.; Frandsen, P.B.; Donath, A.; Liu, S.; Machida, R.; Misof, B.; Peters, R.S.; Shimizu, S.; Zhou, X.; et al. Phylogenomics changes our understanding about earwig evolution. Syst. Entomol. 2020, 45, 516–526. [Google Scholar] [CrossRef]
- Flook, P.K.; Klee, S.; Rowell, C.H.F. Combined molecular phylogenetic analysis of the Orthoptera (Arthropoda, Insecta) and implications for their higher systematics. Syst. Biol. 1999, 48, 233–253. [Google Scholar] [CrossRef] [Green Version]
- Castro, L.R.; Dowton, M. The position of the Hymenoptera within the Holometabola as inferred from the mitochondrial genome of Perga condei (Hymenoptera: Symphyta: Pergidae). Mol. Phylogenet. Evol. 2005, 34, 469–479. [Google Scholar] [CrossRef] [PubMed]
- Galtier, N.; Nabholz, B.; Glémin, S.; Hurst, G.D.D. Mitochondrial DNA as a marker of molecular diversity: A reappraisal. Mol. Ecol. 2009, 18, 4541–4550. [Google Scholar] [CrossRef] [PubMed]
- Kocarek, P.; John, V.; Hulva, P. When the body hides the ancestry: Phylogeny of morphologically modified epizoic earwigs based on molecular evidence. PLoS ONE 2013, 8, e66900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarvis, K.J.; Haas, F.; Whiting, M.F. Phylogeny of earwigs (Insecta: Dermaptera) based on molecular and morphological evidence: Reconsidering the classification of Dermaptera. Syst. Entomol. 2005, 30, 442–453. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family | Species | Length (bp) | A + T% | Accession Number | Reference |
|---|---|---|---|---|---|
| Haplodiplatyidae | Haplodiplatys aotouensis Ma & Chen, 1991 (Fujian) | 16,134 | 71.7 | ON186792 | this study |
| Haplodiplatys aotouensis Ma & Chen, 1991 (Jiangxi) | 16,222 | 71.9 | ON186793 | this study | |
| Apachyidae | Apachyus feae de Bormans, 1894 | 19,029 | 61.2 | MW291948 | [20] |
| Diplatyidae | Diplatys flavicollis Shiraki, 1907 | 12,950 | 73.5 | MW291949 | [20] |
| Pygidicranidae | Challia fletcheri Burr, 1904 | 20,456 | 72.6 | NC_018538 | [17] |
| Anisolabididae | Euborellia arcanum Matzke & Kočárek, 2015 | 16,087 | 68.3 | KX673196 | [22] |
| Forficulidae | Eudohrnia metallica (Dohrn, 1865) | 16,324 | 58.7 | KX091853 | GenBank |
| Paratimomenus flavocapitatus Shiraki,1906 | 15,677 | 67.4 | KX091861 | GenBank | |
| Perlidae (Plecoptera) | Kamimuria chungnanshana Wu, 1938 | - | - | NC_028076 | [23] |
| Gene | Position (bp) | Size (bp) | Strand | Intergenic Nucleotides | Anticodon | Start/Stop Codons | A + T% |
|---|---|---|---|---|---|---|---|
| trnI | 119−188; 119−188 | 70; 70 | J | 0; 0 | GAT; GAT | 72.9; 72.9 | |
| trnM | 200−271; 200−271 | 72; 72 | J | 11; 11 | CAT; CAT | 75.0; 75.0 | |
| nad2 | 272−1270; 272−1270 | 999; 999 | J | 0; 0 | ATT/TAA; ATT/TAA | 70.4; 70.4 | |
| trnW | 1310−1388; 1310−1388 | 79; 79 | J | 39; 39 | TCA; TCA | 73.4; 73.4 | |
| cox1 | 1446−2945; 1446−2945 | 1500; 1500 | J | 57; 57 | ATT/TAA; ATT/TAA | 65.9; 65.9 | |
| trnL2 | 2957−3023; 2957−3023 | 67; 67 | J | 11; 11 | TAA; TAA | 68.7; 68.7 | |
| cox2 | 3134−3724; 3134−3724 | 591; 591 | J | 110; 110 | ATT/TAG; ATT/TAG | 67.2; 67.3 | |
| trnK | 3732−3797; 3732−3797 | 66; 66 | J | 7; 7 | CTT; CTT | 63.6; 63.6 | |
| trnD | 3797−3865; 3797−3865 | 69; 69 | J | −1; −1 | GTC; GTC | 76.8; 76.8 | |
| atp8 | 3866−4039; 3866−4039 | 174; 174 | J | 0; 0 | ATT/TAA; ATT/TAA | 72.4; 72.4 | |
| atp6 | 4033−4710; 4033−4710 | 678; 678 | J | −7; −7 | ATG/TAA; ATG/TAA | 70.5; 70.5 | |
| cox3 | 4769−5557; 4770−5558 | 789; 789 | J | 58; 58 | ATG/TAA; ATG/TAA | 65.4; 65.4 | |
| trnG | 5598−5665; 5599−5669 | 68; 68 | J | 40; 40 | TCC; TCC | 79.4; 79.4 | |
| nad3 | 5666−6019; 5667−6020 | 354; 354 | J | 0; 0 | ATT/TAA; ATT/TAA | 69.8; 69.8 | |
| trnA | 6130−6196; 6139−6205 | 67; 67 | J | 110; 118 | TGC; TGC | 80.6; 80.6 | |
| trnN | 6216−6289; 6225−6298 | 74; 74 | N | 19; 19 | GTT; GTT | 78.4; 78.4 | |
| trnE | 6338−6406; 6349−6417 | 69; 69 | J | 48; 50 | TTC; TTC | 75.4; 75.4 | |
| trnY | 6467−6537; 6478−6548 | 71; 71 | N | 60; 60 | GTA; GTA | 83.1; 83.1 | |
| trnC | 6624−6690; 6635−6701 | 67; 67 | N | 86; 86 | GCA; GCA | 86.6; 86.6 | |
| trnQ | 6700−6768; 6711−6779 | 69; 69 | N | 9; 9 | TTG; TTG | 76.8; 76.8 | |
| trnS1 | 6816−6884; 6845−6913 | 69; 69 | J | 47; 65 | GCT; GCT | 72.5; 72.5 | |
| trnR | 6885−6950; 6914−6979 | 66; 66 | J | 0; 0 | TCG; TCG | 74.2; 74.2 | |
| trnF | 6987−7053; 7016−7082 | 67; 67 | J | 36; 36 | GAA; GAA | 83.6; 83.6 | |
| nad5 | 7130−8872; 7159−8901 | 1743; 1743 | N | 76; 76 | ATC/TAA; ATC/TAA | 71.1; 71.1 | |
| trnH | 8873−8941; 8902−8970 | 69; 69 | N | 0; −3 | GTG; GTG | 79.7; 79.7 | |
| nad4 | 8954−10303; 8983−10332 | 1350; 1350 | N | 12; 12 | ATG/TAA; ATG/TAA | 70.2; 70.2 | |
| nad4l | 10303−10590; 10332−10619 | 288; 288 | N | −1; −1 | ATG/TAA; ATG/TAA | 71.2; 71.2 | |
| trnT | 10599−10667; 10628−10696 | 69; 69 | J | 8; 8 | TGT; TGT | 76.8; 76.8 | |
| trnP | 10668−10734; 10697−10763 | 67; 67 | N | 0; 0 | TGG; TGG | 76.1; 76.1 | |
| nad6 | 10737−11237; 10766−11266 | 501; 501 | J | 2; 2 | ATT/TAG; ATT/TAG | 73.1; 73.3 | |
| cytb | 11270−12409; 11299−12438 | 1140; 1140 | J | 32; 32 | ATG/TAA; ATG/TAA | 69.9; 69.9 | |
| trnS2 | 12438−12508; 12467−12537 | 71; 71 | J | 28; 28 | TGA; TGA | 84.5; 84.5 | |
| nad1 | 12687−13631; 12716−13660 | 945; 945 | N | 178; 178 | ATT/TAA; ATT/TAA | 66.2; 66.2 | |
| trnL1 | 13632−13700; 13661−13729 | 69; 69 | N | 0; 0 | TAG; TAG | 81.2; 81.2 | |
| rrnL | 13701−15108; 13730−15137 | 1408; 1408 | N | 0; 0 | − | 75.4; 75.4 | |
| trnV | 15109−15181; 15138−15210 | 73; 73 | N | 0; 0 | TAC; TAC | 72.6; 72.6 | |
| rrnS | 15182−15995; 15211−16025 | 814; 815 | N | 0; 0 | − | 75.4; 75.5 | |
| CR | 15996−16134; 16026−16222 | 139; 197 | J | 0; 0 | − | 77.0; 83.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, H.-L.; Chen, S.; Chen, Q.-D.; Pu, D.-Q.; Chen, Z.-T.; Liu, Y.-Y.; Liu, X. The First Mitochondrial Genomes of the Family Haplodiplatyidae (Insecta: Dermaptera) Reveal Intraspecific Variation and Extensive Gene Rearrangement. Biology 2022, 11, 807. https://doi.org/10.3390/biology11060807
Liu H-L, Chen S, Chen Q-D, Pu D-Q, Chen Z-T, Liu Y-Y, Liu X. The First Mitochondrial Genomes of the Family Haplodiplatyidae (Insecta: Dermaptera) Reveal Intraspecific Variation and Extensive Gene Rearrangement. Biology. 2022; 11(6):807. https://doi.org/10.3390/biology11060807
Chicago/Turabian StyleLiu, Hong-Ling, Song Chen, Qing-Dong Chen, De-Qiang Pu, Zhi-Teng Chen, Yue-Yue Liu, and Xu Liu. 2022. "The First Mitochondrial Genomes of the Family Haplodiplatyidae (Insecta: Dermaptera) Reveal Intraspecific Variation and Extensive Gene Rearrangement" Biology 11, no. 6: 807. https://doi.org/10.3390/biology11060807
APA StyleLiu, H. -L., Chen, S., Chen, Q. -D., Pu, D. -Q., Chen, Z. -T., Liu, Y. -Y., & Liu, X. (2022). The First Mitochondrial Genomes of the Family Haplodiplatyidae (Insecta: Dermaptera) Reveal Intraspecific Variation and Extensive Gene Rearrangement. Biology, 11(6), 807. https://doi.org/10.3390/biology11060807