
Phylogenomics Resolves the Phylogeny of Theaceae by Using Low-Copy and Multi-Copy Nuclear Gene Makers and Uncovers a Fast Radiation Event Contributing to Tea Plants Diversity


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Source and Transcriptome Assembly
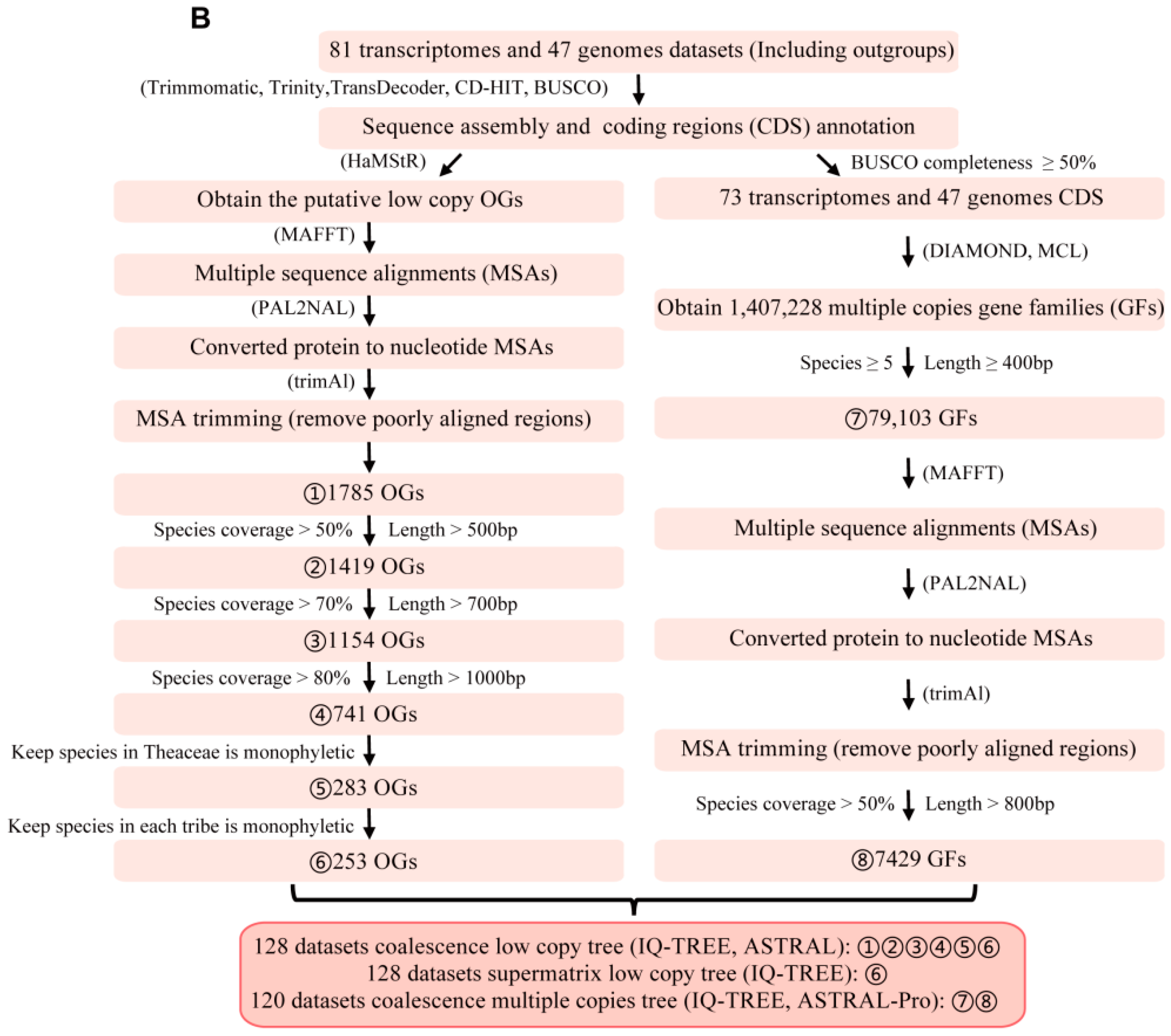
2.2. Orthologs Identification and Gene Sets Filtrations
2.3. Gene Ontology Analyses
2.4. Phylogenetic Analyses
2.5. Divergence Time Estimation
2.6. Speciation Rate Calculation
3. Results
3.1. Identification of Low-Copy Orthologous Genes and Noise Filtrations


3.2. Gene Ontology Analyses Suggested the Housekeeping Function for 1785 Low-Copy Nuclear Genes

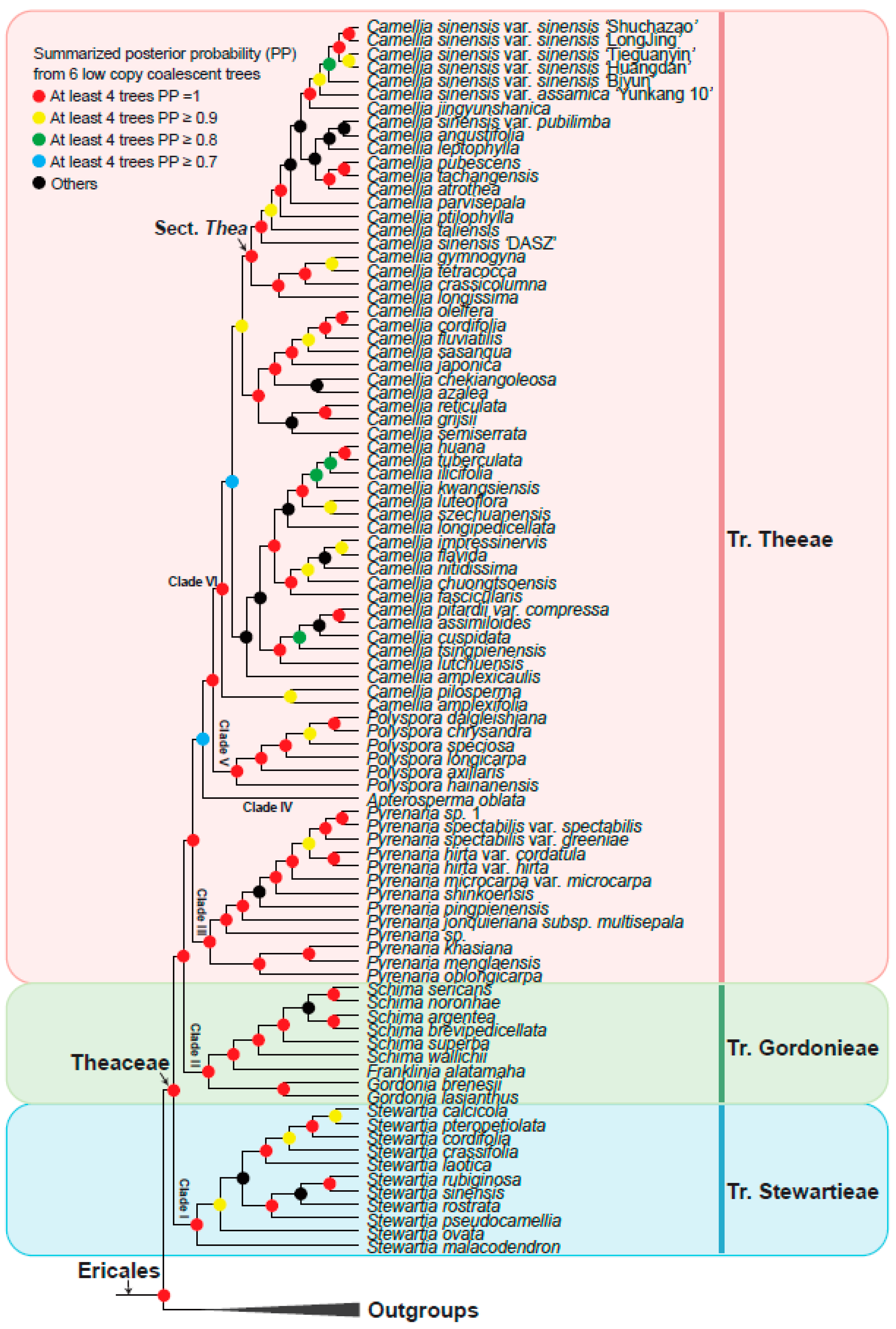
3.3. Theaceae Were Divided into Three Tribes: Theeae, Gordonieae, and Stewartieae

3.4. A Robust Topology at Generic Level of Theaceae
3.5. Phylogeny Comparison from Inference by Low-Copy with Multi-Copy Nuclear Genes
3.6. Molecular Dating Suggested Theaceae Originated Early Than the K-Pg Boundary
3.7. Camellia Specific Fast Radiation Associated with Climate Optimum
4. Discussion
4.1. Conserved Low-Copy Nuclear Genes with Main Housekeeping Functions Are Effective Markers for Theaceae Phylogeny
4.2. A Robust Theaceae Phylogeny Supported by Extensive Low-Copy and Multi-Copy Nuclear Gene Markers
4.3. Genus Camellia Has a Significant Diversification Shift Rate Related to the Species Radiation with the Mid-Miocene Climatic Optimum
4.4. A Fast Radiation Started from the Most Common Ancestors of Genus Camellia
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Glumac, S.P. The World Flora Online. Available online: http://worldfloraonline.org/ (accessed on 2 September 2021).
- Xia, E.; Tong, W.; Hou, Y.; An, Y.; Chen, L.; Wu, Q.; Liu, Y.; Yu, J.; Li, F.; Li, R.; et al. The reference genome of tea plant and resequencing of 81 diverse accessions provide insights into Iits genome evolution and adaptation. Mol. Plant 2020, 13, 1013–1026. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhang, Y.; Qiu, H.; Guo, Y.; Wan, H.; Zhang, X.; Scossa, F.; Alseekh, S.; Zhang, Q.; Wang, P.; et al. Genome assembly of wild tea tree DASZ reveals pedigree and selection history of tea varieties. Nat. Commun. 2020, 11, 3719. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Feng, H.; Chang, Y.; Ma, C.; Wang, L.; Hao, X.; Cheng, H.; Wang, L.; Cui, P.; Jin, J. Population sequencing enhances understanding of tea plant evolution. Nat. Commun. 2020, 11, 4447. [Google Scholar] [CrossRef]
- Lin, P.; Wang, K.; Wang, Y.; Hu, Z.; Yan, C.; Huang, H.; Ma, X.; Cao, Y.; Long, W.; Liu, W.; et al. The genome of oil-Camellia and population genomics analysis provide insights into seed oil domestication. Genome Biol. 2022, 23, 14. [Google Scholar] [CrossRef] [PubMed]
- Gong, W.; Xiao, S.; Wang, L.; Liao, Z.; Chang, Y.; Mo, W.; Hu, G.; Li, W.; Zhao, G.; Zhu, H.; et al. Chromosome-level genome of Camellia lanceoleosa provides a valuable resource for understanding genome evolution and self-incompatibility. Plant J. 2022, 110, 881–898. [Google Scholar] [CrossRef]
- Shen, T.F.; Huang, B.; Xu, M.; Zhou, P.Y.; Ni, Z.X.; Gong, C.; Wen, Q.; Cao, F.L.; Xu, L.A. The reference genome of Camellia chekiangoleosa provides insights into camellia evolution and tea oil biosynthesis. Hortic. Res. 2022, 9, uhab083. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zeng, Q.; del Mar Contreras, M.; Wang, L. Profiling and quantification of phenolic compounds in Camellia seed oils: Natural tea polyphenols in vegetable oil. Food Res. Int. 2017, 102, 184–194. [Google Scholar] [CrossRef]
- Fu, M.; Yang, X.; Zheng, J.; Wang, L.; Yang, X.; Tu, Y.; Ye, J.; Zhang, W.; Liao, Y.; Cheng, S.; et al. Unraveling the regulatory mechanism of color diversity in Camellia japonica petals by integrative transcriptome and metabolome analysis. Front. Plant Sci. 2021, 12, 1119. [Google Scholar] [CrossRef]
- Rose, J.P.; Kleist, T.J.; Löfstrand, S.D.; Drew, B.T.; Schoenenberger, J.; Sytsma, K.J. Phylogeny, historical biogeography, and diversification of angiosperm order Ericales suggest ancient Neotropical and East Asian connections. Mol. Phylogen. Evol. 2018, 122, 59–79. [Google Scholar] [CrossRef]
- Kubo, K.-I.; Entani, T.; Takara, A.; Wang, N.; Fields, A.M.; Hua, Z.; Toyoda, M.; Kawashima, S.-I.; Ando, T.; Isogai, A.; et al. Collaborative non-self recognition system in S-RNase–based self-incompatibility. Science 2010, 330, 796–799. [Google Scholar] [CrossRef]
- Prince, L.M.; Parks, C.R. Phylogenetic relationships of Theaceae inferred from chloroplast DNA sequence data. Am. J. Bot. 2001, 88, 2309–2320. [Google Scholar] [CrossRef] [PubMed]
- Rao, M.; Steinbauer, M.J.; Xiang, X.; Zhang, M.; Mi, X.; Zhang, J.; Ma, K.; Svenning, J.C. Environmental and evolutionary drivers of diversity patterns in the tea family (Theaceae s.s.) across China. Ecol. Evol. 2018, 8, 11663–11676. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.-X.; Yang, J.-B.; Lei, L.-G.; Li, D.-Z.; Yoshino, H.; Ikeda, T. Reassessing the relationships between Gordoniaand Polyspora (Theaceae) based on the combined analyses of molecular data from the nuclear, plastid and mitochondrial genomes. Plant Syst. Evol. 2004, 248, 45–55. [Google Scholar] [CrossRef]
- Wang, Y.; He, H.; Min, T.; Zhou, L.; Fritsch, P. The phylogenetic position of Apterosperma (Theaceae) based on morphological and karyotype characters. Plant Syst. Evol. 2006, 260, 39–52. [Google Scholar] [CrossRef]
- Yu, X.Q.; Gao, L.M.; Soltis, D.E.; Soltis, P.S.; Yang, J.B.; Fang, L.; Yang, S.X.; Li, D.Z. Insights into the historical assembly of East Asian subtropical evergreen broadleaved forests revealed by the temporal history of the tea family. New Phytol. 2017, 215, 1235–1248. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.-Q.; Drew, B.T.; Yang, J.-B.; Gao, L.-M.; Li, D.-Z. Comparative chloroplast genomes of eleven Schima (Theaceae) species: Insights into DNA barcoding and phylogeny. PLoS ONE 2017, 12, e0178026. [Google Scholar] [CrossRef] [Green Version]
- Yan, Y.; Davis, C.C.; Dimitrov, D.; Wang, Z.; Rahbek, C.; Borregaard, M.K. Phytogeographic history of the tea family inferred through high-resolution phylogeny and fossils. Syst. Biol. 2021, 70, 1256–1271. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhao, L.; Folk, R.A.; Zhao, J.-L.; Zamora, N.A.; Yang, S.-X.; Soltis, D.E.; Soltis, P.S.; Gao, L.-M.; Peng, H. Phylotranscriptomics of Theaceae: Generic level relationships, reticulation and whole-genome duplication. Ann. Bot. 2022, 129, 457–471. [Google Scholar] [CrossRef]
- Zhang, W.; Kan, S.-L.; Zhao, H.; Li, Z.-Y.; Wang, X.-Q. Molecular phylogeny of tribe Theeae (Theaceae s.s.) and its implications for generic delimitation. PLoS ONE 2014, 9, e98133. [Google Scholar] [CrossRef] [Green Version]
- Tang, C.Q. The Subtropical Vegetation of Southwestern China: Plant Distribution, Diversity and Ecology; Springer: Berlin/Heidelberg, Germany, 2015; Volume 11. [Google Scholar]
- LI, M.M.; LI, J.H.; Del Tredici, P.; Corajod, J.; FU, C.X. Phylogenetics and biogeography of Theaceae based on sequences of plastid genes. J. Syst. Evol. 2013, 51, 396–404. [Google Scholar] [CrossRef]
- Zhang, L.; Chen, F.; Zhang, X.; Li, Z.; Zhao, Y.; Lohaus, R.; Chang, X.; Dong, W.; Ho, S.Y.W.; Liu, X.; et al. The water lily genome and the early evolution of flowering plants. Nature 2020, 577, 79–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Zhang, R.; Jiang, K.-W.; Qi, J.; Hu, Y.; Guo, J.; Zhu, R.; Zhang, T.; Egan, A.N.; Yi, T.-S. Nuclear phylotranscriptomics and phylogenomics support numerous polyploidization events and hypotheses for the evolution of rhizobial nitrogen-fixing symbiosis in Fabaceae. Mol. Plant 2021, 14, 748–773. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Zhang, Q.; Sun, R.; Kong, H.; Zhang, N.; Ma, H. Resolution of deep angiosperm phylogeny using conserved nuclear genes and estimates of early divergence times. Nat. Commun. 2014, 5, 4956. [Google Scholar] [CrossRef] [Green Version]
- Xia, E.-H.; Zhang, H.-B.; Sheng, J.; Li, K.; Zhang, Q.-J.; Kim, C.; Zhang, Y.; Liu, Y.; Zhu, T.; Li, W. The tea tree genome provides insights into tea flavor and independent evolution of caffeine biosynthesis. Mol. Plant 2017, 10, 866–877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, C.; Yang, H.; Wang, S.; Zhao, J.; Liu, C.; Gao, L.; Xia, E.; Lu, Y.; Tai, Y.; She, G.; et al. Draft genome sequence of Camellia sinensis var. sinensis provides insights into the evolution of the tea genome and tea quality. Proc. Natl. Acad. Sci. USA 2018, 115, E4151–E4158. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.D.; Zheng, C.; Ma, J.Q.; Jiang, C.K.; Ercisli, S.; Yao, M.Z.; Chen, L. The chromosome-scale genome reveals the evolution and diversification after the recent tetraploidization event in tea plant. Hortic. Res. 2020, 7, 63. [Google Scholar] [CrossRef]
- Zhang, Q.J.; Li, W.; Li, K.; Nan, H.; Shi, C.; Zhang, Y.; Dai, Z.Y.; Lin, Y.L.; Yang, X.L.; Tong, Y.; et al. The chromosome-level reference genome of tea tree unveils recent bursts of non-autonomous LTR retrotransposons in driving genome size evolution. Mol. Plant 2020, 13, 935–938. [Google Scholar] [CrossRef]
- Wang, P.; Yu, J.; Jin, S.; Chen, S.; Yue, C.; Wang, W.; Gao, S.; Cao, H.; Zheng, Y.; Gu, M.; et al. Genetic basis of high aroma and stress tolerance in the oolong tea cultivar genome. Hortic. Res. 2021, 8, 107. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, S.; Shi, L.; Gong, D.; Zhang, S.; Zhao, Q.; Zhan, D.; Vasseur, L.; Wang, Y.; Yu, J.; et al. Haplotype-resolved genome assembly provides insights into evolutionary history of the tea plant Camellia sinensis. Nat. Genet. 2021, 53, 1250–1259. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Zhang, N.; Zhang, Q.; Endress, P.K.; Huang, J.; Ma, H. Resolution of deep eudicot phylogeny and their temporal diversification using nuclear genes from transcriptomic and genomic datasets. New Phytol. 2017, 214, 1338–1354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, Y.; Huang, C.H.; Hu, Y.; Wen, J.; Li, S.; Yi, T.; Chen, H.; Xiang, J.; Ma, H. Evolution of Rosaceae fruit types based on nuclear phylogeny in the context of geological times and genome duplication. Mol. Biol. Evol. 2017, 34, 262–281. [Google Scholar] [CrossRef] [Green Version]
- Qi, X.P.; Kuo, L.Y.; Guo, C.; Li, H.; Li, Z.; Qi, J.; Wang, L.; Hu, Y.; Xiang, J.; Zhang, C.; et al. A well-resolved fern nuclear phylogeny reveals the evolution history of numerous transcription factor families. Mol. Phylogen. Evol. 2018, 127, 961–977. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.-H.; Sun, R.; Hu, Y.; Zeng, L.; Zhang, N.; Cai, L.; Zhang, Q.; Koch, M.A.; Al-Shehbaz, I.; Edger, P.P. Resolution of Brassicaceae phylogeny using nuclear genes uncovers nested radiations and supports convergent morphological evolution. Mol. Biol. Evol. 2016, 33, 394–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [Green Version]
- Reyes-Chin-Wo, S.; Wang, Z.; Yang, X.; Kozik, A.; Arikit, S.; Song, C.; Xia, L.; Froenicke, L.; Lavelle, D.O.; Truco, M.J.; et al. Genome assembly with in vitro proximity ligation data and whole-genome triplication in lettuce. Nat. Commun. 2017, 8, 14953. [Google Scholar] [CrossRef]
- Hirakawa, H.; Sumitomo, K.; Hisamatsu, T.; Nagano, S.; Shirasawa, K.; Higuchi, Y.; Kusaba, M.; Koshioka, M.; Nakano, Y.; Yagi, M.; et al. De novo whole-genome assembly in Chrysanthemum seticuspe, a model species of Chrysanthemums, and its application to genetic and gene discovery analysis. DNA Res. 2019, 26, 195–203. [Google Scholar] [CrossRef] [Green Version]
- Iorizzo, M.; Ellison, S.; Senalik, D.; Zeng, P.; Satapoomin, P.; Huang, J.; Bowman, M.; Iovene, M.; Sanseverino, W.; Cavagnaro, P.; et al. A high-quality carrot genome assembly provides new insights into carotenoid accumulation and asterid genome evolution. Nat. Genet. 2016, 48, 657–666. [Google Scholar] [CrossRef] [Green Version]
- Hosmani, P.S.; Flores-Gonzalez, M.; van de Geest, H.; Maumus, F.; Bakker, L.V.; Schijlen, E.; van Haarst, J.; Cordewener, J.; Sanchez-Perez, G.; Peters, S. An improved de novo assembly and annotation of the tomato reference genome using single-molecule sequencing, Hi-C proximity ligation and optical maps. bioRxiv 2019. [Google Scholar] [CrossRef]
- Kim, S.; Park, M.; Yeom, S.-I.; Kim, Y.-M.; Lee, J.M.; Lee, H.-A.; Seo, E.; Choi, J.; Cheong, K.; Kim, K.-T. Genome sequence of the hot pepper provides insights into the evolution of pungency in Capsicum species. Nat. Genet. 2014, 46, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Ma, T.; Kang, M.; Ai, F.; Zhang, J.; Dong, G.; Liu, J. A high-quality Actinidia chinensis (kiwifruit) genome. Hortic. Res. 2019, 6, 117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowak, M.D.; Russo, G.; Schlapbach, R.; Huu, C.N.; Lenhard, M.; Conti, E. The draft genome of Primula veris yields insights into the molecular basis of heterostyly. Genome Biol. 2015, 16, 12. [Google Scholar] [CrossRef] [Green Version]
- Jaillon, O.; Aury, J.-M.; Noel, B.; Policriti, A.; Clepet, C.; Cassagrande, A.; Choisne, N.; Aubourg, S.; Vitulo, N.; Jubin, C. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 2007, 449, 463–467. [Google Scholar]
- Filiault, D.L.; Ballerini, E.S.; Mandáková, T.; Aköz, G.; Derieg, N.J.; Schmutz, J.; Jenkins, J.; Grimwood, J.; Shu, S.; Hayes, R.D.; et al. The Aquilegia genome provides insight into adaptive radiation and reveals an extraordinarily polymorphic chromosome with a unique history. Elife 2018, 7, e36426. [Google Scholar] [CrossRef]
- Ebersberger, I.; Strauss, S.; von Haeseler, A. HaMStR: Profile hidden markov model based search for orthologs in ESTs. BMC Evol. Biol. 2009, 9, 157. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Capella Gutiérrez, S.; Silla Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Suyama, M.; Torrents, D.; Bork, P. PAL2NAL: Robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006, 34, W609–W612. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Rabiee, M.; Sayyari, E.; Mirarab, S. ASTRAL-III: Polynomial time species tree reconstruction from partially resolved gene trees. BMC Bioinform. 2018, 19, 153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Stichting, C.; Centrum, M.; Dongen, S.V. A Cluster Algorithm for Graphs; Information Systems; CWI: Nampa, ID, USA, 2000. [Google Scholar]
- Enright, A.J.; Dongen, S.V.; Ouzounis, C.A. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002, 30, 1575–1584. [Google Scholar] [CrossRef]
- Zhang, C.; Celine, S.; Molloy, E.K.; Siavash, M. ASTRAL-Pro: Quartet-Based Species-Tree Inference despite Paralogy. Mol. Biol. Evol. 2020, 37, 3292–3307. [Google Scholar] [CrossRef]
- Morris, J.L.; Puttick, M.N.; Clark, J.W.; Edwards, D.; Kenrick, P.; Pressel, S.; Wellman, C.H.; Yang, Z.; Schneider, H.; Donoghue, P.C. The timescale of early land plant evolution. Proc. Natl. Acad. Sci. USA 2018, 115, E2274–E2283. [Google Scholar] [CrossRef] [Green Version]
- Doyle, J.A.; Hotton, C.L. Diversification of early angiosperm pollen in a cladistic context. Pollen Spores Patterns Diversif. 1991, 169, 195. [Google Scholar]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [Green Version]
- Reis, M.d.; Yang, Z. Approximate likelihood calculation on a phylogeny for Bayesian estimation of divergence times. Mol. Biol. Evol. 2011, 28, 2161–2172. [Google Scholar] [CrossRef] [Green Version]
- Rabosky, D.L.; Grundler, M.; Anderson, C.; Title, P.; Shi, J.J.; Brown, J.W.; Huang, H.; Larson, J.G. BAMM tools: An R package for the analysis of evolutionary dynamics on phylogenetic trees. Methods Ecol. Evol. 2014, 5, 701–707. [Google Scholar] [CrossRef]
- Prince, L.M. A brief nomenclatural review of genera and tribes in Theaceae. Aliso J. Syst. Florist. Bot. 2007, 24, 105–121. [Google Scholar] [CrossRef] [Green Version]
- Zachos, J.C.; Dickens, G.R.; Zeebe, R.E. An early Cenozoic perspective on greenhouse warming and carbon-cycle dynamics. Nature 2008, 451, 279–283. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Xu, W.; Hu, Y.; Huang, J.; Zhao, Y.; Zhang, L.; Huang, C.-H.; Ma, H. Phylotranscriptomics in Cucurbitaceae reveal multiple Whole-Genome Duplications and key morphological and molecular innovations. Mol. Plant 2020, 13, 1117–1133. [Google Scholar] [CrossRef]
- Yang, L.; Su, D.; Chang, X.; Foster, C.S.P.; Sun, L.; Huang, C.-H.; Zhou, X.; Zeng, L.; Ma, H.; Zhong, B. Phylogenomic insights into deep phylogeny of angiosperms based on broad nuclear gene sampling. Plant Commun. 2020, 1, 100027. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.H.; Zhang, C.; Liu, M.; Hu, Y.; Gao, T.; Qi, J.; Ma, H. Multiple polyploidization events across asteraceae with two nested events in the early history revealed by nuclear phylogenomics. Mol. Biol. Evol. 2016, 33, 2820–2835. [Google Scholar] [CrossRef] [Green Version]
- Zhang, N.; Zeng, L.; Shan, H.; Ma, H. Highly conserved low-copy nuclear genes as effective markers for phylogenetic analyses in angiosperms. New Phytol. 2012, 195, 923–937. [Google Scholar] [CrossRef]
- Philippe, H.; Delsuc, F.; Brinkmann, H.; Lartillot, N. Phylogenomics. Annu. Rev. Ecol. Evol. Syst. 2005, 36, 541–562. [Google Scholar] [CrossRef]
- Chang, H.T. A taxonomy of the genus Camellia. Acta Sci. Nat. Univ. Sunyatseni 1981, 1, 1–180. [Google Scholar]
- Wang, Y.; Chen, F.; Ma, Y.; Zhang, T.; Sun, P.; Lan, M.; Li, F.; Fang, W. An ancient whole-genome duplication event and its contribution to flavor compounds in the tea plant (Camellia sinensis). Hortic. Res. 2021, 8, 244–248. [Google Scholar] [CrossRef]
- Svenning, J.-C.; Eiserhardt, W.L.; Normand, S.; Ordonez, A.; Sandel, B. The influence of paleoclimate on present-day patterns in biodiversity and ecosystems. Annu. Rev. Ecol. Evol. Syst. 2015, 46, 551–572. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Zhu, X.; Zhao, Y.; Guo, J.; Zhang, T.; Huang, W.; Huang, J.; Hu, Y.; Huang, C.-H.; Ma, H. Phylotranscriptomics Resolves the Phylogeny of Pooideae and Uncovers Factors for Their Adaptive Evolution. Mol. Biol. Evol. 2022, 39, msac026. [Google Scholar] [CrossRef] [PubMed]
- Van de Peer, Y.; Mizrachi, E.; Marchal, K. The evolutionary significance of polyploidy. Nat. Rev. Genet. 2017, 18, 411–424. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.D.; Dangl, J.L. The plant immune system. Nature 2006, 444, 323–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.; Han, B.; Jiao, Y. Genetic contribution of paleopolyploidy to adaptive evolution in angiosperms. Mol. Plant 2020, 13, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Baskin, J.M.; Baskin, C.C.; Huang, Z. More than just a coating: Ecological importance, taxonomic occurrence and phylogenetic relationships of seed coat mucilage. Perspect. Plant Ecol. Evol. Syst. 2012, 14, 434–442. [Google Scholar] [CrossRef]
- Lei, J.; Shen, Z.; Yi, X. Pericarp thickness and seed size determine acorn dispersal of five rodent-dispersed oak species. Acta Theriol. Sin. 2012, 32, 83–89. [Google Scholar]
- Yan, X.H.; Zhou, B.; Yin, Z.F.; Wang, N.; Zhang, Z.G. Reproductive biological characteristics potentially contributed to invasiveness in an alien invasive plant Bidens frondosa. Plant Species Biol. 2016, 31, 107–116. [Google Scholar] [CrossRef]
- Smykal, P.; Gennen, J.; De Bodt, S.; Ranganath, V.; Melzer, S. Flowering of strict photoperiodic Nicotiana varieties in non-inductive conditions by transgenic approaches. Plant Mol. Biol. 2007, 65, 233–242. [Google Scholar] [CrossRef]
- Fourquin, C.; del Cerro, C.; Victoria, F.C.; Vialette-Guiraud, A.; de Oliveira, A.C.; Ferrándiz, C. A change in SHATTERPROOF protein lies at the origin of a fruit morphological novelty and a new strategy for seed dispersal in Medicago genus. Plant Physiol. 2013, 162, 907–917. [Google Scholar] [CrossRef] [Green Version]
- Novaes, E.; Kirst, M.; Chiang, V.; Winter-Sederoff, H.; Sederoff, R. Lignin and biomass: A negative correlation for wood formation and lignin content in trees. Plant Physiol. 2010, 154, 555–561. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, L.; Li, M.; Han, Q.; Qiao, Z.; Hao, Y.; Balbuena, T.S.; Zhao, Y. Phylogenomics Resolves the Phylogeny of Theaceae by Using Low-Copy and Multi-Copy Nuclear Gene Makers and Uncovers a Fast Radiation Event Contributing to Tea Plants Diversity. Biology 2022, 11, 1007. https://doi.org/10.3390/biology11071007
Cheng L, Li M, Han Q, Qiao Z, Hao Y, Balbuena TS, Zhao Y. Phylogenomics Resolves the Phylogeny of Theaceae by Using Low-Copy and Multi-Copy Nuclear Gene Makers and Uncovers a Fast Radiation Event Contributing to Tea Plants Diversity. Biology. 2022; 11(7):1007. https://doi.org/10.3390/biology11071007
Chicago/Turabian StyleCheng, Lin, Mengge Li, Qunwei Han, Zhen Qiao, Yanlin Hao, Tiago Santana Balbuena, and Yiyong Zhao. 2022. "Phylogenomics Resolves the Phylogeny of Theaceae by Using Low-Copy and Multi-Copy Nuclear Gene Makers and Uncovers a Fast Radiation Event Contributing to Tea Plants Diversity" Biology 11, no. 7: 1007. https://doi.org/10.3390/biology11071007
APA StyleCheng, L., Li, M., Han, Q., Qiao, Z., Hao, Y., Balbuena, T. S., & Zhao, Y. (2022). Phylogenomics Resolves the Phylogeny of Theaceae by Using Low-Copy and Multi-Copy Nuclear Gene Makers and Uncovers a Fast Radiation Event Contributing to Tea Plants Diversity. Biology, 11(7), 1007. https://doi.org/10.3390/biology11071007