
Primary-like Human Hepatocytes Genetically Engineered to Obtain Proliferation Competence as a Capable Application for Energy Metabolism Experiments in In Vitro Oncologic Liver Models




Abstract
:Simple Summary
Abstract
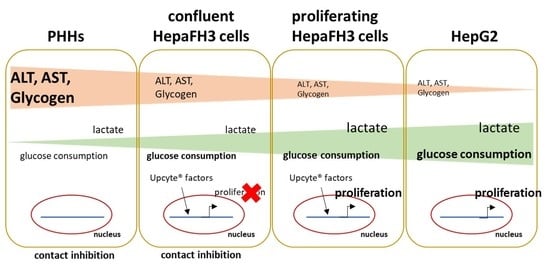
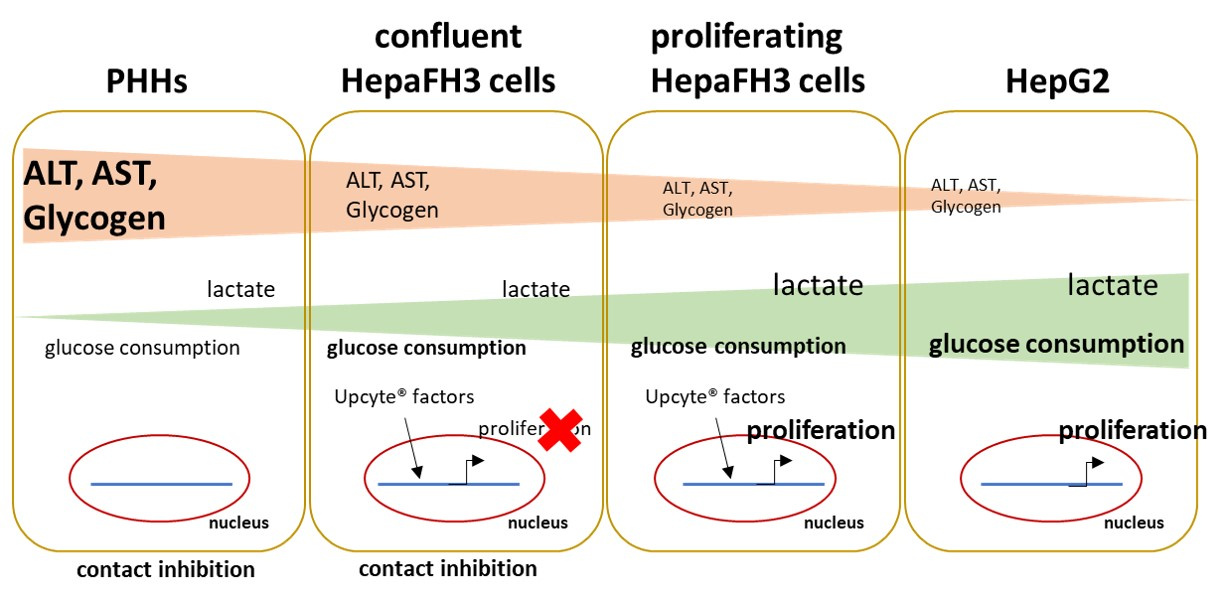
1. Introduction
2. Materials and Methods
2.1. Chemicals and Material
2.2. Isolation of Primary Human Hepatocytes
2.3. Generation of Upcyte Hepatocytes
2.4. In Vitro Cell Culture and Induction of Steatosis
2.5. Oil Red O Staining
2.6. Sulforhodamine B Protein Staining
2.7. Western Blot Analysis
2.8. Quantification of Total Protein
2.9. Cellular Metabolic Activity
2.10. Glucose
2.11. Glycogen
2.12. Urea
2.13. ALT/AST
2.14. Albumin
2.15. Lactate Assay
2.16. Statistical Analysis
3. Results
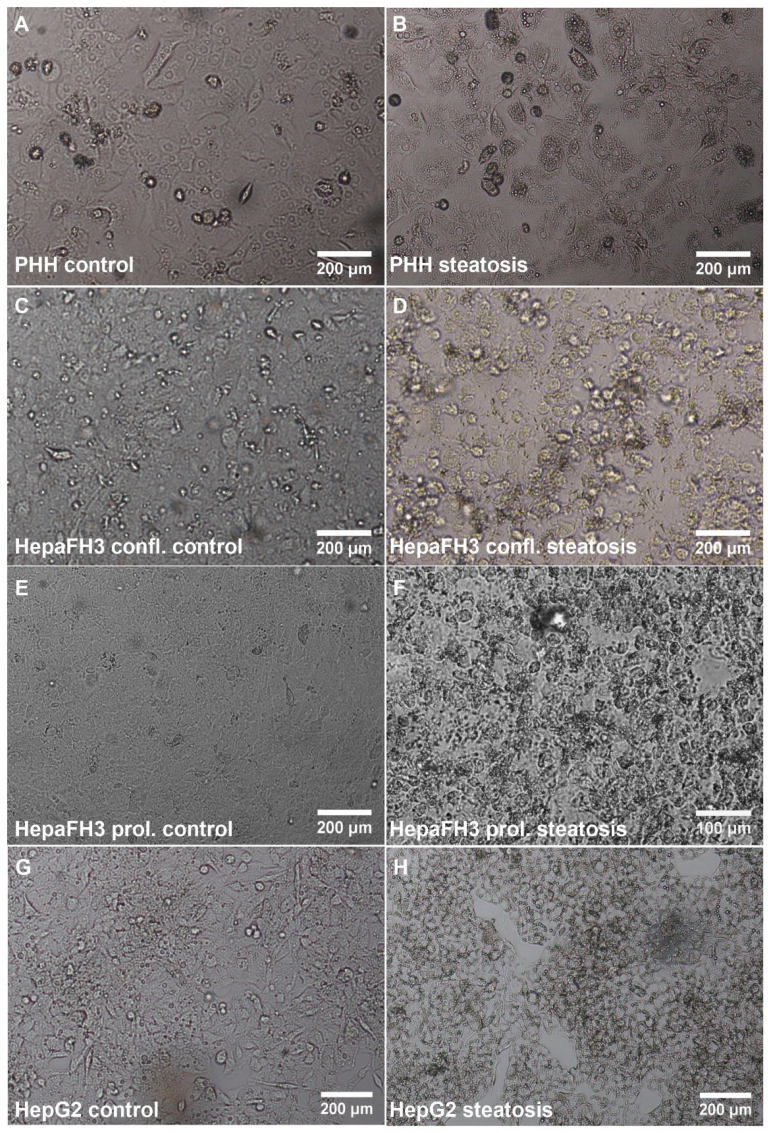
3.1. HepaFH3 Show Rather Morphologic Similarities to HepG2 Cells Than to PHHs
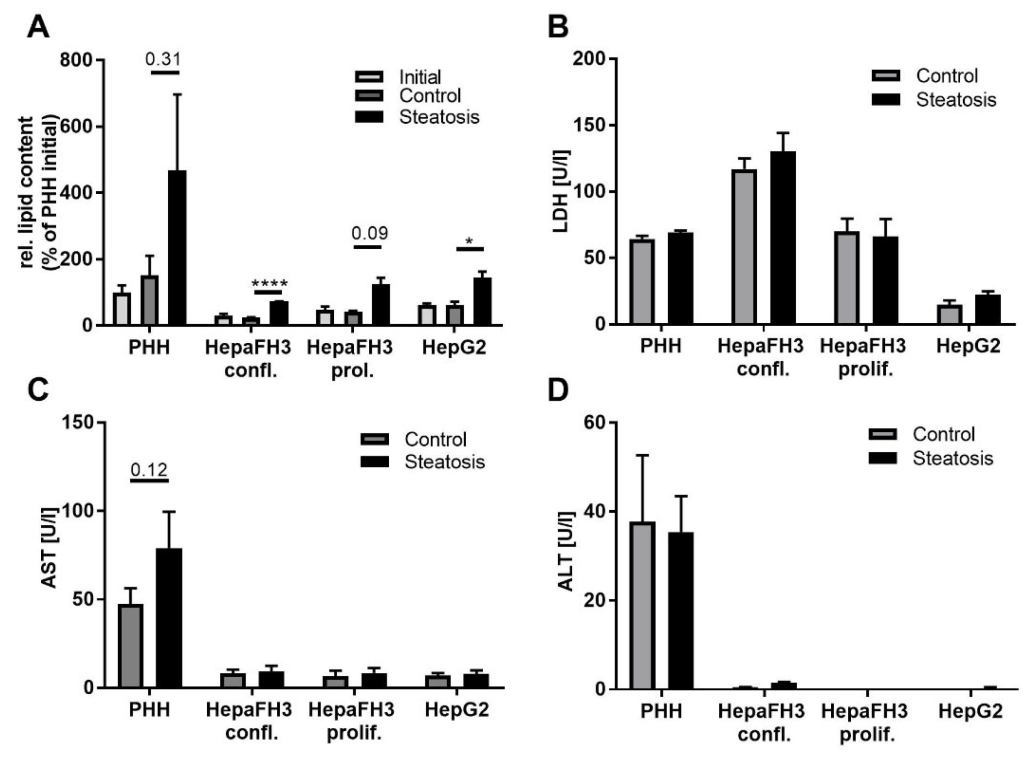
3.2. PHHs, HepaFH3 and HepG2 Cells Exhibit the Same Ability of Lipid Storage
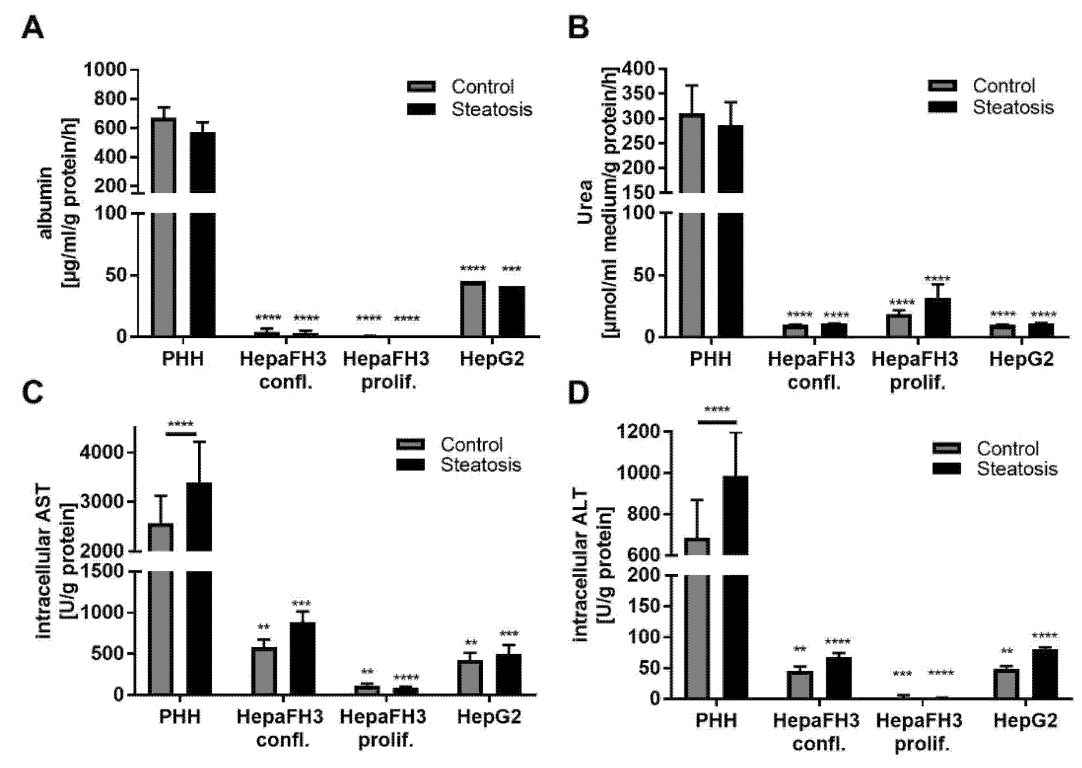
3.3. HepaFH3 and HepG2 Cells Show Strongly Diminished Levels of Basic Hepatocyte Markers Compared to PHHs
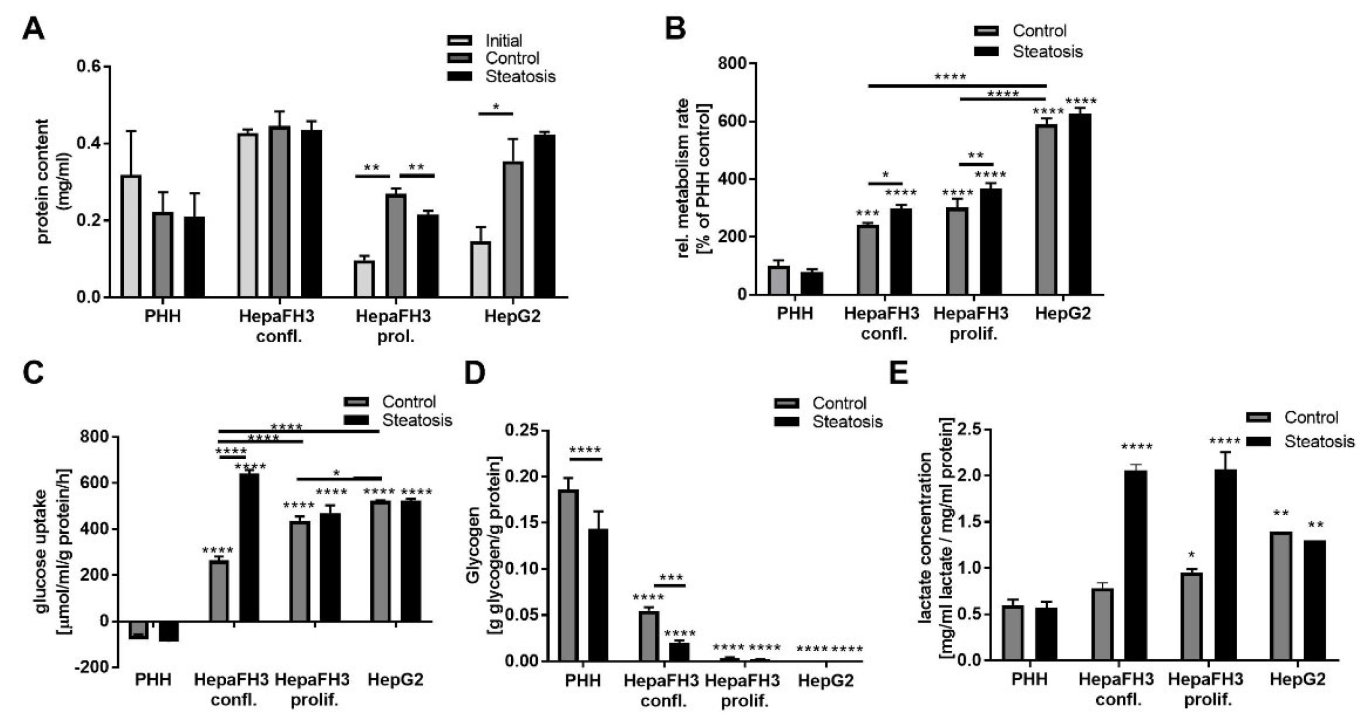
3.4. Proliferative Rate and Glucose Metabolism Are Elevated in Proliferating HepaFH3 and HepG2 Cells Compared to Confluent HepaFH3 and PHH
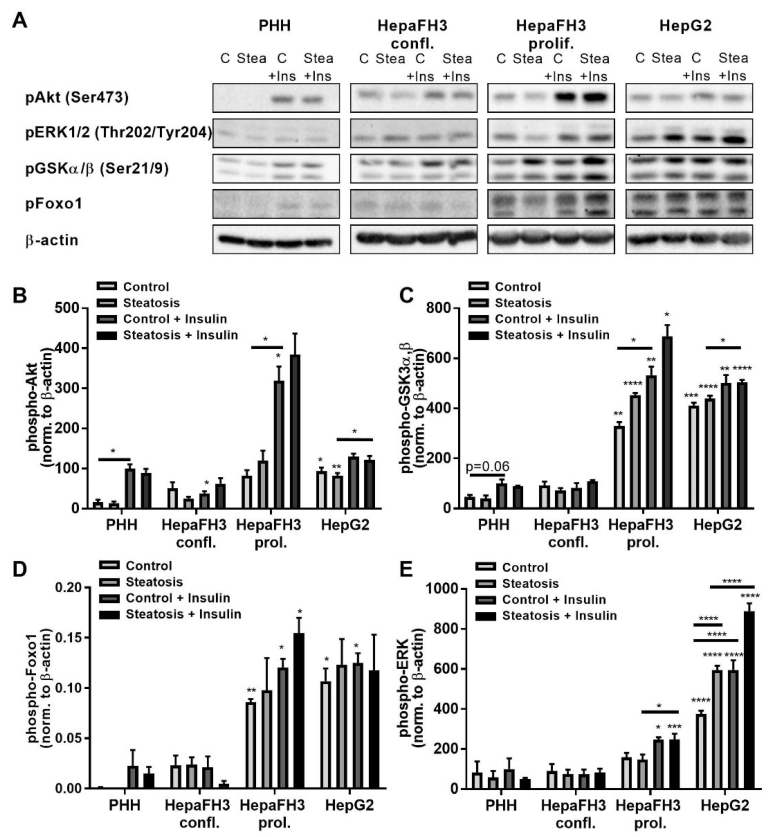
3.5. Increased Phosphorylation of AKT Kinase, ERK and Foxo1 of Proliferating HepaFH3 and HepG2 Cells Suggests Increased Proliferation and Survival
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, M.J.; Houlihan, D.D.; Bentham, L.; Shaw, J.C.; Cramb, R.; Olliff, S.; Gill, P.S.; Neuberger, J.M.; Lilford, R.J.; Newsome, P.N. Presence and severity of non-alcoholic fatty liver disease in a large prospective primary care cohort. J. Hepatol. 2012, 56, 234–240. [Google Scholar] [CrossRef] [PubMed]
- McPherson, S.; Hardy, T.; Henderson, E.; Burt, A.D.; Day, C.P.; Anstee, Q.M. Evidence of NAFLD progression from steatosis to fibrosing-steatohepatitis using paired biopsies: Implications for prognosis and clinical management. J. Hepatol. 2015, 62, 1148–1155. [Google Scholar] [CrossRef] [PubMed]
- Wong, V.W.-S.; Wong, G.L.-H.; Choi, P.C.-L.; Chan, A.W.-H.; Li, M.K.-P.; Chan, H.-Y.; Chim, A.M.-L.; Yu, J.; Sung, J.J.-Y.; Chan, H.L.-Y. Disease progression of non-alcoholic fatty liver disease: A prospective study with paired liver biopsies at 3 years. Gut 2010, 59, 969–974. [Google Scholar] [CrossRef]
- Stine, J.G.; Wentworth, B.J.; Zimmet, A.; Rinella, M.E.; Loomba, R.; Caldwell, S.H.; Argo, C.K. Systematic review with meta-analysis: Risk of hepatocellular carcinoma in non-alcoholic steatohepatitis without cirrhosis compared to other liver diseases. Aliment. Pharmacol. Ther. 2018, 48, 696–703. [Google Scholar] [CrossRef]
- Huang, D.Q.; El-Serag, H.B.; Loomba, R. Global epidemiology of NAFLD-related HCC: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 223–238. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Brown, R.; Wahl, R. Overexpression of glut-1 glucose transporter in human breast cancer an immunohistochemical study. Cancer 1993, 72, 2979–2985. [Google Scholar] [CrossRef]
- Nowak, N.; Kulma, A.; Gutowicz, J. Up-regulation of Key Glycolysis Proteins in Cancer Development. Open Life Sci. 2018, 13, 569–581. [Google Scholar] [CrossRef]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Dai, H.; Wu, Y.; Li, B.; Yi, J.; Xu, C.; Wu, X. In Vitro and in vivo mechanism of hepatocellular carcinoma inhibition by β-TCP nanoparticles. Int. J. Nanomed. 2019, 14, 3491–3502. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.H.; Lee, C.M.; Ha, S.H.; Lee, J.; Jang, K.Y.; Park, S.H. Induction of cell cycle arrest and apoptosis by tomentosin in hepatocellular carcinoma HepG2 and Huh7 cells. Hum. Exp. Toxicol. 2021, 40, 231–244. [Google Scholar] [CrossRef]
- Arzumanian, V.A.; Kiseleva, O.I.; Poverennaya, E.V. The Curious Case of the HepG2 Cell Line: 40 Years of Expertise. Int. J. Mol. Sci. 2021, 22, 13135. [Google Scholar] [CrossRef]
- Gómez-Lechón, M.J.; Donato, M.T.; Martínez-Romero, A.; Jiménez, N.; Castell, J.V.; O’Connor, J.-E. A human hepatocellular in vitro model to investigate steatosis. Chem. Biol. Interact. 2007, 165, 106–116. [Google Scholar] [CrossRef]
- Molinaro, A.; Becattini, B.; Solinas, G. Insulin signaling and glucose metabolism in different hepatoma cell lines deviate from hepatocyte physiology toward a convergent aberrant phenotype. Sci. Rep. 2020, 10, 12031. [Google Scholar] [CrossRef]
- Skolik, R.A.; Solocinski, J.; Konkle, M.E.; Chakraborty, N.; Menze, M.A. Global changes to HepG2 cell metabolism in response to galactose treatment. Am. J. Physiol. Cell Physiol. 2021, 320, C778–C793. [Google Scholar] [CrossRef]
- Gómez-Lechón, M.J.; Castell, J.V.; Donato, M.T. An update on metabolism studies using human hepatocytes in primary culture. Expert Opin. Drug Metab. Toxicol. 2008, 4, 837–854. [Google Scholar] [CrossRef]
- Garnier, D.; Li, R.; Delbos, F.; Fourrier, A.; Collet, C.; Guguen-Guillouzo, C.; Chesné, C.; Nguyen, T.H. Expansion of human primary hepatocytes in vitro through their amplification as liver progenitors in a 3D organoid system. Sci. Rep. 2018, 8, 8222. [Google Scholar] [CrossRef]
- Rose, S.; Ezan, F.; Cuvellier, M.; Bruyère, A.; Legagneux, V.; Langouët, S.; Baffet, G. Generation of proliferating human adult hepatocytes using optimized 3D culture conditions. Sci. Rep. 2021, 11, 515. [Google Scholar] [CrossRef] [PubMed]
- Ramboer, E.; De Craene, B.; De Kock, J.; Vanhaecke, T.; Berx, G.; Rogiers, V.; Vinken, M. Strategies for immortalization of primary hepatocytes. J. Hepatol. 2014, 61, 925–943. [Google Scholar] [CrossRef] [PubMed]
- Fukaya, K.-I.; Asahi, S.; Nagamori, S.; Sakaguchi, M.; Gao, C.; Miyazaki, M.; Namba, M. Establishment of a human hepatocyte line (OUMS-29) having CYP 1A1 and 1A2 activities from fetal liver tissue by transfection of SV40 LT. In Vitro Cell Dev. Biol. Anim. 2001, 37, 266. [Google Scholar] [CrossRef]
- Tsuruga, Y.; Kiyono, T.; Matsushita, M.; Takahashi, T.; Kasai, H.; Todo, S. Establishment of Immortalized Human Hepatocytes by Introduction of HPV16 E6/E7 and hTERT as Cell Sources for Liver Cell-Based Therapy. Cell Transplant. 2008, 17, 1083–1094. [Google Scholar] [CrossRef] [PubMed]
- Burkard, A.; Dähn, C.; Heinz, S.; Zutavern, A.; Sonntag-Buck, V.; Maltman, D.; Przyborski, S.; Hewitt, N.J.; Braspenning, J. Generation of proliferating human hepatocytes using Upcyte® technology: Characterisation and applications in induction and cytotoxicity assays. Xenobiotica 2012, 42, 939–956. [Google Scholar] [CrossRef] [PubMed]
- Herzog, N.; Hansen, M.; Miethbauer, S.; Schmidtke, K.-U.; Anderer, U.; Lupp, A.; Sperling, S.; Seehofer, D.; Damm, G.; Scheibner, K.; et al. Primary-like human hepatocytes genetically engineered to obtain proliferation competence display hepatic differentiation characteristics in monolayer and organotypical spheroid cultures. Cell Biol. Int. 2016, 40, 341–353. [Google Scholar] [CrossRef] [PubMed]
- Rajan, N.; Habermehl, J.; Coté, M.-F.; Doillon, C.J.; Mantovani, D. Preparation of ready-to-use, storable and reconstituted type I collagen from rat tail tendon for tissue engineering applications. Nat. Protoc. 2006, 1, 2753–2758. [Google Scholar] [CrossRef]
- Kegel, V.; Deharde, D.; Pfeiffer, E.; Zeilinger, K.; Seehofer, D.; Damm, G. Protocol for Isolation of Primary Human Hepatocytes and Corresponding Major Populations of Non-parenchymal Liver Cells. J. Vis. Exp. 2016, 109, e53069. [Google Scholar] [CrossRef]
- Damm, G.; Schicht, G.; Zimmermann, A.; Rennert, C.; Fischer, N.; Kießig, M.; Wagner, T.; Kegel, V.; Seehofer, D. Effect of glucose and insulin supplementation on the isolation of primary human hepatocytes. EXCLI J. 2019, 18, 1071–1091. [Google Scholar] [CrossRef] [PubMed]
- Braspenning, J.; Holder, S.; Kuepper, H. Propagation of Primary Cells and Use Therof. Patent EP2185690, 19 May 2010. [Google Scholar]
- Seidemann, L.; Krüger, A.; Kegel-Hübner, V.; Seehofer, D.; Damm, G. Influence of Genistein on Hepatic Lipid Metabolism in an In Vitro Model of Hepatic Steatosis. Molecules 2021, 26, 1156. [Google Scholar] [CrossRef]
- Mooli, R.G.R.; Rodriguez, J.; Takahashi, S.; Solanki, S.; Gonzalez, F.J.; Ramakrishnan, S.K.; Shah, Y.M. Hypoxia via ERK Signaling Inhibits Hepatic PPARα to Promote Fatty Liver. Cell. Mol. Gastroenterol. Hepatol. 2021, 12, 585–597. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.-L.; Pan, Q.; Cao, H.-X.; Xin, F.-Z.; Zhao, Z.-H.; Yang, R.-X.; Zeng, J.; Zhou, H.; Fan, J.-G. Lipotoxic Hepatocyte-Derived Exosomal MicroRNA 192-5p Activates Macrophages Through Rictor/Akt/Forkhead Box Transcription Factor O1 Signaling in Nonalcoholic Fatty Liver Disease. Hepatology 2020, 72, 454–469. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.V.; Tan, A.S. Characterization of the cellular reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT): Subcellular localization, substrate dependence, and involvement of mitochondrial electron transport in MTT reduction. Arch. Biochem. Biophys. 1993, 303, 474–482. [Google Scholar] [CrossRef] [PubMed]
- Zawada, R.J.X.; Kwan, P.; Olszewski, K.L.; Llinas, M.; Huang, S.-G. Quantitative determination of urea concentrations in cell culture medium. Biochem. Cell Biol. 2009, 87, 541–544. [Google Scholar] [CrossRef]
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in cancer: Mechanisms and advances in clinical trials. Mol. Cancer 2019, 18, 26. [Google Scholar] [CrossRef]
- Welsh, G.I.; Wilson, C.; Proud, C.G. GSK3: A SHAGGY frog story. Trends Cell Biol. 1996, 6, 274–279. [Google Scholar] [CrossRef]
- Hermida, M.A.; Dinesh Kumar, J.; Leslie, N.R. GSK3 and its interactions with the PI3K/AKT/mTOR signalling network. Adv. Biol. Regul. 2017, 65, 5–15. [Google Scholar] [CrossRef]
- Guo, Y.-J.; Pan, W.-W.; Liu, S.-B.; Shen, Z.-F.; Xu, Y.; Hu, L.-L. ERK/MAPK signalling pathway and tumorigenesis. Exp. Ther. Med. 2020, 19, 1997–2007. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Matsuzaki, J.; Katsuda, T.; Saito, Y.; Saito, H.; Ochiya, T. Generation of functional human hepatocytes in vitro: Current status and future prospects. Inflamm. Regen. 2019, 39, 13. [Google Scholar] [CrossRef]
- Levy, G.; Bomze, D.; Heinz, S.; Ramachandran, S.D.; Noerenberg, A.; Cohen, M.; Shibolet, O.; Sklan, E.; Braspenning, J.; Nahmias, Y. Long-term culture and expansion of primary human hepatocytes. Nat. Biotechnol. 2015, 33, 1264–1271. [Google Scholar] [CrossRef]
- Tolosa, L.; Gómez-Lechón, M.J.; López, S.; Guzmán, C.; Castell, J.V.; Donato, M.T.; Jover, R. Human Upcyte Hepatocytes: Characterization of the Hepatic Phenotype and Evaluation for Acute and Long-Term Hepatotoxicity Routine Testing. Toxicol. Sci. 2016, 152, 214–229. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, S.D.; Vivarès, A.; Klieber, S.; Hewitt, N.J.; Muenst, B.; Heinz, S.; Walles, H.; Braspenning, J. Applicability of second-generation upcyte® human hepatocytes for use in CYP inhibition and induction studies. Pharmacol. Res. Perspect. 2015, 3, e00161. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Lum, J.J.; Hatzivassiliou, G.; Thompson, C.B. The biology of cancer: Metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008, 7, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, A.M.; Fabris, R.; Bassetto, F.; Serra, R.; Leturque, A.; Federspil, G.; Girard, J.; Vettor, R. Hyperlactatemia reduces muscle glucose uptake and GLUT-4 mRNA while increasing (E1alpha)PDH gene expression in rat. Am. J. Physiol. 1999, 276, E922–E929. [Google Scholar] [CrossRef]
- Choi, C.S.; Kim, Y.-B.; Lee, F.N.; Zabolotny, J.M.; Kahn, B.B.; Youn, J.H. Lactate induces insulin resistance in skeletal muscle by suppressing glycolysis and impairing insulin signaling. Am. J. Physiol. Endocrinol. Metab. 2002, 283, E233–E240. [Google Scholar] [CrossRef]
- Farzaneh, Z.; Vosough, M.; Agarwal, T.; Farzaneh, M. Critical signaling pathways governing hepatocellular carcinoma behavior; small molecule-based approaches. Cancer Cell Int. 2021, 21, 208. [Google Scholar] [CrossRef]
- Wang, Z.; Cui, X.; Hao, G.; He, J. Aberrant expression of PI3K/AKT signaling is involved in apoptosis resistance of hepatocellular carcinoma. Open Life Sci. 2021, 16, 1037–1044. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, P.D.; Delgado, E.R.; Alencastro, F.; Leek, M.P.; Roy, N.; Weirich, M.P.; Stahl, E.C.; Otero, P.A.; Chen, M.I.; Brown, W.K.; et al. The Polyploid State Restricts Hepatocyte Proliferation and Liver Regeneration in Mice. Hepatology 2019, 69, 1242–1258. [Google Scholar] [CrossRef]
- Yang, S.; Pang, L.; Dai, W.; Wu, S.; Ren, T.; Duan, Y.; Zheng, Y.; Bi, S.; Zhang, X.; Kong, J. Role of Forkhead Box O Proteins in Hepatocellular Carcinoma Biology and Progression (Review). Front. Oncol. 2021, 11, 667730. [Google Scholar] [CrossRef]
- Wondisford, A.R.; Xiong, L.; Chang, E.; Meng, S.; Meyers, D.J.; Li, M.; Cole, P.A.; He, L. Control of Foxo1 gene expression by co-activator P300. J. Biol. Chem. 2014, 289, 4326–4333. [Google Scholar] [CrossRef]
- Park, S.; Park, J.-H.; Jung, H.-J.; Jang, J.-H.; Ahn, S.; Kim, Y.; Suh, P.-G.; Chae, S.; Yoon, J.H.; Ryu, S.H.; et al. A secretome profile indicative of oleate-induced proliferation of HepG2 hepatocellular carcinoma cells. Exp. Mol. Med. 2018, 50, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.; Jeong, D.-W.; Park, J.-W.; Lee, K.-W.; Fukuda, J.; Chun, Y.-S. Fatty-acid-induced FABP5/HIF-1 reprograms lipid metabolism and enhances the proliferation of liver cancer cells. Commun. Biol. 2020, 3, 638. [Google Scholar] [CrossRef] [PubMed]
- Rennert, C.; Heil, T.; Schicht, G.; Stilkerich, A.; Seidemann, L.; Kegel-Hübner, V.; Seehofer, D.; Damm, G. Prolonged Lipid Accumulation in Cultured Primary Human Hepatocytes Rather Leads to ER Stress than Oxidative Stress. Int. J. Mol. Sci. 2020, 21, 7097. [Google Scholar] [CrossRef] [PubMed]
- Giulitti, F.; Petrungaro, S.; Mandatori, S.; Tomaipitinca, L.; de Franchis, V.; D’Amore, A.; Filippini, A.; Gaudio, E.; Ziparo, E.; Giampietri, C. Anti-tumor Effect of Oleic Acid in Hepatocellular Carcinoma Cell Lines via Autophagy Reduction. Front. Cell Dev. Biol. 2021, 9, 629182. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Zhu, M.; Liu, X.; Chen, X.; Yuan, Y.; Li, L.; Liu, J.; Lu, Y.; Cheng, J.; Chen, Y. Oleic acid ameliorates palmitic acid induced hepatocellular lipotoxicity by inhibition of ER stress and pyroptosis. Nutr. Metab. 2020, 17, 11. [Google Scholar] [CrossRef]
- Nishiwaki, M.; Toyoda, M.; Oishi, Y.; Ishida, S.; Horiuchi, S.-I.; Makino-Itou, H.; Kimura, T.; Ohno, S.-I.; Ohkura, T.; Enosawa, S.; et al. Immortalization of human hepatocytes from biliary atresia with CDK4R24C, cyclin D1, and TERT for cytochrome P450 induction testing. Sci. Rep. 2020, 10, 17503. [Google Scholar] [CrossRef]
- Giri, S.; Bader, A. Immortalization of Human Fetal Hepatocyte by Ectopic Expression of Human Telomerase Reverse Transcriptase, Human Papilloma Virus (E7) and Simian Virus 40 Large T (SV40 T) Antigen Towards Bioartificial Liver Support. J. Clin. Exp. Hepatol. 2014, 4, 191–201. [Google Scholar] [CrossRef]
- Marion, M.-J.; Hantz, O.; Durantel, D. The HepaRG cell line: Biological properties and relevance as a tool for cell biology, drug metabolism, and virology studies. Methods Mol. Biol. 2010, 640, 261–272. [Google Scholar] [CrossRef]
- Cerec, V.; Glaise, D.; Garnier, D.; Morosan, S.; Turlin, B.; Drenou, B.; Gripon, P.; Kremsdorf, D.; Guguen-Guillouzo, C.; Corlu, A. Transdifferentiation of hepatocyte-like cells from the human hepatoma HepaRG cell line through bipotent progenitor. Hepatology 2007, 45, 957–967. [Google Scholar] [CrossRef]
- Lübberstedt, M.; Müller-Vieira, U.; Mayer, M.; Biemel, K.M.; Knöspel, F.; Knobeloch, D.; Nüssler, A.K.; Gerlach, J.C.; Zeilinger, K. HepaRG human hepatic cell line utility as a surrogate for primary human hepatocytes in drug metabolism assessment in vitro. J. Pharmacol. Toxicol. Methods 2011, 63, 59–68. [Google Scholar] [CrossRef]
- Lin, H.; Wong, G.L.-H.; Whatling, C.; Chan, A.W.-H.; Leung, H.H.-W.; Tse, C.-H.; Shu, S.S.-T.; Chim, A.M.-L.; Lai, J.C.-T.; Yip, T.C.-F.; et al. Association of genetic variations with NAFLD in lean individuals. Liver Int. 2022, 42, 149–160. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Donor | Age | Sex (w/m/d) | Diagnosis | Secondary Diagnosis |
|---|---|---|---|---|
| Donor 1 | 32 | m | Brain metastasis | n.k. |
| Donor 2 | 49 | w | Small bowel tumor with liver metastasis | n.k. |
| Donor 3 | 60 | w | Klatskin tumor | hyperthyroidism |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scheffschick, A.; Babel, J.; Sperling, S.; Nerusch, J.; Herzog, N.; Seehofer, D.; Damm, G. Primary-like Human Hepatocytes Genetically Engineered to Obtain Proliferation Competence as a Capable Application for Energy Metabolism Experiments in In Vitro Oncologic Liver Models. Biology 2022, 11, 1195. https://doi.org/10.3390/biology11081195
Scheffschick A, Babel J, Sperling S, Nerusch J, Herzog N, Seehofer D, Damm G. Primary-like Human Hepatocytes Genetically Engineered to Obtain Proliferation Competence as a Capable Application for Energy Metabolism Experiments in In Vitro Oncologic Liver Models. Biology. 2022; 11(8):1195. https://doi.org/10.3390/biology11081195
Chicago/Turabian StyleScheffschick, Andrea, Jonas Babel, Sebastian Sperling, Julia Nerusch, Natalie Herzog, Daniel Seehofer, and Georg Damm. 2022. "Primary-like Human Hepatocytes Genetically Engineered to Obtain Proliferation Competence as a Capable Application for Energy Metabolism Experiments in In Vitro Oncologic Liver Models" Biology 11, no. 8: 1195. https://doi.org/10.3390/biology11081195
APA StyleScheffschick, A., Babel, J., Sperling, S., Nerusch, J., Herzog, N., Seehofer, D., & Damm, G. (2022). Primary-like Human Hepatocytes Genetically Engineered to Obtain Proliferation Competence as a Capable Application for Energy Metabolism Experiments in In Vitro Oncologic Liver Models. Biology, 11(8), 1195. https://doi.org/10.3390/biology11081195