
Anti-Prion Systems Block Prion Transmission, Attenuate Prion Generation, Cure Most Prions as They Arise and Limit Prion-Induced Pathology in Saccharomyces cerevisiae

Abstract
:Simple Summary
Abstract
1. Introduction
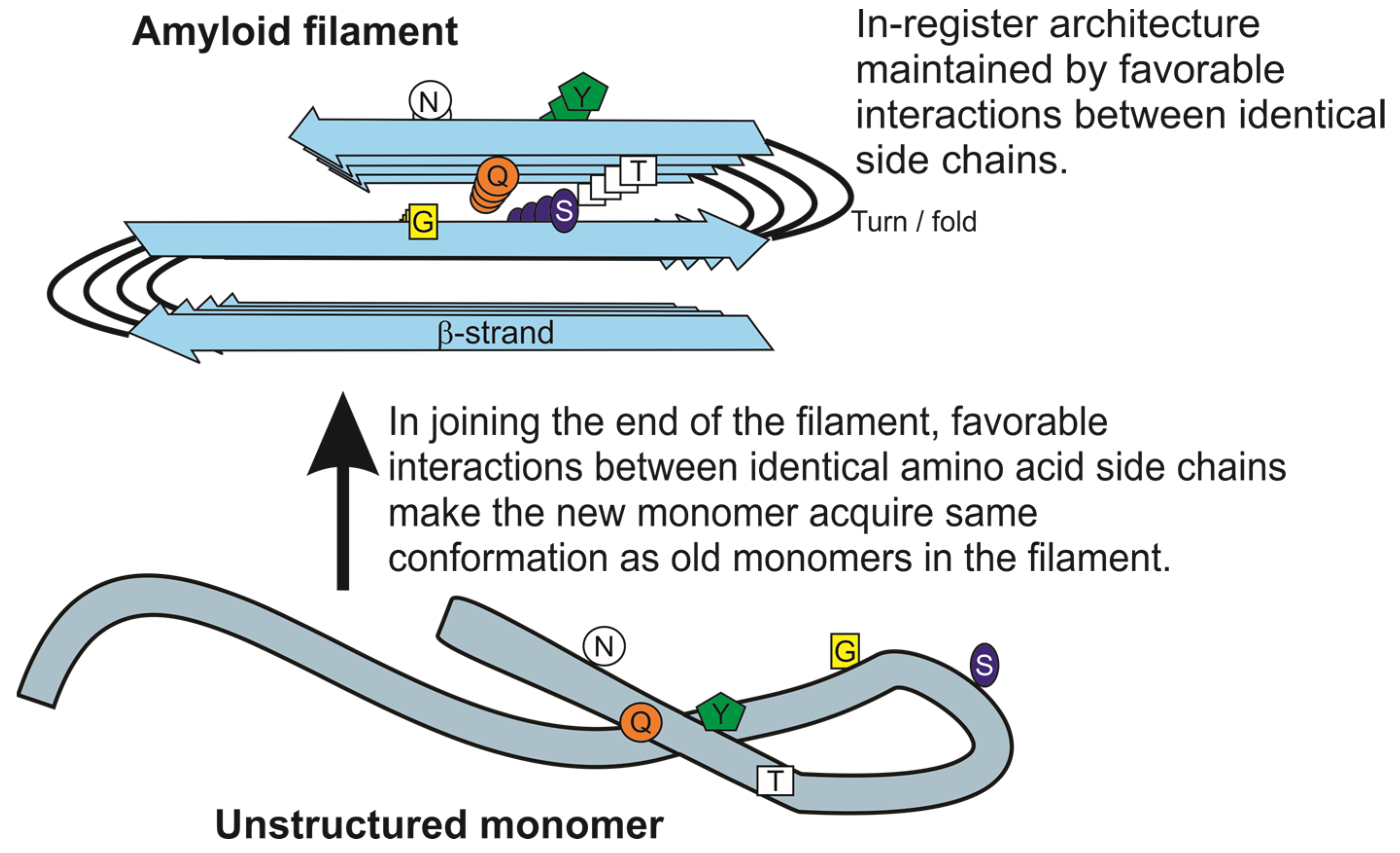
2. Prion Protein Polymorphisms Impede Infection
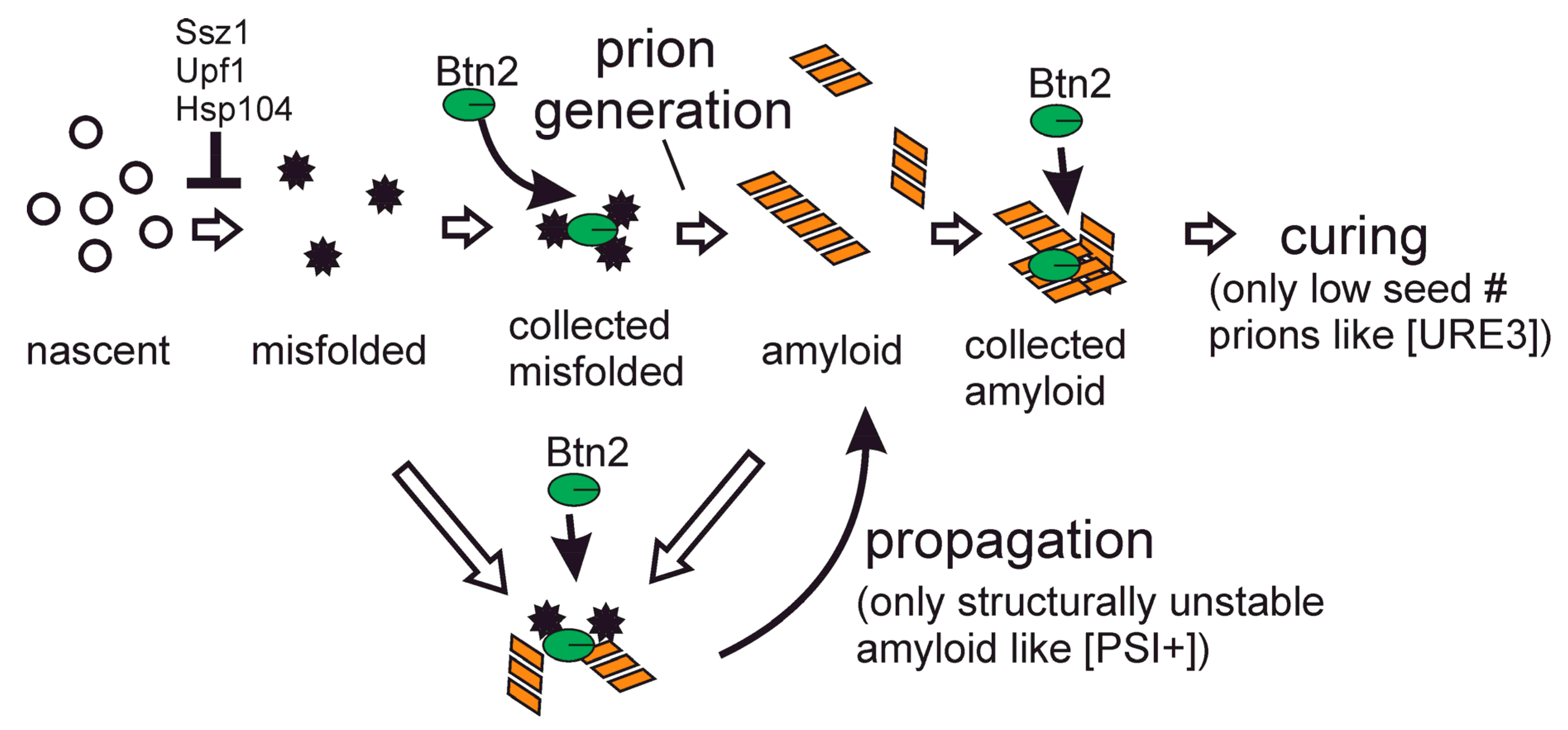
3. Btn2 Sequesters Prion Amyloids, Curing Most [URE3] Isolates
4. Ribosome-Associated Chaperones Cure Many [Psi+] Variants and Block Formation of Others
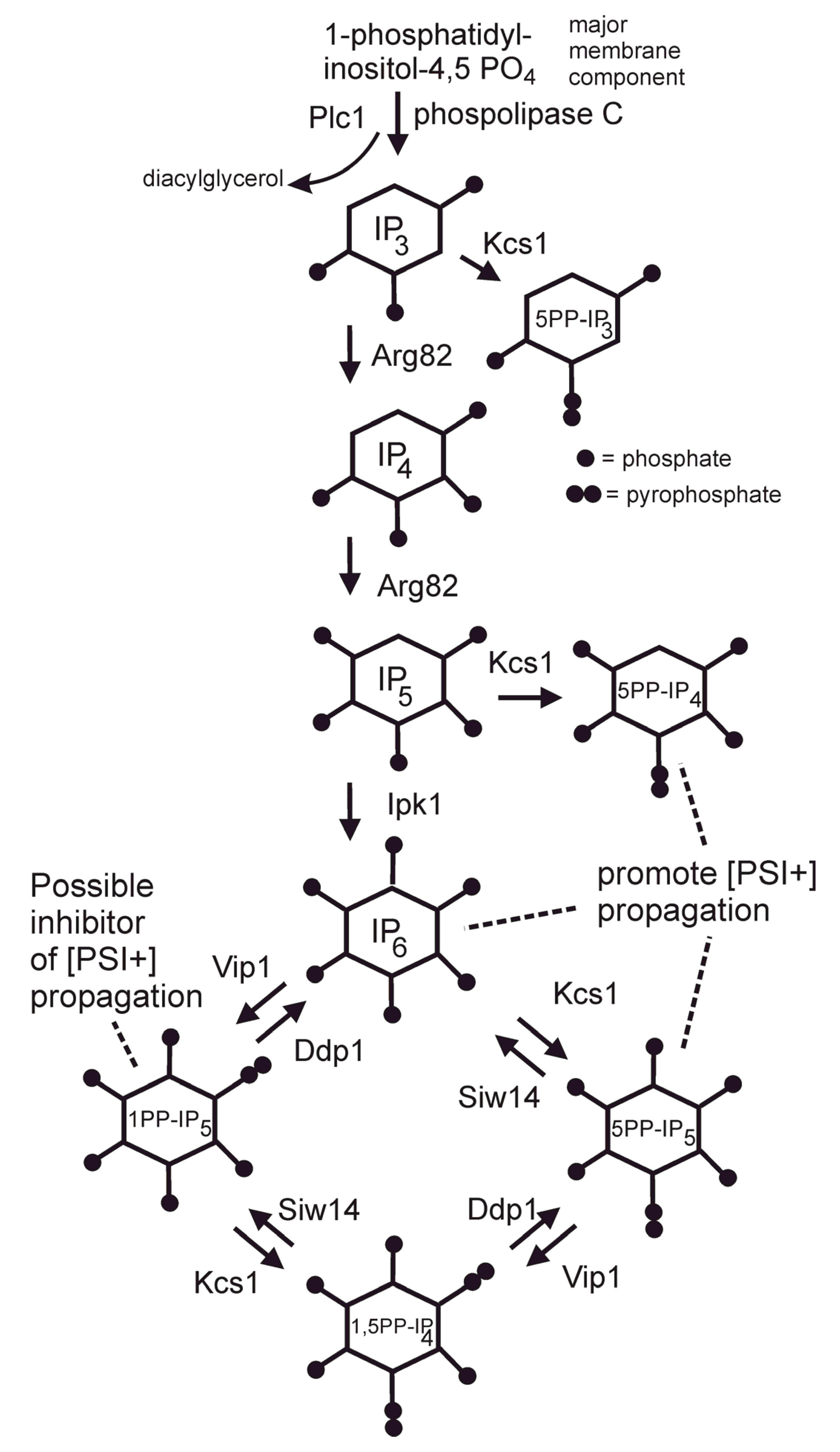
5. Siw14 Curing of [PSI+] and Inositol Polyphosphates
6. Normal Levels of Hsp104 Cure Many [PSI+] Variants and Decrease Formation of Others
7. Upf Proteins Cure Most [PSI+] Variants by Association with Sup35
8. Ribosome-Associated Chaperones, Nonsense-Mediated Decay Factors and Hsp104 Together Repress [PSI+] Prion Emergence ~5000-fold
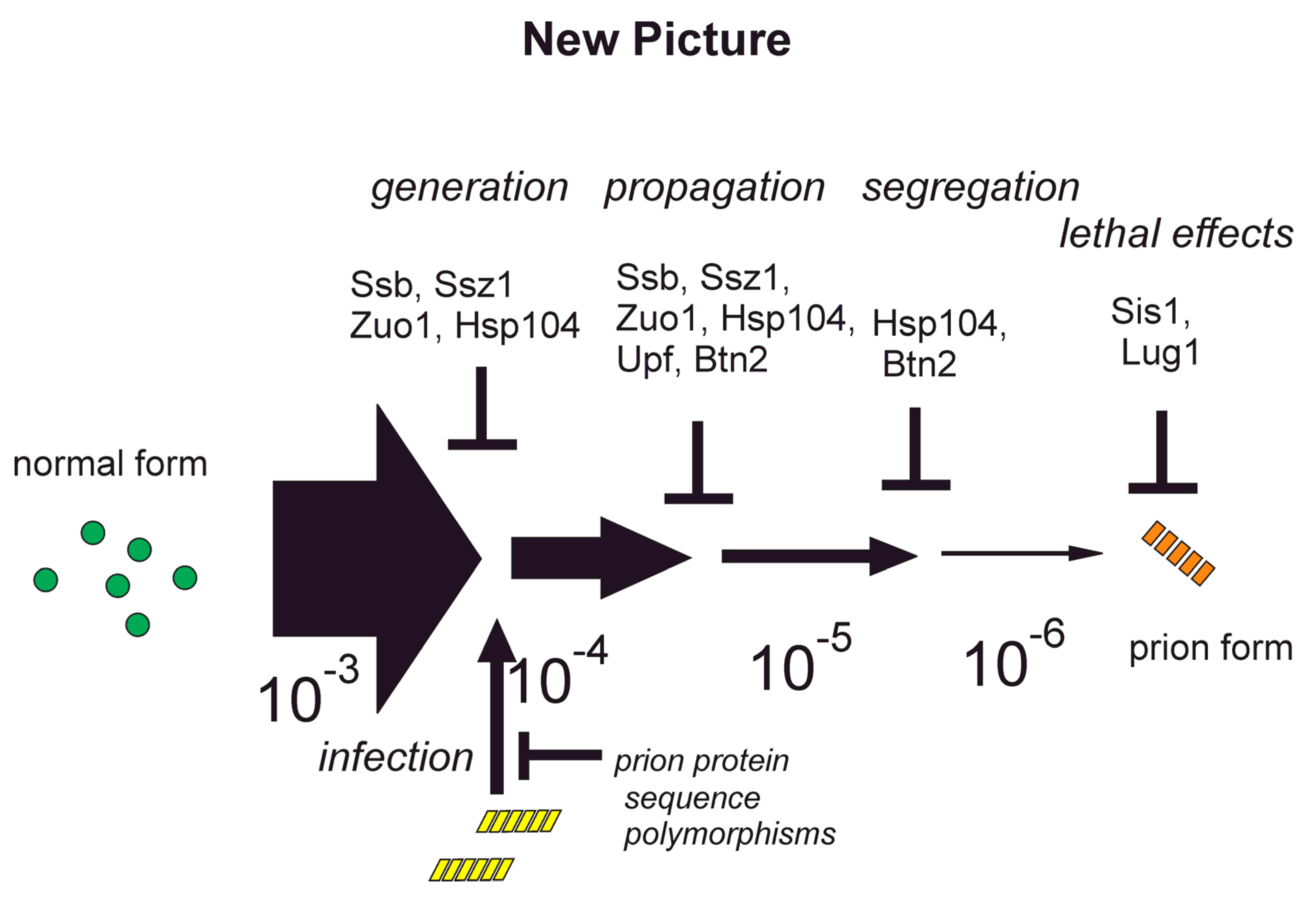
9. Anti-prion Systems Turn a Tsunami of Prions into a Slow Drip
10. Why do Btn2 and Cur1 Affect [PSI+] The Opposite of Their Effects on [URE3]?
11. Sis1 Reduces [PSI+] Pathogenesis by Keeping Some SUP35 Soluble (Active)
12. Lug1 Prevents Lethality of Otherwise Mild [URE3] Variants
13. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wickner, R.B. [URE3] as an altered URE2 protein: Evidence for a prion analog in S. cerevisiae. Science 1994, 264, 566–569. [Google Scholar] [CrossRef]
- Cox, B.S. PSI, a cytoplasmic suppressor of super-suppressor in yeast. Heredity 1965, 20, 505–521. [Google Scholar] [CrossRef]
- Lacroute, F. Non-Mendelian mutation allowing ureidosuccinic acid uptake in yeast. J. Bacteriol. 1971, 106, 519–522. [Google Scholar] [CrossRef]
- Zhouravleva, G.; Frolova, L.; LeGoff, X.; LeGuellec, R.; Inge-Vectomov, S.; Kisselev, L.; Philippe, M. Termination of translation in eukaryotes is governed by two interacting polypeptide chain release factors, eRF1 and eRF3. EMBO J. 1995, 14, 4065–4072. [Google Scholar] [CrossRef]
- Stansfield, I.; Jones, K.M.; Kushnirov, V.V.; Dagkesamanskaya, A.R.; Poznyakovski, A.I.; Paushkin, S.V.; Nierras, C.R.; Cox, B.S.; Ter-Avanesyan, M.D.; Tuite, M.F. The products of the SUP45 (eRF1) and SUP35 genes interact to mediate translation termination in Saccharomyces cerevisiae. EMBO J. 1995, 14, 4365–4373. [Google Scholar] [CrossRef]
- Liebman, S.W.; Chernoff, Y.O. Prions in yeast. Genetics 2012, 191, 1041–1072. [Google Scholar] [CrossRef]
- Wickner, R.B.; Shewmaker, F.; Bateman, D.A.; Edskes, H.E.; Gorkovskiy, A.; Dayani, Y.; Bezsonov, E.E. Yeast prions: Structure, biology and prion-handling systems. Microbiol. Mol. Biol. Rev. 2015, 79, 1–17. [Google Scholar] [CrossRef]
- Drillien, R.; Lacroute, F. Ureidosuccinic acid uptake in yeast and some aspects of its regulation. J. Bacteriol. 1972, 109, 203–208. [Google Scholar] [CrossRef]
- Derkatch, I.L.; Bradley, M.E.; Zhou, P.; Chernoff, Y.O.; Liebman, S.W. Genetic and environmental factors affecting the de novo appearance of the [PSI+] prion in Saccharomyces cerevisiae. Genetics 1997, 147, 507–519. [Google Scholar] [CrossRef]
- Sondheimer, N.; Lindquist, S. Rnq1: An epigenetic modifier of protein function in yeast. Mol. Cell 2000, 5, 163–172. [Google Scholar] [CrossRef]
- Derkatch, I.L.; Bradley, M.E.; Hong, J.Y.; Liebman, S.W. Prions affect the appearance of other prions: The story of [PIN]. Cell 2001, 106, 171–182. [Google Scholar] [CrossRef]
- Shewmaker, F.; Wickner, R.B.; Tycko, R. Amyloid of the prion domain of Sup35p has an in-register parallel β-sheet structure. Proc. Natl. Acad. Sci. USA 2006, 103, 19754–19759. [Google Scholar] [CrossRef] [PubMed]
- Baxa, U.; Wickner, R.B.; Steven, A.C.; Anderson, D.; Marekov, L.; Yau, W.-M.; Tycko, R. Characterization of β-sheet structure in Ure2p1-89 yeast prion fibrils by solid state nuclear magnetic resonance. Biochemistry 2007, 46, 13149–13162. [Google Scholar] [CrossRef]
- Wickner, R.B.; Dyda, F.; Tycko, R. Amyloid of Rnq1p, the basis of the [PIN+] prion, has a parallel in-register β-sheet structure. Proc. Natl. Acad. Sci. USA 2008, 105, 2403–2408. [Google Scholar] [CrossRef] [PubMed]
- Margittai, M.; Langen, R. Fibrils with parallel in-register structure constitute a major class of amyloid fibrils: Molecular insights from electron paramagnetic resonance spectroscopy. Q. Rev. Biophys. 2008, 41, 265–297. [Google Scholar] [CrossRef]
- Ngo, S.; Gu, L.; Guo, Z. Hierarchical organization in the amyloid core of yeast prion protein Ure2. J. Biol. Chem. 2011, 286, 29691–29699. [Google Scholar] [CrossRef]
- Wang, J.; Park, G.; Lee, Y.K.; Nguyen, M.; San Fung, T.; Lin, T.Y.; Hsu, F.; Guo, Z. Spin label scanning reveals likely locations of beta-strands of the Ure2 prion domain. ACS Omega 2020, 5, 5984–5993. [Google Scholar] [CrossRef]
- Derkatch, I.L.; Chernoff, Y.O.; Kushnirov, V.V.; Inge-Vechtomov, S.G.; Liebman, S.W. Genesis and variability of [PSI] prion factors in Saccharomyces cerevisiae. Genetics 1996, 144, 1375–1386. [Google Scholar] [CrossRef]
- Schlumpberger, M.; Prusiner, S.B.; Herskowitz, I. Induction of distinct [URE3] yeast prion strains. Mol. Cell Biol. 2001, 21, 7035–7046. [Google Scholar] [CrossRef]
- Carta, M.; Aguzzi, A. Molecular foundations of prion strain diversity. Curr. Opin. Neurobiol. 2022, 72, 22–31. [Google Scholar] [CrossRef]
- Bateman, D.; Wickner, R.B. The [PSI+] prion exists as a dynamic cloud of variants. PLoS Genet. 2013, 9, e1003257. [Google Scholar] [CrossRef] [PubMed]
- Kimberlin, R.H.; Cole, S.; Walker, C.A. Temporary and permanent modifications to a single strain of mouse scrapie on transmission to rats and hamsters. J. Gen. Virol. 1987, 68, 1875–1881. [Google Scholar] [CrossRef] [PubMed]
- McGlinchey, R.; Kryndushkin, D.; Wickner, R.B. Suicidal [PSI+] is a lethal yeast prion. Proc. Natl. Acad. Sci. USA 2011, 108, 5337–5341. [Google Scholar] [CrossRef] [PubMed]
- Wickner, R.B.; Edskes, H.K.; Shewmaker, F.; Nakayashiki, T. Prions of fungi: Inherited structures and biological roles. Nat. Rev. Microbiol. 2007, 5, 611–618. [Google Scholar] [CrossRef]
- Toyama, B.H.; Kelly, M.J.; Gross, J.D.; Weissman, J.S. The structural basis of yeast prion strain variants. Nature 2007, 449, 233–237. [Google Scholar] [CrossRef]
- Dergalev, A.A.; Alexandrovg, A.; Ivannikov, R.I.; Ter-Avanesyan, M.D.; Kushnirov, V.V. Yeast Sup35 prion structure: Two types, four parts, many variants. Int. J. Mol. Sci. 2019, 20, 2633. [Google Scholar] [CrossRef]
- Kraus, A.; Hoyt, F.; Schwartz, C.L.; Hansen, B.; Artikis, E.; Hughson, A.G.; Raymond, G.J.; Race, B.; Baron, G.S.; Caughey, B. High-resolution structure and strain comparison of infectious mammalian prions. Mol. Cell 2021, 81, 4540–4551.e6. [Google Scholar] [CrossRef]
- Antzutkin, O.N.; Balbach, J.J.; Leapman, R.D.; Rizzo, N.W.; Reed, J.; Tycko, R. Multiple quantum solid-state NMR indicates a parallel, not antiparallel, organization of beta-sheets in Alzheimer’s beta-amyloid fibrils. Proc. Natl. Acad. Sci. USA 2000, 97, 13045–13050. [Google Scholar] [CrossRef]
- Petkova, A.T.; Leapman, R.D.; Guo, Z.; Yau, W.M.; Mattson, M.P.; Tycko, R. Self-propagating, molecular-level polymorphism in Alzheimer’s beta-amyloid fibrils. Science 2005, 307, 262–265. [Google Scholar] [CrossRef]
- Jaunmuktane, Z.; Mead, S.; Ellis, M.; Wadsworth, J.D.; Nicoll, A.J.; Kenny, J.; Launchbury, F.; Linehan, J.; Richard-Loendt, A.; Walker, A.S.; et al. Evidence for human transmission of amyloid-β pathology and cerebral amyloid angiopathy. Nature 2015, 525, 247–250. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, A.; Morales-Scheihing, D.; Salvadores, N.; Moreno-Gonzales, I.; Gonzales, C.; Taylor-Presse KMendez, N.; Shahnawaz, M.; Gaber, A.O.; Sabek, O.M.; Fraga, D.W.; et al. Inductionn of IAPP amyloid deposition and associated diabetic abnormalities by a prion-like mechanism. J. Exper. Med. 2017, 214, 2591–2610. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kwon, S.-H.; Kam, T.-I.; Panicker, N.; Karuppagounder, S.S.; Lee, S.; Lee, J.H.; Kim, W.R.; Kook, M.; Foss, C.A.; et al. Transneuronal propagation of pathologic α-synuclein from the gut to the brain models Parkinson’s disease. Neuron 2019, 103, 627–641. [Google Scholar] [CrossRef] [PubMed]
- Sampson, T.R.; Challis, C.; Jain, N.; Moiseyenko, A.; Ladinsky, M.S.; Shastri, G.G.; Thron, T.; Needham, B.D.; Horvath, I.; Debelius, J.W.; et al. A gut bacterial amyloid promotes α−synuclein aggregation and motor impairment in mice. Elife 2020, 9, e53111. [Google Scholar] [CrossRef] [PubMed]
- Wickner, R.B.; Bezsonov, E.E.; Son, M.; Ducatez, M.; DeWilde, M.; Edskes, H.E. Anti-prion systems in yeast and inositol polyphosphates. Biochemistry 2018, 57, 1285–1292. [Google Scholar] [CrossRef]
- Coustou, V.; Deleu, C.; Saupe, S.; Begueret, J. The protein product of the het-s heterokaryon incompatibility gene of the fungus Podospora anserina behaves as a prion analog. Proc. Natl. Acad. Sci. USA 1997, 94, 9773–9778. [Google Scholar] [CrossRef]
- Ritter, C.; Maddelein, M.L.; Siemer, A.B.; Luhrs, T.; Ernst, M.; Meier, B.H.; Saupe, S.J.; Riek, R. Correlation of structural elements and infectivity of the HET-s prion. Nature 2005, 435, 844–848. [Google Scholar] [CrossRef]
- Wasmer, C.; Lange, A.; Van Melckebeke, H.; Siemer, A.B.; Riek, R.; Meier, B.H. Amyloid fibrils of the HET-s(218-279) prion form a beta solenoid with a triangular hydrophobic core. Science 2008, 319, 1523–1526. [Google Scholar] [CrossRef]
- Lund, P.M.; Cox, B.S. Reversion analysis of [psi-] mutations in Saccharomyces cerevisiae. Genet. Res. 1981, 37, 173–182. [Google Scholar] [CrossRef]
- Nakayashiki, T.; Kurtzman, C.P.; Edskes, H.K.; Wickner, R.B. Yeast prions [URE3] and [PSI+] are diseases. Proc. Natl. Acad. Sci. USA 2005, 102, 10575–10580. [Google Scholar] [CrossRef]
- Halfmann, R.; Jarosz, D.F.; Jones, S.K.; Chang, A.; Lancster, A.K.; Lindquist, S. Prions are a common mechanism for phenotypic inheritance in wild yeasts. Nature 2012, 482, 363–368. [Google Scholar] [CrossRef] [Green Version]
- Kelly, A.C.; Shewmaker, F.P.; Kryndushkin, D.; Wickner, R.B. Sex, prions and plasmids in yeast. Proc. Natl. Acad. Sci. USA 2012, 109, E2683–E2690. [Google Scholar] [CrossRef] [PubMed]
- Kushnirov, V.V.; Ter-Avanesyan, M.D.; Didichenko, S.A.; Smirnov, V.N.; Chernoff, Y.O.; Derkach, I.L.; Novikova, O.N.; Inge-Vechtomov, S.G.; Neistat, M.A.; Tolstorukov, I.I. Divergence and conservation of SUP2 (SUP35) gene of yeasts Pichia pinus and Saccharomyces cerevisiae. Yeast 1990, 6, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Kushnirov, V.V.; Kochneva-Pervukhova, N.V.; Cechenova, M.B.; Frolova, N.S.; Ter-Avanesyan, M.D. Prion properties of the Sup35 protein of yeast Pichia methanolica. EMBO J. 2000, 19, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Santoso, A.; Chien, P.; Osherovich, L.Z.; Weissman, J.S. Molecular basis of a yeast prion species barrier. Cell 2000, 100, 277–288. [Google Scholar] [CrossRef]
- Jensen, M.A.; True, H.L.; Chernoff, Y.O.; Lindquist, S. Molecular population genetics and evolution of a prion-like protein in Saccharomyces cerevisiae. Genetics 2001, 159, 527–535. [Google Scholar] [CrossRef]
- Edskes, H.K.; Wickner, R.B. Conservation of a portion of the S. cerevisiae Ure2p prion domain that interacts with the full–length protein. Proc. Natl. Acad. Sci. USA 2002, 99 (Suppl. 4), 16384–16391.
- Resende, C.G.; Outeiro, T.F.; Sands, L.; Lindquist, S.; Tuite, M.F. Prion protein gene polymorphisms in Saccharomyces cerevisiae. Mol. Microbiol. 2003, 49, 1005–1017. [Google Scholar] [CrossRef]
- Edskes, H.E.; Khamar, H.J.; Winchester, C.-L.; Greenler, A.J.; Zhou, A.; McGlinchey, R.P.; Gorkovskiy, A.; Wickner, R.B. Sporadic distribution of prion-forming ability of Sup35p from yeasts and fungi. Genetics 2014, 198, 605–616. [Google Scholar] [CrossRef]
- Shewmaker, F.; Mull, L.; Nakayashiki, T.; Masison, D.C.; Wickner, R.B. Ure2p function is enhanced by its prion domain in Saccharomyces cerevisiae. Genetics 2007, 176, 1557–1565. [Google Scholar] [CrossRef]
- Hosoda, N.; Kobayashii, T.; Uchida, N.; Funakoshi, Y.; Kikuchi, Y.; Hoshino, S.; Katada, T. Translation termination factor eRF3 mediates mRNA decay through the regulation of deadenylation. J. Biol. Chem. 2003, 278, 38287–38291. [Google Scholar] [CrossRef]
- Li, X.; Kandel, E.R.; Derkatch, I.L. Functional role of Tia1/Pub1 and Sup35 prion domains: Directing protein synthesis machinery to the tubulin cytoskeleton. Mol. Cell 2014, 55, 305–318. [Google Scholar] [CrossRef] [Green Version]
- Bateman, D.A.; Wickner, R.B. [PSI+] prion transmission barriers protect Saccharomyces cerevisiae from infection: Intraspecies ‘species barriers’. Genetics 2012, 190, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Mead, S.; Stumpf, M.P.; Whitfield, J.; Beck, J.A.; Poulter, M.; Campbell, T.; Uphill, J.B.; Goldstein, D.; Alpers, M.; Fisher, E.M.; et al. Balancing selection at the prion protein gene consistent with prehistoric kurulike epidemics. Science 2003, 300, 640–643. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Newnam, G.P.; Chernoff, Y.O. Prion species barrier between the closely related yeast proteins is detected despite coaggregation. Proc. Natl. Acad. Sci. USA 2007, 104, 2791–2796. [Google Scholar] [CrossRef] [PubMed]
- Edskes, H.K.; McCann, L.M.; Hebert, A.M.; Wickner, R.B. Prion variants and species barriers among Saccharomyces Ure2 proteins. Genetics 2009, 181, 1159–1167. [Google Scholar] [CrossRef] [PubMed]
- Kryndushkin, D.; Shewmaker, F.; Wickner, R.B. Curing of the [URE3] prion by Btn2p, a Batten disease-related protein. EMBO J. 2008, 27, 2725–2735. [Google Scholar] [CrossRef]
- Wickner, R.B.; Beszonov, E.; Bateman, D.A. Normal levels of the antiprion proteins Btn2 and Cur1 cure most newly formed [URE3] prion variants. Proc. Natl. Acad. Sci. USA 2014, 111, E2711–E2720. [Google Scholar] [CrossRef]
- Kryndushkin, D.; Ihrke, G.; Piermartiri, T.C.; Shewmaker, F. A yeast model of optineurin proteinopathy reveals a unique aggregation pattern associated with cellular toxicity. Mol. Microbiol. 2012, 86, 1531–1547. [Google Scholar] [CrossRef]
- Malinovska, L.; Kroschwald, S.; Munder, M.C.; Richter, D.; Alberti, S. Molecular chaperones and stress-inducible protein-sorting factors coordinate the spaciotemporal distribution of protein aggregates. Mol. Biol. Cell 2012, 23, 3041–3056. [Google Scholar] [CrossRef]
- Ho, C.T.; Grousi, T.; Shatz, O.; Jawed, A.; Ruger-Herreros, C.; Semmelink, M.; Zahn, R.; Richter, K.; Bukau, B.; Mogk, A. Cellular sequestrases maintain basal Hsp70 capacity ensuring balanced proteostasis. Nat. Commun. 2019, 10, 4851. [Google Scholar] [CrossRef]
- Specht, S.; Miller, S.B.M.; Mogk, A.; Bukau, B. Hsp42 is required for sequestration of protein aggregates into deposition sites in Saccharomyces cerevisiae. J. Cell Biol. 2011, 195, 617–629. [Google Scholar] [CrossRef]
- Bezsonov, E.E.; Edskes, H.E.; Wickner, R.B. Innate immunity to yeast prions: Btn2p and Cur1p curing of the [URE3] prion is prevented by 60S ribosomal protein deficiency or ubiquitin/proteasome system overactivity. Genetics 2021, 217, iyab013. [Google Scholar] [CrossRef] [PubMed]
- Barbitoff, Y.A.; Matveenko, A.G.; Moskalnko, S.E.; Zemlyanko, O.M.; Newnam, G.P.; Patel, A.; Chernova, T.A.; Chernoff, Y.O.; Zhouravleva, G.A. To CURe or not to CURe? Differential effects of the chaperone sorting factor Cur1 on yeast prions are mediated by the chaperone Sis1. Mol. Microbiol. 2017, 105, 242–257. [Google Scholar] [CrossRef] [PubMed]
- Wickner, R.B.; Edskes, H.E.; Son, M.; Wu, S.; Niznikiewicz, M. Innate immunity to prions: Anti-prion systems turn a tsunami of prions into a slow drip. Curr. Genet. 2021, 67, 833–847. [Google Scholar] [CrossRef] [PubMed]
- Son, M.; Wickner, R.B. Anti-prion systems in yeast cooperate to cure or prevent the generation of nearly all [PSI+] and [URE3] prions. Proc. Natl. Acad. Sci. USA 2022, 119, e2205500119. [Google Scholar] [CrossRef]
- Edskes, H.E.; Stroobant, E.E.; DeWilde, M.; Bezsonov, E.E.; Wickner, R.B. Proteasome control of [URE3] prion propagation by degradation of anti-prion proteins Cur1 and Btn2 in Saccharomyces cerevisiae. Genetics 2021, 218, iyab037. [Google Scholar] [CrossRef]
- Nelson, R.J.; Ziegilhoffer, T.; Nicolet, C.; Werner-Washburne, M.; Craig, E.A. The translation machinery and 70 kDal heat shock protein cooperate in protein synthesis. Cell 1992, 71, 97–105. [Google Scholar] [CrossRef]
- Zhang, Y.; Valentín Gesé, G.; Conz, C.; Lapouge, K.; Kopp, J.; Wölfle, T.; Rospert, S.; Sinning, I. The ribosome-associated complex RAC serves in a relay that directs nascent chains to Ssb. Nat. Comm. 2020, 11, 1504. [Google Scholar] [CrossRef]
- Chernoff, Y.O.; Newnam, G.P.; Kumar, J.; Allen, K.; Zink, A.D. Evidence for a protein mutator in yeast: Role of the Hsp70-related chaperone Ssb in formation, stability and toxicity of the [PSI+] prion. Mol. Cell Biol. 1999, 19, 8103–8112. [Google Scholar] [CrossRef]
- Amor, A.J.; Castanzo, D.T.; Delany, S.P.; Selechnik, D.M.; van Ooy, A.; Cameron, D.M. The ribosome-associated complex antagonizes prion formation in yeast. Prion 2015, 9, 144–164. [Google Scholar] [CrossRef]
- Kiktev, D.A.; Melomed, M.M.; Lu, C.D.; Newnam, G.P.; Chernoff, Y.O. Feedback control of prion formation and propagation by the ribosome-associated chaperone complex. Mol. Microbiol. 2015, 96, 621–632. [Google Scholar] [CrossRef] [Green Version]
- Son, M.; Wickner, R.B. Normal levels of ribosome-associated chaperones cure two groups of [PSI+] variants. Proc. Natl. Acad. Sci. USA 2020, 117, 26298–26306. [Google Scholar] [CrossRef] [PubMed]
- Manogaran, A.L.; Kirkland, K.T.; Liebman, S.W. An engineered nonsense URA3 allele provides a versatile system to detect the presence, absence and appearance of the [PSI+] prion in Saccharomyces cerevisiae. Yeast 2006, 23, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Wickner, R.B.; Kelly, A.C.; Bezsonov, E.E.; Edskes, H.E. Prion propagation is controlled by inositol polyphosphates. Proc. Natl. Acad. Sci. USA 2017, 114, E8402–E8410. [Google Scholar] [CrossRef]
- Steidle, E.A.; Chong, L.S.; Wu, M.; Crooke, E.; Fiedler, D.; Resnick, A.C.; Rolfes, R.J. A novel inositol pyrophosphate phosphatase in Saccharomyces cerevisiae: Siw14 protein selectively cleaves the β-phosphate from 5-diphosphoinositol pentakisphosphate (5PP-IP5). J. Biol. Chem. 2016, 291, 6772–6783. [Google Scholar] [CrossRef]
- Tsui, M.M.; York, J.D. Roles of inositol phosphates and inositol pyrophosphates in development, cell signaling and nuclear processes. Adv. Enzym. Regul. 2010, 50, 324–337. [Google Scholar] [CrossRef]
- Lee, B.; Park, S.J.; Hong, S.; Kim, K.; Kim, S. Inositol polyphosphate multikinase signaling: Multifaceted functions in health and disease. Mol. Cells 2021, 44, 187–194. [Google Scholar] [CrossRef]
- Wu, M.; Chong, L.S.; Perlman, D.H.; Resnick, A.C.; Fiedler, D. Inositol polyphosphates intersect with signaling and metabolic networks via two distinct mechanisms. Proc. Natl. Acad. Sci. USA 2016, 113, E6757–E6765. [Google Scholar] [CrossRef]
- Steidle, E.A.; Morrisette, V.A.; Fujimaki, K.; Chong, L.; Resnick, A.C.; Capaldi, A.P.; Rolfes, R.J. The InsP7 phosphatase Siw14 regulates inositol pyrophosphate levels to control localization of the general stress response transcription factor Msn2. J. Biol. Chem. 2020, 295, 2043–2056. [Google Scholar] [CrossRef] [PubMed]
- Chernoff, Y.O.; Lindquist, S.L.; Ono, B.-I.; Inge-Vechtomov, S.G.; Liebman, S.W. Role of the chaperone protein Hsp104 in propagation of the yeast prion-like factor [psi+]. Science 1995, 268, 880–884. [Google Scholar] [CrossRef]
- Glover, J.R.; Lindquist, S. Hsp104, Hsp70, and Hsp40: A novel chaperone system that rescues previously aggregated proteins. Cell 1998, 94, 73–82. [Google Scholar] [CrossRef] [Green Version]
- Tessarz, P.; Mogk, A.; Bukau, B. Substrate threading through the central pore of the Hsp104 chaperone as a common mechanism for protein disaggregation and prion propagation. Mol. Microbiol. 2008, 68, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, P.C.; Ness, F.; Edwards, S.R.; Cox, B.S.; Tuite, M.F. The elimination of the yeast [PSI+] prion by guanidine hydrochloride is the result of Hsp104 inactivation. Mol. Microbiol. 2001, 40, 1357–1369. [Google Scholar] [CrossRef] [PubMed]
- Jung, G.; Masison, D.C. Guanidine hydrochloride inhibits Hsp104 activity in vivo: A possible explanation for its effect in curing yeast prions. Curr. Microbiol. 2001, 43, 7–10. [Google Scholar] [CrossRef]
- Moriyama, H.; Edskes, H.K.; Wickner, R.B. [URE3] prion propagation in Saccharomyces cerevisiae: Requirement for chaperone Hsp104 and curing by overexpressed chaperone Ydj1p. Mol. Cell Biol. 2000, 20, 8916–8922. [Google Scholar] [CrossRef]
- Reidy, M.; Miot, M.; Masison, D.C. Prokaryotic chaperones support yeast prions and thermotolerance and define disaggregation machinery interactions. Genetics 2012, 192, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Chernoff, Y.O.; Ono, B.-I. Dosage-dependent modifiers of PSI-dependent omnipotent suppression in yeast. In Protein Synthesis and Targeting in Yeast; Brown, A.J.P., Tuite, M.F., McCarthy, J.E.G., Eds.; Springer: Berlin, Germany, 1992; pp. 101–107. [Google Scholar]
- Helsen, C.W.; Glover, J.R. Insight into molecular basis of curing of [PSI+] prion by overexpression of 104-kDa heat shock protein (Hsp104). J. Biol. Chem. 2012, 287, 542–556. [Google Scholar] [CrossRef]
- Winkler, J.; Tyedmers, J.; Bukau, B.; Mogk, A. Hsp70 targets Hsp100 chaperones to substrates for protein disaggregation and prion fragmentation. J. Cell Biol. 2012, 198, 387–404. [Google Scholar] [CrossRef]
- Park, Y.N.; Zhou, X.; Yim, Y.I.; Todor, H.; Ellerbrock, R.; Reidy, M.; Eisenberg, E.; Masison, D.C.; Greene, L.E. Hsp104 overexpression cures Saccharomyces cerevisiae [PSI+] by causing dissolution of the prion seeds. Eukaryot. Cell 2014, 13, 635–647. [Google Scholar] [CrossRef]
- Ness, F.; Cox, B.; Wonwigkam, J.; Naeimi, W.R.; Tuite, M.F. Over-expression of the molecular chaperone Hsp104 in Saccharomyces cerevisiae results in the malpartition of [PSI+] propagons. Mol. Microbiol. 2017, 104, 125–143. [Google Scholar] [CrossRef]
- Hung, G.C.; Masison, D.C. N-terminal domain of yeast Hsp104 chaperone is dispensable for thermotolerance and prion propagation but necessary for curing prions by Hsp104 overexpression. Genetics 2006, 173, 611–620. [Google Scholar] [CrossRef] [Green Version]
- Gorkovskiy, A.; Reidy, M.; Masison, D.C.; Wickner, R.B. Hsp104 at normal levels cures many [PSI+] variants in a process promoted by Sti1p, Hsp90 and Sis1p. Proc. Natl. Acad. Sci. USA 2017, 114, E4193–E4202. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-W.; Kushnirov, V.V.; King, C.-Y. Mutable yeast prion variants are stabilized by a defective Hsp104 chaperone. Mol. Microbiol. 2021, 115, 774–788. [Google Scholar] [CrossRef] [PubMed]
- Son, M.; Wickner, R.B. Nonsense-mediated mRNA decay factors cure most [PSI+] prion variants. Proc. Natl. Acad. Sci. USA 2018, 115, E1184–E1193. [Google Scholar] [CrossRef]
- He, F.; Jacobson, A. Nonsense-mediated mRNA decay: Degradation of defective transcripts is only part of the story. Ann. Rev. Genet. 2015, 49, 339–366. [Google Scholar] [CrossRef] [PubMed]
- Czaplinski, K.; Ruiz-Echevarria, M.J.; Paushkin, S.V.; Han, X.; Weng, Y.; Perlick, H.A.; Dietz, H.C.; Ter-Avanesyan, M.D.; Peltz, S.W. The surveillance complex interacts with the translation release factors to enhance termination and degrade aberrant mRNAs. Genes Dev. 1998, 12, 1665–1677. [Google Scholar] [CrossRef]
- Weng, Y.; Czaplinski, K.; Peltz, S.W. Identification and characterization of mutations in the UPF1 gene that affect nonsense suppression and the formation of the Upf protein complex but not mRNA turnover. Mol. Cell Biol. 1996, 16, 5491–5506. [Google Scholar] [CrossRef]
- Weng, Y.; Czaplinski, K.; Peltz, S.W. Genetic and biochemical characterization of mutations in the ATPasse and helicase region of the Upf1 protein. Mol. Cell Biol. 1996, 16, 5477–5490. [Google Scholar] [CrossRef]
- He, F.; Ganesan, R.; Jacobson, A. Intra- and Intermolecular regulatory interactions in Upf1p, the RNA helicase central to nonsense-mediated mRNA decay in yeast. Mol. Cell Biol. 2013, 33, 4672–4684. [Google Scholar] [CrossRef]
- Urakov, V.N.; Mitkevich, O.V.; Dergalev, A.A.; Ter-Avanesyan, M.D. The Pub1 and Upf1 proteins act in concert to protect yeast from toxicity of the [PSI+] prion. Int. J. Mol. Sci. 2018, 19, 3663. [Google Scholar] [CrossRef]
- Kirkland, P.A.; Reidy, M.; Masison, D.C. Functions of yeast Hsp40 chaperone Sis1p dispensable for prion propagation but important for prion curing and protection from prion toxicity. Genetics 2011, 188, 565–577. [Google Scholar] [CrossRef] [Green Version]
- Kumar, J.; Reidy, M.; Masison, D.C. Yeast J-protein Sis1 prevents prion toxicity by maintaining prion protein solubility. Genetics 2021, 219, iyab129. [Google Scholar] [CrossRef]
- Edskes, H.K.; Mukhamedova, M.; Edskes, B.K.; Wickner, R.B. Hermes transposon mutagenesis shows [URE3] prion pathology prevented by a ubiquitin-targeting protein: Evidence for carbon/nitrogen assimilation cross-talk and a second function for Ure2p in Saccharomyces cerevisiae. Genetics 2018, 209, 789–800. [Google Scholar] [CrossRef]
- Seol, J.H.; Shevchenko, A.; Shevchenko, A.; Deshales, R.J. Skip1 forms multiple protein complexes, including RAVE, a regulator of V-ATPase assembly. Nat. Cell. Biol. 2001, 3, 384–391. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Chien, P.; Naber, N.; Cooke, R.; Weissman, J.S. Conformational variations in an infectious protein determine prion strain differences. Nature 2004, 428, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Baxa, U.; Ross, P.D.; Wickner, R.B.; Steven, A.C. The N-terminal prion domain of Ure2p converts from an unfolded to a thermally resistant conformation. J. Mol. Biol. 2004, 339, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Cox, B.S.; Ness, F.; Tuite, M.F. Analysis of the generation and segregation of propagons: Entities that propagate the [PSI+] prion in yeast. Genetics 2003, 165, 23–33. [Google Scholar] [CrossRef]
- Luke, M.M.; Sutton, A.; Arndt, K.T. Characterization of SIS1, a Saccharomyces cerevisiae homolog of bacterial dnaJ proteins. J. Cell Biol. 1991, 114, 623–638. [Google Scholar] [CrossRef]
- Higurashi, T.; Hines, J.K.; Sahi, C.; Aron, R.; Craig, E.A. Specificity of the J-protein Sis1 in the propagation of 3 yeast prions. Proc. Natl. Acad. Sci. USA 2008, 105, 16596–16601. [Google Scholar] [CrossRef]
- Harris, J.M.; Nguyen, P.P.; Patel, M.J.; Sporn, Z.A.; Hines, J.K. Functional diversification of Hsp40: Distinct J-protein functional requirements for two prions allow for chaperone-dependent prion selection. PLoS Genet. 2014, 10, e41004510. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Anti-Prion Components | Target Prion(s) | Fold New Prions | Mechanism of Action | Ref. |
|---|---|---|---|---|
| Btn2 | [URE3] | 5× | Sequesters amyloid filaments and other misfolded proteins | [56,57] |
| Cur1 | [URE3] | 5× | ??? | [56] |
| Ssb1/2, Ssz1, Zuo1 | [PSI+] | 10–15× | Ribosome-associated chaperones that assure proper folding of nascent proteins | [69,72] |
| Upf1, Upf2, Upf3 | [PSI+] | 10–15× | Nonsense-mediated decay components that complex with Sup35 directly blocking amyloid formation | [95] |
| Hsp104 | [PSI+], [URE3] | 13× | Controversial | [80,93] |
| Siw14 | [PSI+] | 2× | Pyrophosphatase specific for 5-inositol pyrophosphates that promote [PSI+] propagation by an unknown mechanism | [74] |
| Prion protein polymorphism | [PSI+], [URE3],… | Intraspecies transmission barrier due to sequence difference of prion protein | [52,54] | |
| Sis1 | [PSI+] | Prevents [PSI+] filaments from depleting too much of the essential Sup35 protein | [102,103] | |
| Lug1 | [URE3] | F-box protein that prevents lethality of Ure2 deficiency | [104,105] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wickner, R.B.; Edskes, H.K.; Son, M.; Wu, S. Anti-Prion Systems Block Prion Transmission, Attenuate Prion Generation, Cure Most Prions as They Arise and Limit Prion-Induced Pathology in Saccharomyces cerevisiae. Biology 2022, 11, 1266. https://doi.org/10.3390/biology11091266
Wickner RB, Edskes HK, Son M, Wu S. Anti-Prion Systems Block Prion Transmission, Attenuate Prion Generation, Cure Most Prions as They Arise and Limit Prion-Induced Pathology in Saccharomyces cerevisiae. Biology. 2022; 11(9):1266. https://doi.org/10.3390/biology11091266
Chicago/Turabian StyleWickner, Reed B., Herman K. Edskes, Moonil Son, and Songsong Wu. 2022. "Anti-Prion Systems Block Prion Transmission, Attenuate Prion Generation, Cure Most Prions as They Arise and Limit Prion-Induced Pathology in Saccharomyces cerevisiae" Biology 11, no. 9: 1266. https://doi.org/10.3390/biology11091266
APA StyleWickner, R. B., Edskes, H. K., Son, M., & Wu, S. (2022). Anti-Prion Systems Block Prion Transmission, Attenuate Prion Generation, Cure Most Prions as They Arise and Limit Prion-Induced Pathology in Saccharomyces cerevisiae. Biology, 11(9), 1266. https://doi.org/10.3390/biology11091266