1. Introduction
Protein–protein interactions (PPIs) are the basis for understanding most cellular events in biological systems. Several methods have been used to identify protein–protein associations, to study and understand a cell’s physiological activities, such as signal transduction, transcriptional regulation, and metabolic and regulatory pathways, and even to investigate therapeutic targets. Experimental methods, such as biochemical methods in cell cultures [
1] and living organisms [
2], have been used to determine direct interactions in order to evaluate binding affinities in real time [
3], examine pathogens’ virulence [
4], quantify and visualize PPIs in cells and tissues [
5], and understand the nature of PPIs during biogenesis reactions [
6]. Moreover, molecular methodologies have been included to detect specific PPIs and develop antifungals that disrupt virulence [
7], characterize and screen protein–protein complexes in a model antibody-antigen system [
8], map and quantify effector–host PPIs during an infection [
9], and detect and characterize PPIs in vivo and in vitro assays [
10]. Finally, genetic approaches have been used to identify phase mutations (G2/M or G1/S) regulated by protein–protein interactions on eukaryotic cells [
11], understand the cellular construction of nanostructures through protein–protein interactions [
12], identify the physical interactions and screen mutation function of some enzymes in a yeast network [
13], and detect genetic interactions as potential anticancer therapeutic targets [
14]. However, most of them are time-consuming and expensive.
Additionally, in silico approaches allow for modeling molecular interactions [
15], testing conformational changes of protein–protein docking and protein–DNA docking [
16], detecting enzyme activity [
17], structurally characterizing two different molecules [
18], and even designing new therapeutics. In the same way, datasets are used to identify functional interactions and detect likely PPIs [
19], infer functionally similar genes, and understand the pathogenesis of the disease [
20]. Nevertheless, experimental and computational methods are individually designed and are carried out for specific interactions.
Machine learning (ML) techniques have been widely used for characterizing sequences of PPIs [
21,
22], considering the amino acid residue as the interaction site [
23] and transforming biological sequences into numerical representations [
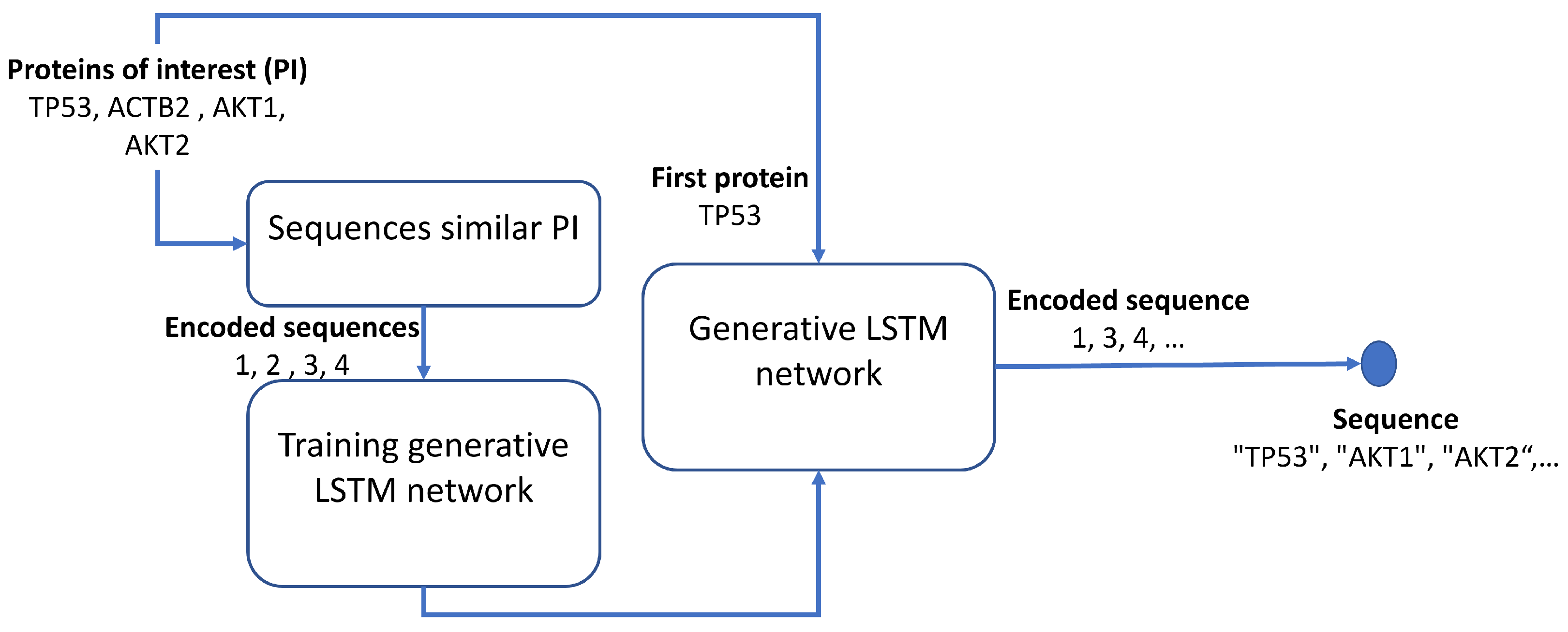
24], thus increasing the number of proteins analyzed simultaneously and optimizing time and resources. In most cases, different ML approaches have been used in computing PPI networks, using well-known physiochemical properties and evolutionary profiles. However, to the best of our knowledge, all investigations have focused on well-known networks or specific targets, not on identifying relevant proteins with partial or null knowledge of the interaction network. This work aims to generate a relevant protein sequence based on bidirectional Long-Short Term Memory (LSTM) without knowledge of their specific interactions. The proposed approach has roots in the complex network analysis pursuing two purposes: to give evidence that several PPI networks are fractal but not scale-free and to extract the relevant proteins based on fractality. The relevant protein sequences (extracted from known PPI networks in which target proteins partake) are the cornerstone to building a bidirectional LSTM network; the LSTM will then generate a sequence based on target proteins.
Related work and preliminaries that underpin this research will be introduced below, followed by a presentation of the methodology and the results. The discussion and conclusion will be given afterwards.
2. Related Work
ML techniques have been used on molecular and cellular levels to model, identify, and predict binding interactions. The support vector machine algorithm has been used to predict interactions of a pair of proteins [
25,
26]. Deep-learning neural networks have been employed to design novel peptides [
27]. On the other hand, based on physical and semantic information about amino acids, the support vector machine classifies the sequences (of a fixed length
n and a set of 20 amino acids) [
28] as positive (they exist) or negative. Similarly, for predicting host–pathogen PPIs, an LSTM was developed to identify the positive sequence of amino acids [
29]. The approaches in [
28,
29] have a high accuracy, of more than 0.98. An LSTM can also identify matches of PPIs from four different species with prediction accuracies of more than 0.92 (rodent: 0.92; bacterium: 0.96; fly: 0.98; nematode: 0.99) [
30]. In the same way, PPIs of primary amino acid sequences across species were identified in [
31]. The deep neural network provides the probability that a pair of proteins interact, and these candidate interactions are compared with those that occur to evaluate the performance. The precision ranged from 0.51 to 0.58, and the recall ranged from 0.22 to 0.54, depending on the species.
Furthermore, classical ML algorithms such as naive Bayes and the support vector machine have been employed to differentiate expressed genes [
32,
33] and validate gene biomarkers [
34]. These approaches used relevant nodes from PPI networks that usually are extracted based on centrality measures such as node degree, closeness, and betweenness. Moreover, ML tools have helped to classify diseases [
35] and prognostic mutations [
36] and detect molecular diseases [
37] based on PPI, as well as to identify infectious diseases and the PPIs between humans and viruses [
38,
39,
40]. Furthermore, clustering methods on PPI networks have been employed to construct hierarchy trees and detect functional modules [
41].
Complex network analysis, such as the fractal dimension of PPI networks, has been employed to detect the sets of PPIs that form subnetworks. In this approach, the fractal dimension is the clustering metric that considers the number of nodes and edges in the boxes computed by the sandbox algorithm [
42]. Furthermore, the fuzzy fractal dimension of PPI networks has been used to identify the essential proteins in PPI networks [
43]. The crucial scale-free property of the dementia and hereditary Parkinson’s PPI networks emerges when the vital proteins are deleted from them, revealing their importance not only in the biological process but also in the network’s topology [
44].
The previous work shows that the ML approaches infer potential interactions, validate previous results, and analyze PPI networks. Deep learning techniques, such as LSTM, and classical machine learning, such as support vector machine algorithms, have shown that they can classify sequences and discern between positive and negative PPIs. Nevertheless, they cannot create new long sequences, as is the purpose of this work. The new unknown PPI sequences obtained by computational methods could help biologists to guide investigations and reduce research time, experiments, and laboratory consumables, leading to the development, design, and discovery of effective drugs acting on these new interactions.
5. Results
The scale-free analysis shows that only four PPI network nodes’ degrees follow a power law with a cutoff distribution; 161 were exponential, and for 30 PPI networks, there is no sufficient statistical evidence supporting a choice between exponential and log-normal models. Finally, for 281 PPI networks, the node’s degree follows a log-normal distribution; see
Table S2.
Table 2 summarizes these results, showing that most PPI networks follow a log-normal distribution.
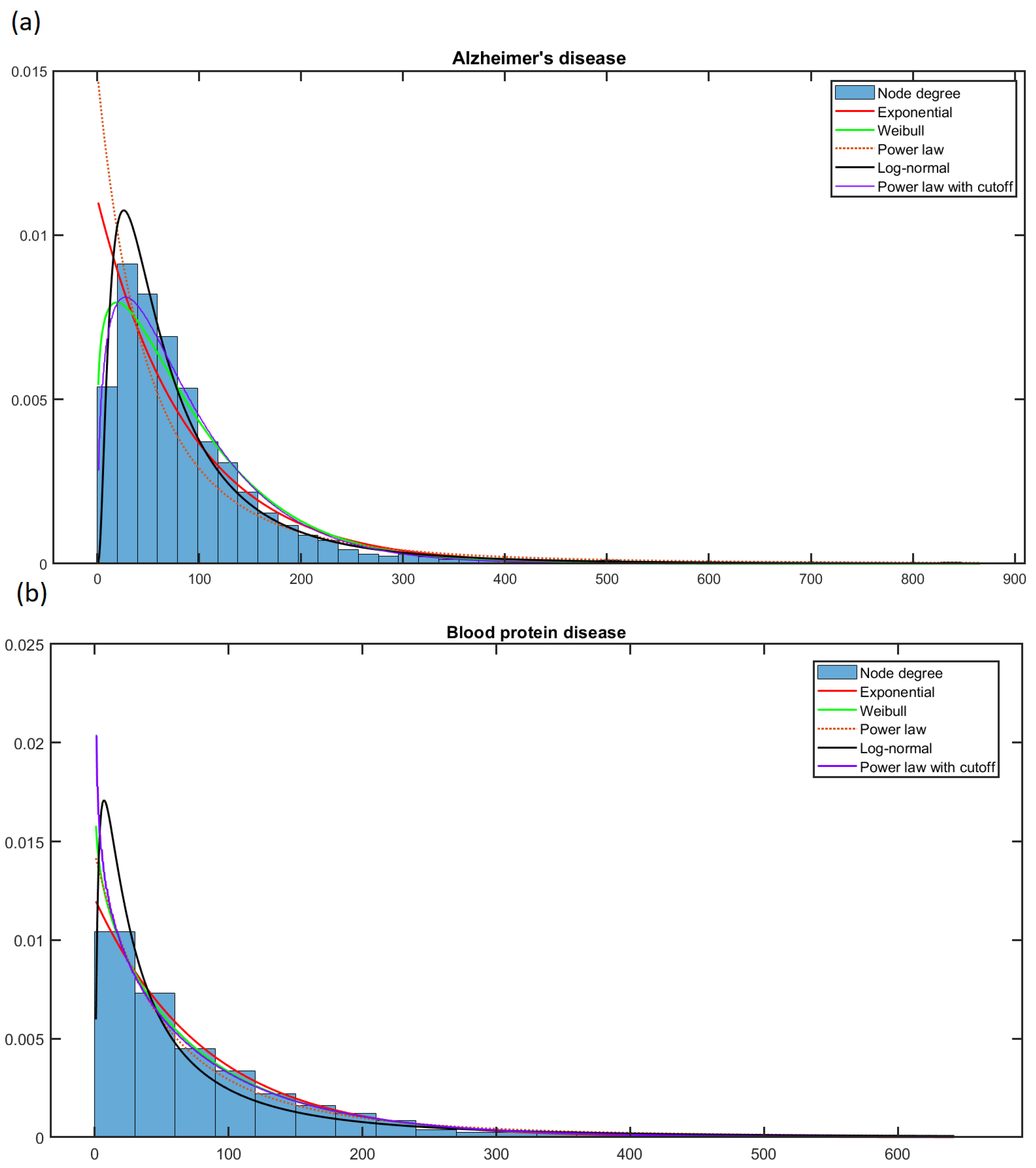
Figure 4 shows the fit of five models for the node degree of (a) Alzheimer’s and (b) Blood protein (hyperproteinemia and hypoproteinemia) diseases. Moreover,
Figure S1 of Supplementary Material shows the fit of several models for the node degree probability distribution of (a) Endocarditis and (b) the Gilles de la Tourette syndrome network.
Figure 4 and
Figure S1 reveal that selecting the best without the AIC is rather difficult. The scale-free analysis results show that 92.88% of the degree distributions of the PPI networks in this work follow a kind of exponential distribution (exponential and log-normal); thus, they are not scale-free. These results undermine the use of the maximum degree-based attack since it is the preferable method for obtaining relevant nodes if the network has the scale-free property.
The fractal analysis shows that the box-covering of 57.74% of PPI networks best fits the delayed exponential function, that of 20.29% is best for the delayed fractal, and that of only 2.30% is best for the exponential function. The number of networks that cannot be differentiated between exponential or delayed exponential or between exponential or fractal is reported in
Table 3. Thus, fractal networks (20.29%) are not as rare as self-similar ones (0.84%). Moreover, the self-similar and fractal analyses suggest that fractality and self-similarity cannot coexist in the PPI networks; see
Tables S2 and S3.
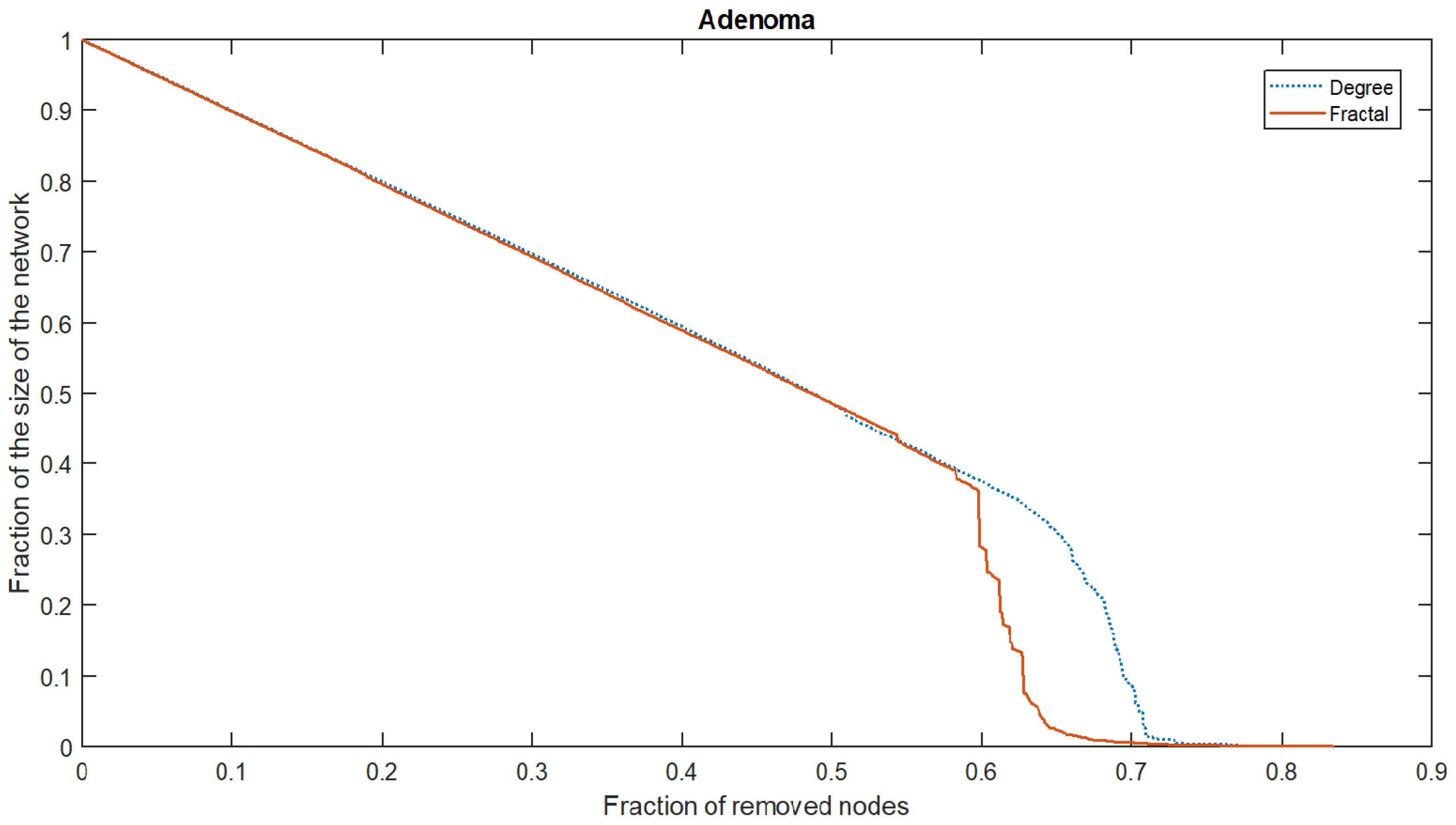
The box-covering of the PPI networks is mostly of the exponential type (78.87–57.74% delayed exponential, 2.30% exponential, and 18.83% exponential or delayed exponential), and 20.29% is of the delayed fractal type; meanwhile, only four cannot be classified in one of the previous sets. These results, in conjunction with the self-similar analysis, suggest that the fractal method for obtaining the relevant proteins of the PPI network is the most suitable, since it obtains good results in fractal and non-fractal networks. For more evidence supporting this, the relevant proteins obtained by the maximum degree-based method and their correlation with the resilience of the PPI network (measured by AURC) [
46] were compared with that obtained by the fractal method. The regression LSTM network was employed for this purpose. An example of the AURC is shown in
Figure 5. The fraction of nodes removed was plotted vs. the fraction of the size of the largest component in the network. Initially, the size is 1, and the fraction of the removed nodes is 0. For a resilient network, the AURC will be approximately 0.5, since the resilience curve will be a straight line with a slope of
. On the contrary, an AURC closer to 0 means that the network’s resilience is poor. The AURC of the fractal and maximum degree-based attacks on the same network provide a measure of their effectiveness that can be compared; for example, the method with the lowest AURC is the most effective at destroying the network. The AURC was computed when the relevant nodes were obtained by the fractal and maximum degree-based methods. A
t-test shows that the fractal method obtained a lower AURC (
,
) compared with the maximum degree-based method (
,
),
. Hence, the proteins obtained by the fractal method are more suitable for maintaining the cohesion of the PPI network.
The MAPE, MAE, and RMSE of the regression of the protein sequences and the AURC obtained by the fractal method are significantly lower than those of the maximum degree-based method. On the other hand, the
of the fractal method is higher than the
of the maximum degree-based method; see
Table 4. The previous results support the finding that the sequences extracted by the fractal method are suitable for maintaining the cohesion of PPI networks. Furthermore, these protein sequences are highly correlated with the PPI network’s resilience.
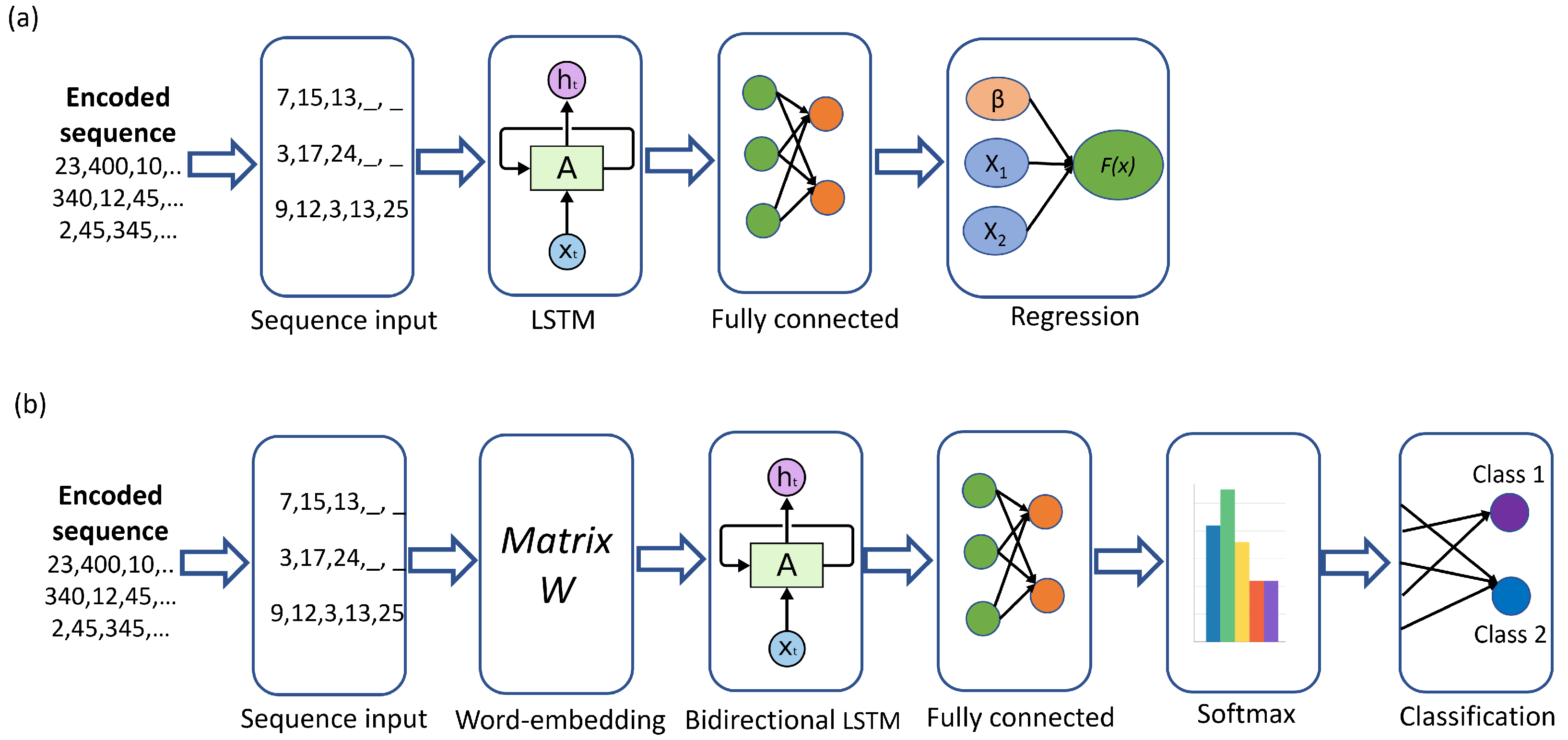
The bidirectional LSTM network was then trained, as described above. The accuracy of the generation process was tested using the protein sequences grouped in Immune, Metabolism, Motor, Nerve, and Bone functions; see
Table 1. The generated and the original sequences were compared in terms of the Jaccard measure and Levenshtein distance. Both sequences were expected to be identical, so the Jaccard value was 1 in this case, and the Levenshtein distance was 0. Since the length of the generated sequences varied from 2 to the length of the original sequence, those with a length of less than 2 were discarded for this analysis. The first
n (length of the generated sequence) proteins were taken from the original sequences to be compared with the generated one, since the latter is usually shorter than the original one. This occurs since the bidirectional LSTM cannot produce a confident set of new proteins, and the generation process stops.
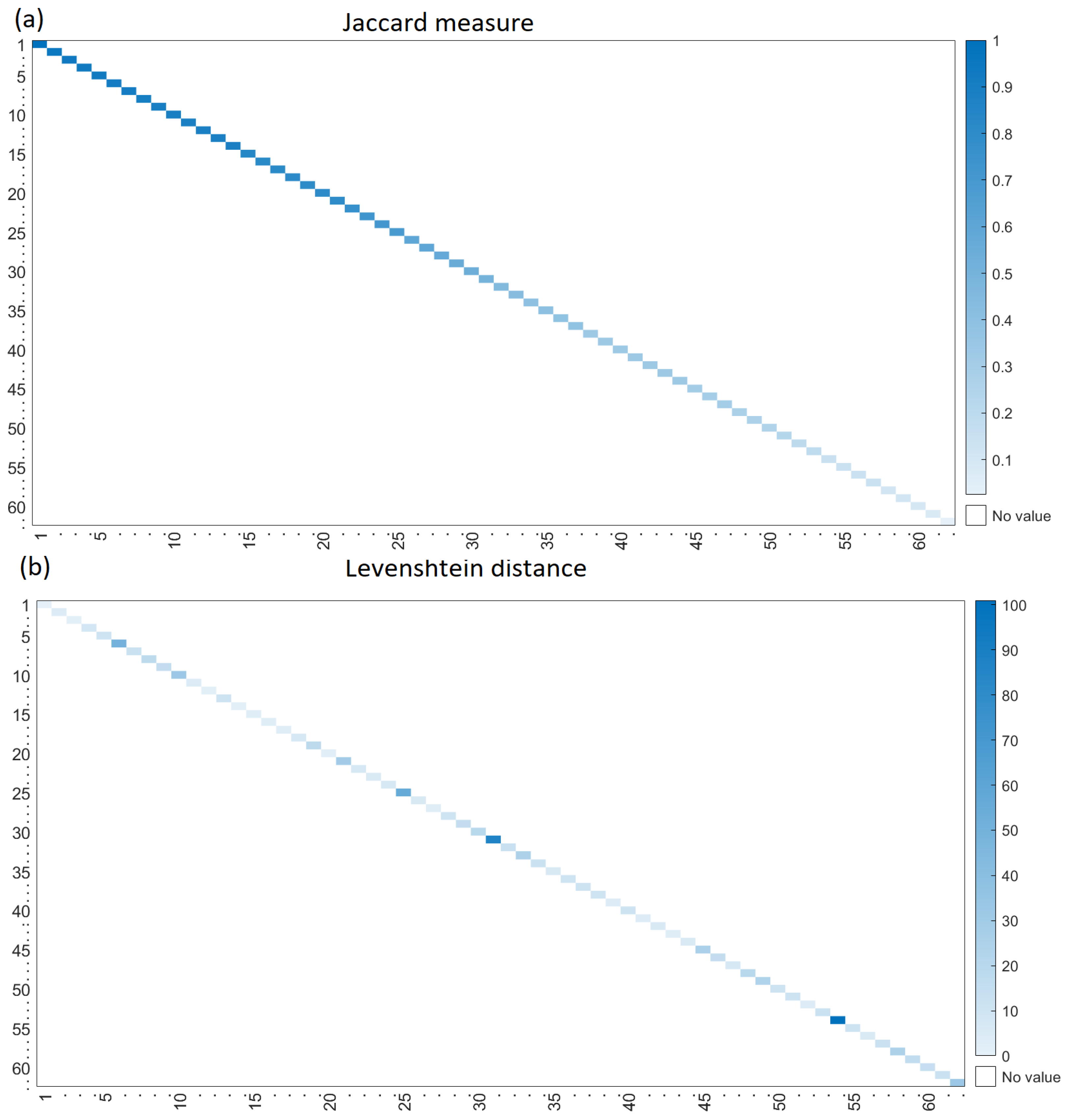
Figure 6a shows the Jaccard measure between the real and generated sequences of Bone. In this heat map, intense blue means that the generated sequence contains several proteins that are also in real sequences. The Levenshtein distance is shown in
Figure 6b; intense blue means that many operations such as proteins deletion and insertion transform the generated sequence into a real sequence.
Figure 6 supports the idea that the generated sequences contain many proteins of the real sequence (intense blue in the Jaccard heat map) and that the proteins in the sequences are in the true positions (light blue in the Levenshtein heat map). Hence, the generated and real sequences match in terms of the proteins and their positions. The plots for the remaining functions are in the
Supplementary Material; see
Figures S2–S5.
Table 5 summarizes the Jaccard measure, the Levenshtein distance, and the length ratio of generated and real sequences grouped by the function of human organs.
The results of
Table 5 show that the generated sequences of Bone contain about 50% of the proteins (Jaccard measure) in the original sequence; meanwhile, the proteins in the generated sequences of Motor are about 24% of the proteins contained in the original sequence. Furthermore, the Levenshtein distance is the erroneous relevance forecasted (position in the sequence); it ranges from 12 to 22. For example, the Levenshtein distance of the generated sequence
G and the real
R is 7; see the Brachydactyly type D network in
Table 6. The first four proteins match in both sequences; however, the five in
G differ from those in the same position in
R.
G can be converted into
R by (1) inserting “RAB7A” and replacing the proteins in the (2) sixth, (3) seventh, (4) eighth, (5) ninth, (6) tenth, and (7) eleventh positions. In practice, this information determines the cost of finding the true protein sequence, which can help researchers in guiding experimental design, understanding pathogenesis, and finding key points for new therapeutics. Finally, the length ratio is the percentage of the total proteins of the real sequence generated. In general, the bidirectional LSTM produced sequences with a length from 14% to 21% of the original sequences.
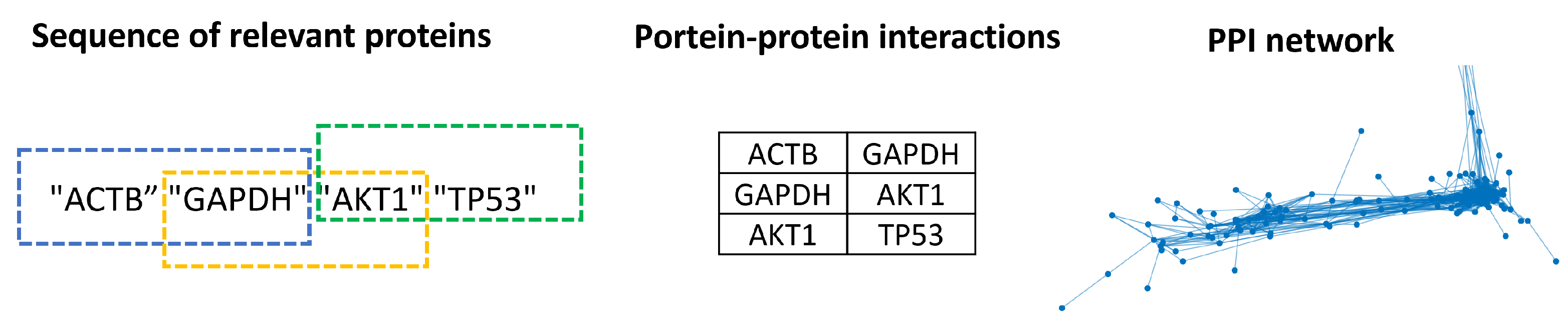
Finally, the sequences of the relevant proteins extracted from PPI networks (by the fractal method) and generated by bidirectional LSTM contained spurious interactions. For example, the first two proteins in a relevant protein sequence could not be directly connected in the network from which it was extracted. Although our approach does not try to identify positive and negative interactions, those contained in the sequences are tested using real accuracy and random accuracy [
79]. First, the sequences were fragmented in pairs, as in [
76]. Let “ACTB”, “GAPDH”, “AKT1”, and “TP53” be a sequence of relevant nodes; the first PPI is “ACTB” and “GAPDH”, and the second is “GAPDH” and “AKT1”; see the middle of
Figure 7 for the resulting PPI. The PPIs of the relevant protein sequences are then tested to determine if they are in the network where it was extracted (an arc between these proteins must exist in the network). Meanwhile, the PPI generated by the bidirectional LSTM was tested to determine whether it belonged to the set of PPI network groups by the function of the human organs used to train the bidirectional LSTM.
Table 7 demonstrated that the mean accuracy (0.949) (see the
column) of extracted protein sequences is similar to that of the generated ones (0.9486) (see
). These true PPIs extracted from PPI networks were also learned by the bidirectional LSTM producing a low rate of spurious PPIs. Furthermore, the fractal method extracts a high number of true PPIs, even though it was not designed for this objective. This low number of spurious PPIs is reflected in the high values of random accuracy; see
in the generated and extracted columns of
Table 7. Random accuracy is the classification rate of the hypothetical random model [
79]. For example, if an extremely biased model classifies each current PPI as true, then the number of correct classifications of spurious PPIs will be zero, and the correct classification of true PPIs will equal its number; hence, the random accuracy depends on how balanced the spurious and true PPIs are. In the balanced data (where 50% of PPIs are spurious and 50% are true), our biased model will obtain an accuracy of 0.5, equivalent to a random classification.
6. Discussion and Conclusions
This study introduces an approach for generating a relevant protein sequence based on bidirectional LSTM with partial knowledge of true PPIs. The general aim of the framework was to conduct scale-free and fractal analysis to determine the topology of PPI networks. The results demonstrate that a handful of PPI networks are self-similar or fractal, but both cannot coexist (the union of scale-free networks (
Table S2) and of fractal networks (
Table S3) is empty). The hub repulsion is a feature that causes the emergence of fractality [
53,
80] but is not the only one. On the other hand, the Barabasi–Albert [
81] model generates scale-free networks but not fractal ones. In Kuang et al. [
82], the model proposed by Song et al. [
53] was extended to conciliate these two approaches. Their results show that the scale-free property and fractality coexist in some networks, with hub attraction and a high clustering coefficient for each box (a property that the Songs networks do not have [
83]). This result coincides with the work of Ikeda [
84], wherein a network model was proposed to generate fractal and scale-free networks based on a high clustering local property. The PPI networks have hub repulsion, meaning that the most important proteins are not directly linked to others but through proteins with fewer connections generating the fractal property. On the other hand, in less analyzed scale-free PPI networks, the hubs are linked to each other directly, but the non-hub nodes in the boxes are poorly connected, preventing the fractal property from emerging. In summary, research on fractal PPI disease networks should focus on the interactions between the non-hubs of the boxes. However, scale-free PPI disease networks must center on the hubs.
Furthermore, based on these results, the fractal attack was selected over the maximum degree-based method for extracting relevant proteins. The sequences extracted by the fractal method are highly correlated with the resilience (measured by the AURC) of the PPI networks, and the fractal extraction produces an average of 94.9% of true PPI sequences. This remarkable feature is also presented in the sequences generated by the bidirectional LSTM, which reaches approximately 94.8% of true PPIs and is comparable with previous studies [
30,
76].
The generated PPI sequences contain an average of 39.5% of proteins that are in the original sequence (the Jaccard measure), and the bidirectional LSTM was able to generate about 25 proteins per sequence by only using the extracted sequences obtained by the fractal method. The ratio between the generated and original sequences of proteins was 17%. This means that large sequences were produced with partial PPI information, given that the mean number of proteins in the original sequences is 303.95 (
). Moreover, these sequences of proteins (that are ordered from high to low relevance) can drive the search for true but unknown PPIs. The results show that the proposed method relies on the sample PPI networks selected to produce the new sequences; thus, it requires careful selection. The results demonstrate that the spurious PPIs in the sequences (extracted and produced) originated from the fractal method, which was only designed to find relevant nodes, such as in [
43]. This paves the way toward the creation of an ad hoc algorithm that reduces false PPIs but finds the essential proteins. The automatic generation of PPI sequences can be a powerful tool for understanding biological processes without limitations such as costs, resources, and time.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}