
Mitogenomic Features and Evolution of the Nile River Dominant Tilapiine Species (Perciformes: Cichlidae)
 , ,
, , 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and DNA Extraction
2.2. Library Preparation Mitogenome Sequencing, Assembly, and Annotation
2.3. Sequence Analysis of the Nile Tilapiine Mitogenomes
2.4. Phylogenetic Analysis
3. Results
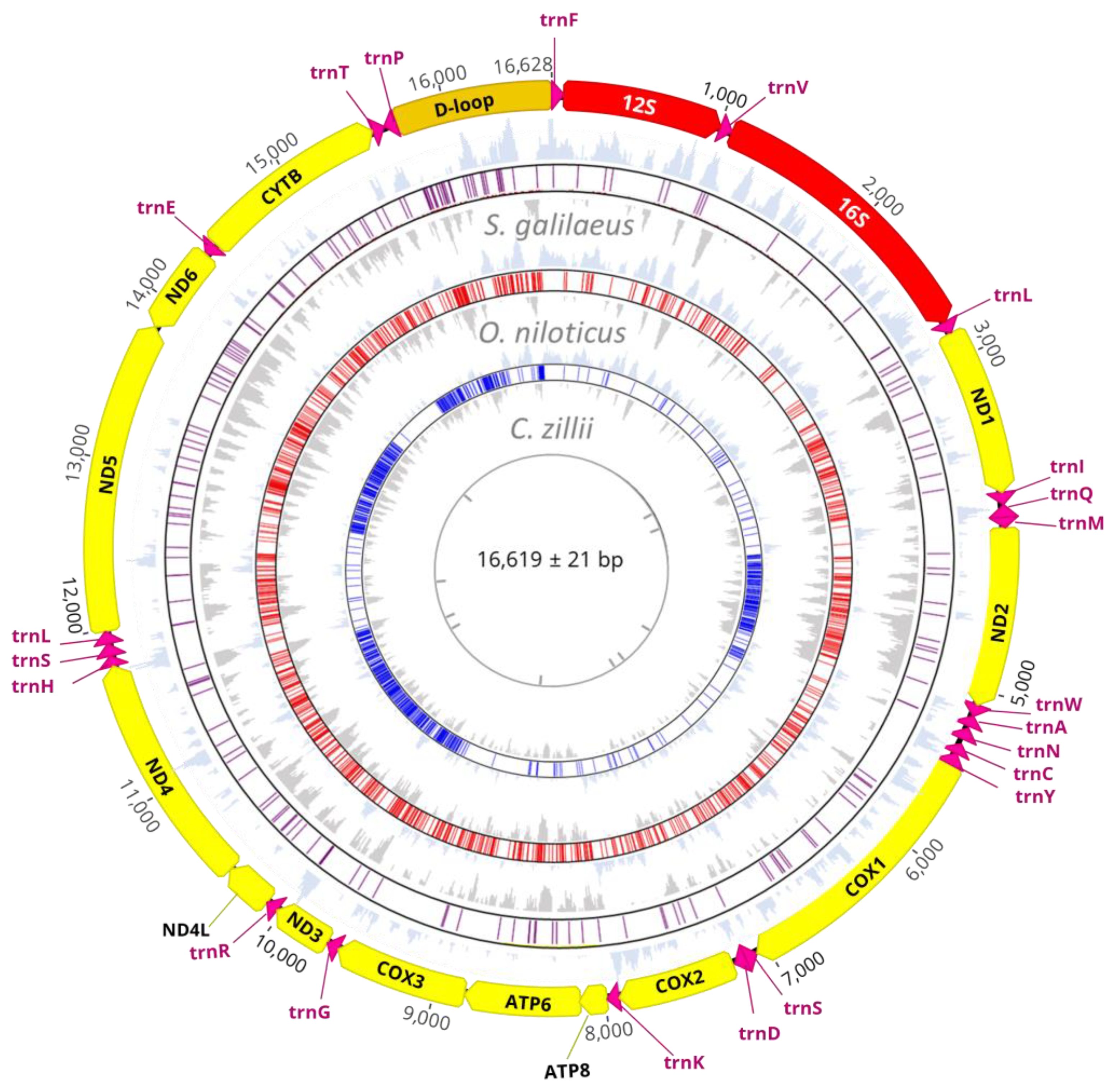
3.1. Mitogenome Structure and Organization
3.2. Polymorphism Assessment
3.2.1. Tandem Repeats
3.2.2. Genetic Diversity
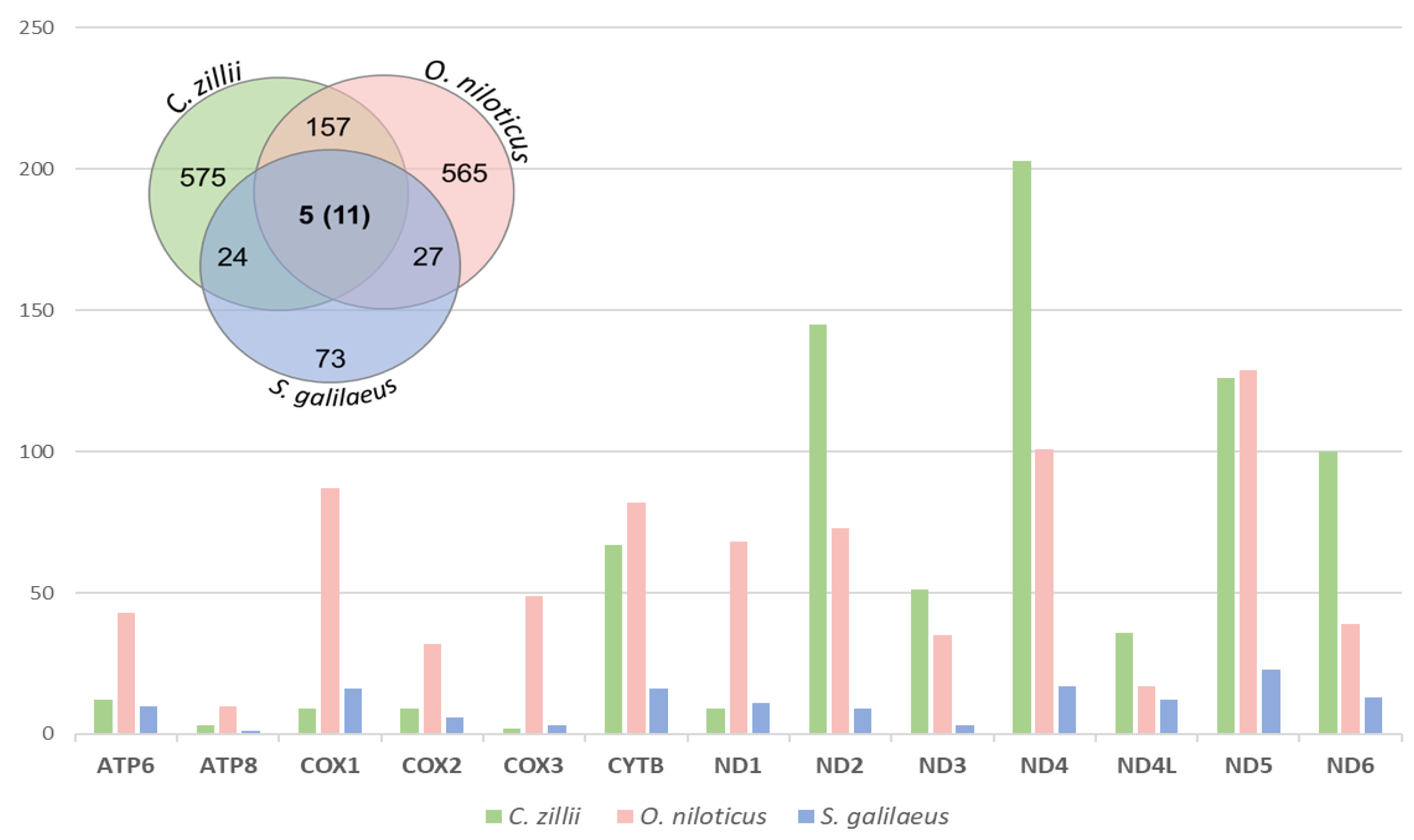
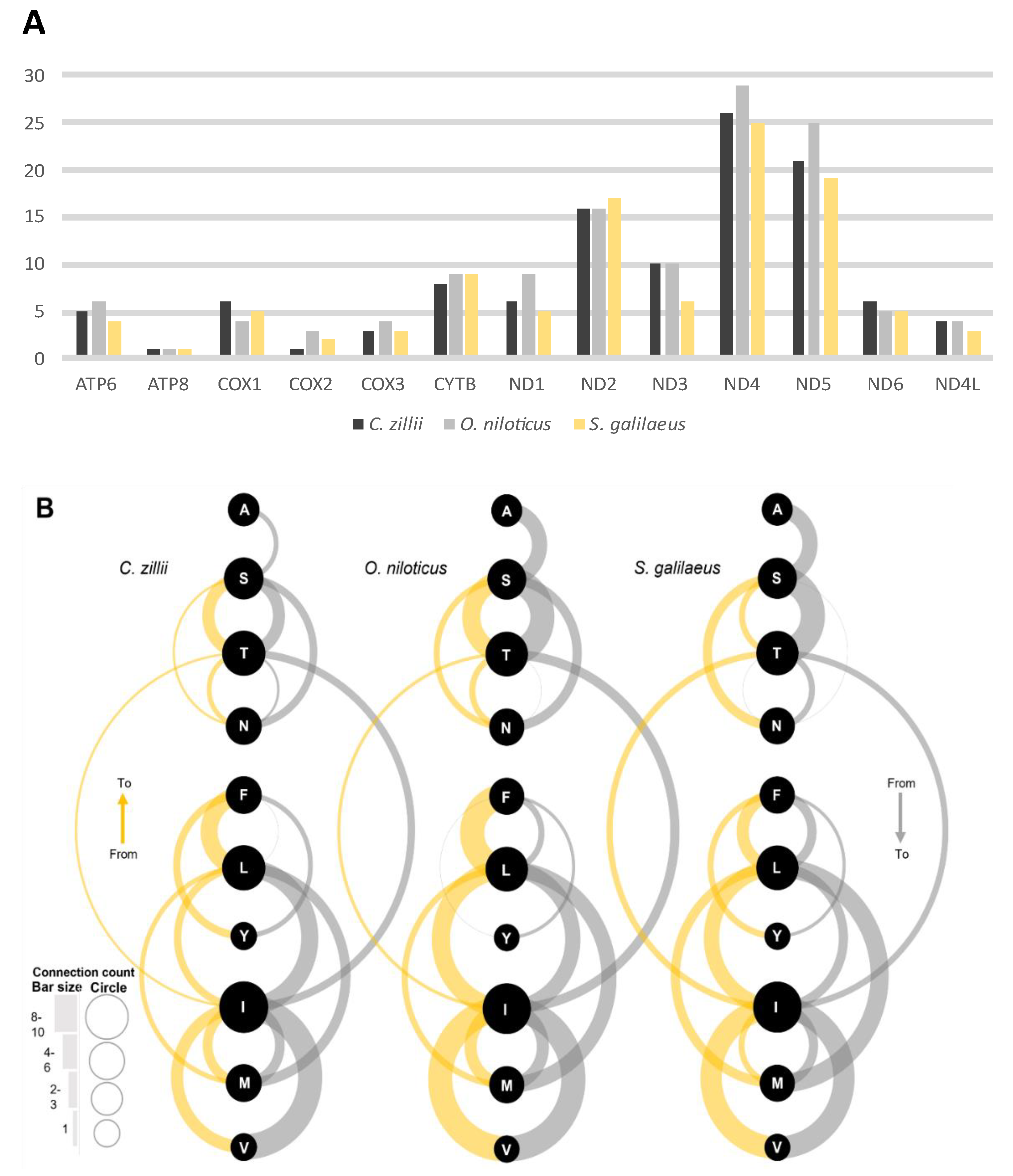
3.2.3. SNPs and Indels
3.3. Molecular Evolution of CDS
3.3.1. Neutrality Test
3.3.2. Substitution Rates of Protein-Coding Genes
3.3.3. Amino Acid Change
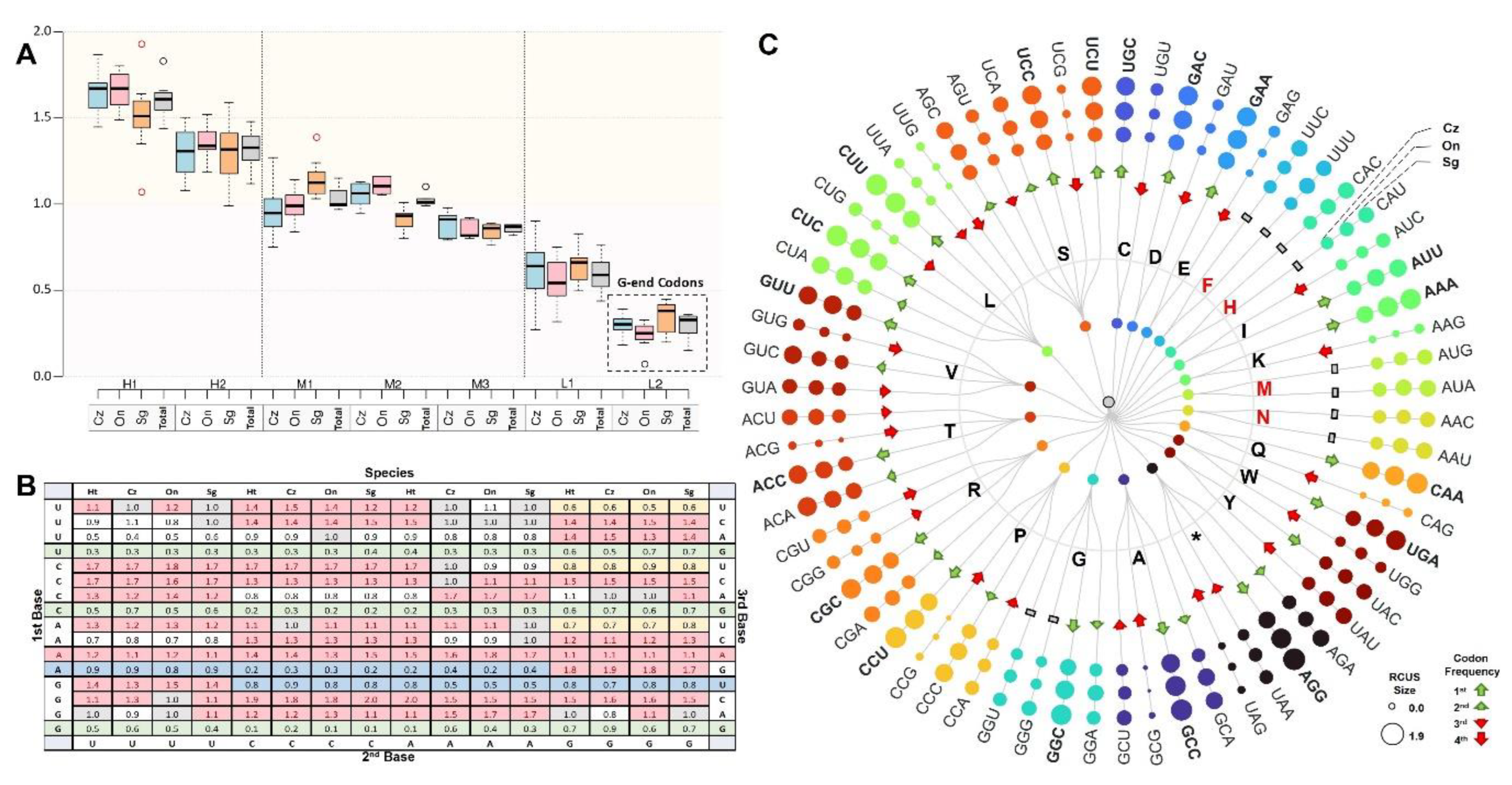
3.3.4. Codon Usage Pattern
3.4. Phylogenetic Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Salzburger, W. Understanding Explosive Diversification through Cichlid Fish Genomics. Nat. Rev. Genet 2018, 19, 705–717. [Google Scholar] [CrossRef] [PubMed]
- Seehausen, O. African Cichlid Fish: A Model System in Adaptive Radiation Research. Proc. R Soc. B 2006, 273, 1987–1998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunz, A.R.; Schliewen, U.K. Molecular Phylogeny and Revised Classification of the Haplotilapiine Cichlid Fishes Formerly Referred to as “Tilapia”. Mol. Phylogenetics Evol. 2013, 68, 64–80. [Google Scholar] [CrossRef] [PubMed]
- Fiteha, Y.G.; Magdy, M. The Evolutionary Dynamics of the Mitochondrial TRNA in the Cichlid Fish Family. Biology 2022, 11, 1522. [Google Scholar] [CrossRef] [PubMed]
- Schliewen, U.K.; Stiassny, M.L. Etia Nguti, a New Genus and Species of Cichlid Fish from the River Mamfue, Upper Cross River Basin in Cameroon, West-Central Africa. Ichthyol. Explor. Freshw. 2003, 14, 61–72. [Google Scholar]
- Schwarzer, J.; Misof, B.; Tautz, D.; Schliewen, U.K. The Root of the East African Cichlid Radiations. BMC Evol. Biol. 2009, 9, 186. [Google Scholar] [CrossRef] [Green Version]
- Chapman, F.A. Culture of. Hybrid Tilapia: A Reference Profile; University of Florida Cooperative Extension Service, Institute of Food and Agricultural Sciences: Gainesville, FL, USA, 2000. [Google Scholar]
- McAndrew, B.J. Evolution, Phylogenetic Relationships and Biogeography. In Tilapias: Biology and Exploitation; Beveridge, M.C.M., McAndrew, B.J., Eds.; Springer Netherlands: Dordrecht, The Netherlands, 2000; pp. 1–32. ISBN 978-0-7923-6391-0. [Google Scholar]
- Watanabe, W.O.; Losordo, T.M.; Fitzsimmons, K.; Hanley, F. Tilapia Production Systems in the Americas: Technological Advances, Trends, and Challenges. Rev. Fish Sci. 2002, 10, 465–498. [Google Scholar] [CrossRef]
- Popma, T.J.; Lovshin, L.L. Worldwide Prospects for Commercial Production of Tilapia. International Center for Aquaculture and Aquatic Environments, Auburn University, Alabama 36849. 1996. Available online: http://agrilife.org/fisheries2/files/2013/09/Worldwide-Prospects-for-Commercial-Production-of-Tilapia.pdf. (accessed on 23 December 2022).
- FAO Fisheries & Aquaculture. Available online: https://www.fao.org/fishery/en/statistics/software/fishstatj (accessed on 1 November 2022).
- Fisheries and Aquaculture—Cultured Aquatic Species. Available online: https://www.fao.org/fishery/en/culturedspecies/search/en (accessed on 1 November 2022).
- Nemati Shizari, F. Rapeseed Cake as a Feed Ingredient for Nile Tilapia: Responses to Replacing Protein from Soybean Meal with Rapeseed Cake, and Fine Milling and Autoclaving of the Rapeseed Cake. Master’s Thesis, Norwegian University of Life Sciences, Ås, Norway, 2014. [Google Scholar]
- Elson, J.; Lightowlers, R. Mitochondrial DNA Clonality in the Dock: Can Surveillance Swing the Case? Trends. Genet 2006, 22, 603–607. [Google Scholar] [CrossRef]
- Zhong, L.; Wang, M.; Li, D.; Tang, S.; Zhang, T.; Bian, W.; Chen, X. Complete Mitochondrial Genome of Odontobutis Haifengensis (Perciformes, Odontobutiae): A Unique Rearrangement of TRNAs and Additional Non-Coding Regions Identified in the Genus Odontobutis. Genomics 2018, 110, 382–388. [Google Scholar] [CrossRef]
- Gissi, C.; Iannelli, F.; Pesole, G. Evolution of the Mitochondrial Genome of Metazoa as Exemplified by Comparison of Congeneric Species. Heredity 2008, 101, 301–320. [Google Scholar] [CrossRef] [Green Version]
- Sagan, L. On the Origin of Mitosing Cells. J. Theor. Biol. 1967, 14, 225-IN6. [Google Scholar] [CrossRef]
- He, A.; Luo, Y.; Yang, H.; Liu, L.; Li, S.; Wang, C. Complete Mitochondrial DNA Sequences of the Nile Tilapia (Oreochromis Niloticus) and Blue Tilapia (Oreochromis Aureus): Genome Characterization and Phylogeny Applications. Mol. Biol. Rep. 2011, 38, 2015–2021. [Google Scholar] [CrossRef]
- Kinaro, Z.O.; Xue, L.; Volatiana, J.A. Complete Mitochondrial DNA Sequences of the Victoria Tilapia (Oreochromis Variabilis) and Redbelly Tilapia (Tilapia Zilli): Genome Characterization and Phylogeny Analysis. Mitochondrial DNA Part A 2016, 27, 2455–2457. [Google Scholar] [CrossRef]
- Luo, C.; Yang, P.; Wang, S. The Complete Mitochondrial Genome of Sarotherodon. Galilaeus. (Linnaeus, 1758) (Perciformes: Cichlidae) and Its Phylogenetic Placement. Mitochondrial DNA Part B 2021, 6, 920–921. [Google Scholar] [CrossRef]
- Ali, S.E.; Jansen, M.D.; Mohan, C.V.; Delamare-Deboutteville, J.; Charo-Karisa, H. Key Risk Factors, Farming Practices and Economic Losses Associated with Tilapia Mortality in Egypt. Aquaculture 2020, 527, 735438. [Google Scholar] [CrossRef]
- Trewavas, E. Tilapiine Fishes of the Genera Sarotherodon, Oreochromis, and Danakilia; British Museum (Natural History): London, UK, 1983. [Google Scholar]
- Magdy, M.; Ou, L.; Yu, H.; Chen, R.; Zhou, Y.; Hassan, H.; Feng, B.; Taitano, N.; van der Knaap, E.; Zou, X.; et al. Pan-Plastome Approach Empowers the Assessment of Genetic Variation in Cultivated Capsicum Species. Hortic. Res. 2019, 6, 108. [Google Scholar] [CrossRef] [Green Version]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq—Versatile and Accurate Annotation of Organelle Genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowe, T.M.; Chan, P.P. TRNAscan-SE on-Line: Integrating Search and Context for Analysis of Transfer RNA Genes. Nucleic. Acids. Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An Integrated and Extendable Desktop Software Platform for the Organization and Analysis of Sequence Data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [Green Version]
- Euesden, J.; Lewis, C.M.; O’Reilly, P.F. PRSice: Polygenic Risk Score Software. Bioinformatics 2015, 31, 1466–1468. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Tajima, F. Statistical Method for Testing the Neutral Mutation Hypothesis by DNA Polymorphism. Genetics 1989, 123, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, L.; Zhang, Q.-L.; Zhou, M.-Q.; Wang, X.-T.; Yang, X.-Z.; Yuan, M.-L. Comparative Mitogenomic Analysis of Mirid Bugs (Hemiptera: Miridae) and Evaluation of Potential DNA Barcoding Markers. PeerJ 2017, 5, e3661. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Sharp, P.M.; Tuohy, T.M.F.; Mosurski, K.R. Codon Usage in Yeast: Cluster Analysis Clearly Differentiates Highly and Lowly Expressed Genes. Nucl. Acids. Res. 1986, 14, 5125–5143. [Google Scholar] [CrossRef] [Green Version]
- Mauri, M.; Elli, T.; Caviglia, G.; Uboldi, G.; Azzi, M. RAWGraphs: A Visualisation Platform to Create Open Outputs. In Proceedings of the 12th Biannual Conference on Italian SIGCHI Chapter, Cagliari, Italy, 18 September 2017; ACM: Cagliari, Italy; pp. 1–5. [Google Scholar]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Bbole, I.; Zhao, J.-L.; Tang, S.-J.; Katongo, C. Mitochondrial Genome Annotation and Phylogenetic Placement of Oreochromis Andersonii and O. Macrochir among the Cichlids of Southern Africa. PLoS ONE 2018, 13, e0203095. [Google Scholar] [CrossRef] [Green Version]
- Perna, N.T.; Kocher, T.D. Patterns of Nucleotide Composition at Fourfold Degenerate Sites of Animal Mitochondrial Genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef]
- Lü, Z.; Zhu, K.; Jiang, H.; Lu, X.; Liu, B.; Ye, Y.; Jiang, L.; Liu, L.; Gong, L. Complete Mitochondrial Genome of Ophichthus Brevicaudatus Reveals Novel Gene Order and Phylogenetic Relationships of Anguilliformes. Int. J. Biol. Macromol. 2019, 135, 609–618. [Google Scholar] [CrossRef]
- Wei, S.; Shi, M.; Sharkey, M.J.; van Achterberg, C.; Chen, X. Comparative Mitogenomics of Braconidae (Insecta: Hymenoptera) and the Phylogenetic Utility of Mitochondrial Genomes with Special Reference to Holometabolous Insects. BMC Genom. 2010, 11, 371. [Google Scholar] [CrossRef]
- Wei, S.-J.; Shi, M.; Chen, X.-X.; Sharkey, M.J.; van Achterberg, C.; Ye, G.-Y.; He, J.-H. New Views on Strand Asymmetry in Insect Mitochondrial Genomes. PLoS ONE 2010, 5, e12708. [Google Scholar] [CrossRef] [Green Version]
- Mu, X.; Liu, Y.; Lai, M.; Song, H.; Wang, X.; Hu, Y.; Luo, J. Characterization of the Macropodus Opercularis Complete Mitochondrial Genome and Family Channidae Taxonomy Using Illumina-Based de Novo Transcriptome Sequencing. Gene 2015, 559, 189–195. [Google Scholar] [CrossRef]
- Shan, W.; Tursun, M.; Zhou, S.; Zhang, Y.; Dai, H. Complete Mitochondrial Genome Sequence of Lepus Yarkandensis Günther, 1875 (Lagomorpha, Leporidae): Characterization and Phylogenetic Analysis. ZK 2021, 1012, 135–150. [Google Scholar] [CrossRef]
- Powell, W.; Machray, G.; Provan, J. Polymorphism Revealed by Simple Sequence Repeats. Trends. Plant. Sci. 1996, 1, 215–222. [Google Scholar] [CrossRef]
- Verstrepen, K.J.; Jansen, A.; Lewitter, F.; Fink, G.R. Intragenic Tandem Repeats Generate Functional Variability. Nat. Genet 2005, 37, 986–990. [Google Scholar] [CrossRef] [Green Version]
- Zl, S.; Li, W.; Jin, X.; Zhang, H. A New Nuclear DNA Marker Revealing Both Microsatellite Variations and Single Nucleotide Polymorphic Loci: A Case Study on Classification of Cultivars in Lagerstroemia indica L. J. Microb. Biochem. Technol. 2016, 8, 266–271. [Google Scholar]
- Krebes, L.; Bastrop, R. The Mitogenome of Gammarus Duebeni (Crustacea amphipoda): A New Gene Order and Non-Neutral Sequence Evolution of Tandem Repeats in the Control Region. Comp. Biochem. Physiol. Part. D. Genom. Proteom. 2012, 7, 201–211. [Google Scholar] [CrossRef]
- Zhang, D.; Ding, G.; Wang, G.; Tang, B.; Sun, H. Structure and Variable Numbers of Tandem Repeats (VNTRs) of the Mitochondrial Control Region in Mitten Crab Eriocheir (Crustacean: Brachyura). Mol. Biol. Rep. 2011, 38, 4935–4940. [Google Scholar] [CrossRef]
- Montgomery, S.B.; Goode, D.L.; Kvikstad, E.; Albers, C.A.; Zhang, Z.D.; Mu, X.J.; Ananda, G.; Howie, B.; Karczewski, K.J.; Smith, K.S. The Origin, Evolution, and Functional Impact of Short Insertion–Deletion Variants Identified in 179 Human Genomes. Genome Res. 2013, 23, 749–761. [Google Scholar] [CrossRef] [Green Version]
- de Lima, P.H.C.; Barcelos, R.M.; Klein, R.C.; Vidigal, P.M.P.; Montandon, C.E.; Fabres-Klein, M.H.; Dergam, J.A.; Mafra, C. Sequencing and Comparative Analysis of the Amblyomma Sculptum Mitogenome. Vet. Parasitol. 2017, 247, 121–128. [Google Scholar] [CrossRef]
- Mandal, S.D.; Chhakchhuak, L.; Gurusubramanian, G.; Kumar, N.S. Mitochondrial Markers for Identification and Phylogenetic Studies in Insects—A Review. DNA Barcodes 2014, 2. [Google Scholar] [CrossRef]
- Schmidt, D. The Effect of Population History on the Distribution of the Tajima’s D Statistic. Population English Edition 1–8. Available online: http://www.deenaschmidt.com/TajimasD.pdf (accessed on 23 December 2022).
- Fijarczyk, A.; Babik, W. Detecting Balancing Selection in Genomes: Limits and Prospects. Mol. Ecol. 2015, 24, 3529–3545. [Google Scholar] [CrossRef]
- Zhai, W.; Nielsen, R.; Slatkin, M. An Investigation of the Statistical Power of Neutrality Tests Based on Comparative and Population Genetic Data. Mol. Biol. Evol. 2009, 26, 273–283. [Google Scholar] [CrossRef] [Green Version]
- Bazin, E.; Glémin, S.; Galtier, N. Population Size Does Not Influence Mitochondrial Genetic Diversity in Animals. Science 2006, 312, 570–572. [Google Scholar] [CrossRef]
- Kerr, K.C.R. Searching for Evidence of Selection in Avian DNA Barcodes. Mol. Ecol. Resour. 2011, 11, 1045–1055. [Google Scholar] [CrossRef]
- Meiklejohn, C.D.; Montooth, K.L.; Rand, D.M. Positive and Negative Selection on the Mitochondrial Genome. Trends. Genet. 2007, 23, 259–263. [Google Scholar] [CrossRef]
- Fischer, C.; Koblmüller, S.; Gülly, C.; Schlötterer, C.; Sturmbauer, C.; Thallinger, G.G. Complete Mitochondrial DNA Sequences of the Threadfin Cichlid (Petrochromis trewavasae) and the Blunthead Cichlid (Tropheus moorii) and Patterns of Mitochondrial Genome Evolution in Cichlid Fishes. PLoS ONE 2013, 8, e67048. [Google Scholar] [CrossRef] [Green Version]
- Joseph, J.; Sreeedharan, S.; George, S.; Antony, M.M. The Complete Mitochondrial Genome of an Endemic Cichlid Etroplus Canarensis from Western Ghats, India (Perciformes: Cichlidae) and Molecular Phylogenetic Analysis. Mol. Biol. Rep. 2022, 49, 3033–3044. [Google Scholar] [CrossRef]
- Doud, M.B.; Ashenberg, O.; Bloom, J.D. Site-Specific Amino Acid Preferences Are Mostly Conserved in Two Closely Related Protein Homologs. Mol. Biol. Evol. 2015, 32, 2944–2960. [Google Scholar] [CrossRef]
- Sharp, P.M.; Cowe, E.; Higgins, D.G.; Shields, D.C.; Wolfe, K.H.; Wright, F. Codon Usage Patterns in Escherichia Coli, Bacillus Subtilis, Saccharomyces Cerevisiae, Schizosaccharomyces Pombe, Drosophila Melanogaster and Homo Sapiens; A Review of the Considerable within-Species Diversity. Nucl. Acids. Res. 1988, 16, 8207–8211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Yuan, J.; Liu, J.; Jin, L.; Chen, J.-Q. Codon Usage Bias and Determining Forces in Green Plant Mitochondrial Genomes: Codon Bias in Plant Mitochondrial Genomes. J. Integr. Plant. Biol. 2011, 53, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Ikemura, T. Correlation between the Abundance of Escherichia Coli Transfer RNAs and the Occurrence of the Respective Codons in Its Protein Genes. J. Mol. Biol. 1981, 146, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.N.; Sultana, S. Codon Usage Bias and Purifying Selection Identified in Cirrhinus Reba Mitogenome. J. Adv. Biotechnol. Exp. Ther. 2022, 5, 605–614. [Google Scholar] [CrossRef]
- Cui; Nie; Sun; Xu; Wang; Yu; Song; Yao Comparative and Phylogenetic Analyses of Ginger (Zingiber officinale) in the Family Zingiberaceae Based on the Complete Chloroplast Genome. Plants 2019, 8, 283. [CrossRef] [PubMed] [Green Version]
- Jia, W.; Higgs, P.G. Codon Usage in Mitochondrial Genomes: Distinguishing Context-Dependent Mutation from Translational Selection. Mol. Biol. Evol. 2008, 25, 339–351. [Google Scholar] [CrossRef] [Green Version]
- Blake, W.J.; KÆrn, M.; Cantor, C.R.; Collins, J.J. Noise in Eukaryotic Gene Expression. Nature 2003, 422, 633–637. [Google Scholar] [CrossRef]
- Ingvarsson, P.K. Gene Expression and Protein Length Influence Codon Usage and Rates of Sequence Evolution in Populus Tremula. Mol. Biol. Evol. 2006, 24, 836–844. [Google Scholar] [CrossRef] [Green Version]
- Rao, Y.; Wu, G.; Wang, Z.; Chai, X.; Nie, Q.; Zhang, X. Mutation Bias Is the Driving Force of Codon Usage in the Gallus Gallus Genome. DNA Res. 2011, 18, 499–512. [Google Scholar] [CrossRef] [Green Version]
- Sueoka, N.; Kawanishi, Y. DNA G+C Content of the Third Codon Position and Codon Usage Biases of Human Genes. Gene 2000, 261, 53–62. [Google Scholar] [CrossRef]
- Sharp, P.M.; Emery, L.R.; Zeng, K. Forces That Influence the Evolution of Codon Bias. Phil. Trans. R Soc. B 2010, 365, 1203–1212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, P.G.; Snow, A.A.; Schug, M.D.; Booton, G.C.; Fuerst, P.A. What Molecules Can Tell Us about Populations: Choosing and Using a Molecular Marker. Ecology 1998, 79, 361. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Repeat Class | Repeat unit (Cz, On, Sg) | Repeat Abundances | Percentage Abundance (%) | Location |
|---|---|---|---|---|
| Dinucleotide | TA (2, 6, 10) | 18 | 23.68 | D-loop |
| TG (0, 1, 0) | 1 | 1.32 | D-loop | |
| Trinucleotide | CAA (2, 6, 5) | 13 | 17.11 | trnS2 1 |
| TTA (2, 5, 5) | 12 | 15.79 | D-loop | |
| Tetranucleotide | CCCG (2, 6, 5) | 13 | 17.11 | trnN-trnC |
| Pentanucleotide | AATGC (0, 5, 0) | 5 | 6.58 | D-loop |
| AATAC (1, 0, 5) | 6 | 7.89 | D-loop | |
| TTATT (0, 5, 0) | 5 | 6.58 | D-loop | |
| 7-nucleotide | TTTAATT (0, 0, 3) | 3 | 3.95 | D-loop |
| Total | - | 76 | 100 | - |
| CDS | C. zillii | O. niloticus | S. galilaeus | ||||||
|---|---|---|---|---|---|---|---|---|---|
| dS | dN | ω | dS | dN | ω | dS | dN | ω | |
| ATP6 | 90 | 4 | 0.04 | 42 | 6 | 0.14 | 3 | 1 | 0.33 |
| ATP8 | 11 | 2 | 0.18 | 9 | 3 | 0.33 | 0 | 0 | 0 |
| COX1 | 159 | 7 | 0.04 | 105 | 7 | 0.07 | 2 | 0 | 0 |
| COX2 | 66 | 7 | 0.11 | 41 | 0 | 0.00 | 0 | 0 | 0 |
| COX3 | 73 | 1 | 0.01 | 46 | 0 | 0.00 | 1 | 0 | 0 |
| CYTB | 161 | 7 | 0.04 | 60 | 9 | 0.15 | 5 | 2 | 0.40 |
| ND1 | 113 | 10 | 0.09 | 69 | 7 | 0.10 | 5 | 1 | 0.20 |
| ND2 | 159 | 21 | 0.13 | 41 | 4 | 0.10 | 3 | 1 | 0.33 |
| ND3 | 50 | 10 | 0.20 | 18 | 4 | 0.22 | 1 | 0 | 0 |
| ND4 | 240 | 19 | 0.08 | 50 | 9 | 0.18 | 7 | 0 | 0 |
| ND4L | 45 | 4 | 0.09 | 11 | 0 | 0.00 | 1 | 0 | 0 |
| ND5 | 224 | 30 | 0.13 | 118 | 24 | 0.20 | 7 | 1 | 0.14 |
| ND6 | 76 | 24 | 0.32 | 25 | 6 | 0.24 | 3 | 3 | 1.00 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fiteha, Y.G.; Rashed, M.A.; Ali, R.A.; Abd El-Moneim, D.; Alshanbari, F.A.; Magdy, M. Mitogenomic Features and Evolution of the Nile River Dominant Tilapiine Species (Perciformes: Cichlidae). Biology 2023, 12, 40. https://doi.org/10.3390/biology12010040
Fiteha YG, Rashed MA, Ali RA, Abd El-Moneim D, Alshanbari FA, Magdy M. Mitogenomic Features and Evolution of the Nile River Dominant Tilapiine Species (Perciformes: Cichlidae). Biology. 2023; 12(1):40. https://doi.org/10.3390/biology12010040
Chicago/Turabian StyleFiteha, Yosur G., Mohamed A. Rashed, Ramadan A. Ali, Diaa Abd El-Moneim, Fahad A. Alshanbari, and Mahmoud Magdy. 2023. "Mitogenomic Features and Evolution of the Nile River Dominant Tilapiine Species (Perciformes: Cichlidae)" Biology 12, no. 1: 40. https://doi.org/10.3390/biology12010040
APA StyleFiteha, Y. G., Rashed, M. A., Ali, R. A., Abd El-Moneim, D., Alshanbari, F. A., & Magdy, M. (2023). Mitogenomic Features and Evolution of the Nile River Dominant Tilapiine Species (Perciformes: Cichlidae). Biology, 12(1), 40. https://doi.org/10.3390/biology12010040