
The Co-Association of Enterobacteriaceae and Pseudomonas with Specific Resistant Cucumber against Fusarium Wilt Disease

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cucumber Cultivars
2.2. Preparation of the Foc Spore Suspension
2.3. Resistance Evaluation and Root Sampling
2.4. DNA Extraction and Illumina Miseq Sequencing
2.5. Bioinformatic Analyses of the Amplicon Sequence Dataset
2.6. Isolation and Identification of Culturable Endophytic Bacteria
2.7. Antagonism Test In Vitro
2.8. Suppressive Ability of the Selected Bacteria on CFW in the Pot Experiment
3. Results
3.1. Resistance Levels of the Cucumber Cultivars
3.2. Analyses of the Root Bacterial Microbiota
3.3. Factors Affecting the Root Bacterial Microbiota
3.4. Identification of Differential Root Bacteria across Cultivars
3.5. Antagonistic Ability of the Culturable Bacterial Isolates
3.6. Suppression of CFW by Differential Bacteria
4. Discussion
4.1. Specific Assembly of Root Microbiota Associated with Cultivars and Pathogens
4.2. Co-Association of Specific Bacteria with the Resistant Cucumber
4.3. Enhanced Resistance Exerted by Differential Bacteria
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Choi, K.; Khan, R.; Lee, S.W. Dissection of plant microbiota and plant-microbiome interactions. J. Mocrobiol. 2021, 59, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Finkel, O.M.; Castrillo, G.; Herrera Paredes, S.; Salas González, I.; Dangl, J.L. Understanding and exploiting plant beneficial microbes. Curr. Opin. Plant Biol. 2017, 38, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Tian, B.; Zhang, C.; Ye, Y.; Wen, J.; Wu, Y.; Wang, H.; Li, H.; Cai, S.; Cai, W.; Cheng, Z.; et al. Beneficial traits of bacterial endophytes belonging to the core communities of the tomato root microbiome. Agric. Ecosys. Envion. 2017, 247, 149–156. [Google Scholar] [CrossRef]
- Torres-Cortés, G.; Bonneau, S.; Bouchez, O.; Genthon, C.; Briand, M.; Jacques, M.A.; Barret, M. Functional microbial features driving community assembly during seed germination and emergence. Front. Plant Sci. 2018, 9, 902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, J.A.; Santos-Medellín, C.M.; Liechty, Z.S.; Nguyen, B.; Lurie, E.; Eason, S.; Phillips, G.; Sundaresan, V. Compositional shifts in root-associated bacterial and archaeal microbiota track the plant life cycle in field-grown rice. PLoS Biol. 2018, 16, e2003862. [Google Scholar] [CrossRef] [Green Version]
- Wei, Z.; Gu, Y.; Friman, V.P.; Kowalchuk, G.A.; Xu, Y.C.; Shen, Q.R.; Jousset, A. Initial soil microbiome composition and functioning predetermine future plant health. Sci. Adv. 2019, 5, eaaw0759. [Google Scholar] [CrossRef] [Green Version]
- Abdelfattah, A.; Tack, A.J.M.; Wasserman, B.; Liu, J.; Berg, G.; Norelli, J.; Droby, S.; Wisniewski, M. Evidence for host–microbiome co-evolution in apple. New Phytol. 2022, 234, 2088–2100. [Google Scholar] [CrossRef]
- Bulgarelli, D.; Garrido-Oter, R.; Münch, P.C.; Weiman, A.; Dröge, J.; Pan, Y.; DMcHardy, A.C.; Schulze-Lefert, P. Structure and function of the bacterial root microbiota in wild and domesticated barley. Cell Host Microbe 2015, 17, 392–403. [Google Scholar] [CrossRef] [Green Version]
- Cardinale, M.; Grube, M.; Erlacher, A.; Quehenberger, J.; Berg, G. Bacterial networks and co-occurrence relationships in the lettuce root microbiota. Environ. Microbiol. 2015, 17, 239–252. [Google Scholar] [CrossRef]
- French, E.; Tran, T.; Iyer-Pascuzzi, A.S. Tomato genotype modulates selection and responses to root microbiota. Phytobiomes J. 2020, 4, 4. [Google Scholar] [CrossRef]
- Kinnunen-Grubb, M.; Sapkota, R.; Vignola, M.; Nunes, I.M.; Nicolaisen, M. Breeding selection imposed a differential selective pressure on the wheat root-associated microbiome. FEMS Microbiol. Ecol. 2020, 96, fiaa196. [Google Scholar] [CrossRef] [PubMed]
- Leff, J.W.; Lynch, R.C.; Kane, N.C.; Fierer, N. Plant domestication and the assembly of bacterial and fungal communities associated with strains of the common sunflower, Helianthus annuus. New Phytol. 2017, 214, 412–423. [Google Scholar] [CrossRef] [PubMed]
- Mendes, L.W.; Raaijmakers, J.M.; de Hollander, M.; Mendes, R.; Tsai, S.M. Influence of resistance breeding in common bean on rhizosphere microbiome composition and function. ISME J. 2017, 12, 212–224. [Google Scholar] [CrossRef] [Green Version]
- Shi, S.; Tian, L.; Nasir, F.; Li, X.; Li, W.; Phan Tran, L.S.; Tian, C. Impact of domestication on the evolution of rhizomicrobiome of rice in response to the presence of Magnaporthe oryzae. Plant Physiol. Biochem. 2018, 132, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Zachow, C.; Muller, H.; Tilcher, R.; Berg, G. Differences between the rhizosphere microbiome of Beta vulgaris ssp. maritima—Ancestor of all beet crops—And modern sugar beets. Front. Microbiol. 2014, 5, 415. [Google Scholar] [CrossRef] [Green Version]
- Escudero-Martinez, C.; Bulgarelli, D. Tracing the evolutionary routes of plant-microbiota interactions. Curr. Opin. Microbiol. 2019, 49, 34–40. [Google Scholar] [CrossRef]
- Perez-Jaramillo, J.E.; Carrion, V.J.; de Hollander, M.; Raaijmakers, J.M. The wild side of plant microbiomes. Microbiome 2018, 6, 143. [Google Scholar] [CrossRef] [Green Version]
- Weinert, N.; Piceno, Y.; Ding, G.C.; Meincke, R.; Heuer, H.; Berg, G.; Schloter, M.; Andersen, G.; Smalla, K. PhyloChip hybridization uncovered an enormous bacterial diversity in the rhizosphere of different potato cultivars: Many common and few cultivar-dependent taxa. FEMS Microbiol. Ecol. 2011, 75, 497–506. [Google Scholar] [CrossRef]
- Kwak, M.J.; Kong, H.G.; Choi, K.; Kwon, S.K.; Song, J.Y.; Lee, J.; Lee, P.A.; Choi, S.Y.; Seo, M.; Lee, H.J.; et al. Rhizosphere microbiome structure alters to enable wilt resistance in tomato. Nat. Biotechnol. 2018, 36, 1100–1109. [Google Scholar] [CrossRef]
- Liu, H.; Dong, Y.H.; Sui, Y.Y.; Li, J.G. Soil microbial mechanism of disease resistant sugar beet variety. Chin Agric. Sci. Bull. 2021, 37, 78–86. (In Chinese) [Google Scholar]
- Yao, H.Y.; Wu, F.Z. Soil microbial community structure in cucumber rhizosphere of different resistance cultivars to fusarium wilt. FEMS Microbiol. Ecol. 2010, 72, 456–463. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.Y.; Zhan, G.L.; Zhang, S.S. Impact of cucumber fusarium wilt in rhizosphere microbe flora and enzyme activity in greenhouse. Fujian J. Agric. Sci. 2011, 4, 605–610. (In Chinese) [Google Scholar] [CrossRef]
- Wen, T.; Yuan, J.; He, X.; Lin, Y.; Huang, Q.; Shen, Q. Enrichment of beneficial cucumber rhizosphere microbes mediated by organic acid secretion. Hortic. Res. 2020, 7, 154. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.C.; Jiang, T.; Liu, Y.X.; Bai, Y.C.; Reed, J.; Qu, B.; Goossens, A.; Nützmann, H.W.; Bai, Y.; Osbourn, A. A specialized metabolic network selectively modulates Arabidopsis root microbiota. Science 2019, 364, eaau6389. [Google Scholar] [CrossRef]
- Liu, X.J.; Liang, G.Y.; Yang, H.; Xie, B.; Sun, Q.; Song, J.F.; Li, Y.J. New cucumber cultivar Chuanlv No.11 without bitterness cultivated in South China. J. Changjiang Veget. 2014, 17, 23–24. (In Chinese) [Google Scholar]
- Gu, X.F.; Zhang, S.P.; Wang, Y.; Hu, X.; Wang, L.H. Selection and breeding of a new heat-tolerant cucumber cultivar Zhongnong No.106. China Veget. 2008, 6, 31–33. (In Chinese) [Google Scholar]
- Han, Y.K.; Du, S.L.; Wei, A.M.; Chen, Z.W.; Liu, N.; Zhang, H.P.; Li, B. A new cucumber open field hybrid cultivar ‘Jinyou 409’. Acta Hortic. Sinica 2016, 43, 1627–1628 (In Chinese). (In Chinese) [Google Scholar] [CrossRef]
- Zhang, W.Z.; Li, J.W.; Wang, J. A new cucumber F1 hybrid—‘Jinyou No. 35’. China Veget. 2007, 12, 33–35. (In Chinese) [Google Scholar]
- Huang, X.Q.; Sun, M.H.; Lu, X.H.; Li, S.D. Serial passage through resistant and susceptible cucumber cultivars affects the virulence of Fusarium oxysporum f. sp. cucumerinum. Microbiol. Open 2019, 8, e00641. [Google Scholar] [CrossRef]
- Sun, R.L.; Jing, Y.L.; de Boer, W.; Guo, R.J.; Li, S.D. Dominant hyphae-associated bacteria of Fusarium oxysporum f. sp. cucumerinum in different cropping systems and insight into their functions. Appl. Soil Ecol. 2021, 165, 103977. [Google Scholar] [CrossRef]
- Bulgarelli, D.; Rott, M.; Schlaeppi, K.; Ver Loren van Themaat, E.; Ahmadinejad, N.; Assenza, F.; Rauf, P.; Huettel, B.; Reinhardt, R.; Schmelzer, E.; et al. Revealing structure and assembly cues for Arabidopsis root-inhabiting bacterial microbiota. Nature 2012, 488, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, D.; Lebeis, S.; Paredes, S.; Yourstone, S.; Gehring, J.; Malfatti, S.; Tremblay, J.; Engelbrektson, A.; Kunin, V.; Rio, T.G.; et al. Defining the core Arabidopsis thaliana root microbiome. Nature 2012, 488, 86–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Men, X.; Zhang, G.; Liang, K.C.; Xin, Y.H.; Wang, J.; Li, A.; Zhang, H.; Liu, H.; Wu, L. Assessment of 16S rRNA gene primers for studying bacterial community structure and function of aging flue-cured tobaccos. AMB Expr. 2018, 8, 182. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callahan, B.; McMurdie, P.; Rosen, M.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Jiang, X.T.; Peng, X.; Deng, G.H.; Sheng, H.F.; Wang, Y.; Zhou, H.W.; Tam, N.F.Y. Illumina sequencing of 16S rRNA tag revealed spatial variations of bacterial communities in a mangrove wetland. Microbiol. Ecol. 2013, 66, 96–104. [Google Scholar] [CrossRef]
- Anderson, M.J. A new method for non-parametric multivariate analysis of variance in ecology. Mathematics 2001, 26, 32–46. [Google Scholar] [CrossRef]
- Ballhausen, M.B.; van Veen, J.A.; Hundscheid, M.P.; de Boer, W. Methods for baiting and enriching fungus-feeding (mycophagous) rhizosphere bacteria. Front. Microbiol. 2015, 6, 1416. [Google Scholar] [CrossRef] [Green Version]
- de Boer, W.; Hundscheid, M.P.; Klein Gunnewiek, P.J.; de Ridder-Duine, A.S.; Thion, C.; van Veen, J.A.; van der Wal, A.; van Overbeek, L.S. Antifungal rhizosphere bacteria can increase as response to the presence of saprotrophic fungi. PLoS ONE 2015, 10, e0137988. [Google Scholar] [CrossRef] [PubMed]
- Rudnick, M.B.; van Veen, J.A.; de Boer, W. Baiting of rhizosphere bacteria with hyphae of common soil fungi reveals a diverse group of potentially mycophagous secondary consumers. Soil Biol. Biochem. 2015, 88, 73–82. [Google Scholar] [CrossRef] [Green Version]
- Weisburg, W.C.; Barns, S.M.; Pelletier, D.A.; Lane, D.J. 16S ribosomal DNA amplification for phylogenetic study. J. Bacteriol. 1991, 173, 697–703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, J.A.; Lennon, J.T. Rapid responses of soil microorganisms improve plant fitness in novel environments. Proc. Natl. Acad. Sci. USA 2012, 109, 14058–14062. [Google Scholar] [CrossRef]
- Vančura, V.; Hanzlíková, A. Root exudates of plants-IV. Differences in chemical composition of seed and seedlings exudates. Plant Soil 1972, 36, 271–282. [Google Scholar] [CrossRef]
- Wagner, M.; Lundberg, D.; del Rio, T.; Tringe, S.G.; Dangl, J.L.; Mitchell-Olds, T. Host genotype and age shape the leaf and root microbiomes of a wild perennial plant. Nat. Commun. 2016, 7, 12151. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Guo, R.J.; Li, Y.J.; Hartman, W.H.; Li, S.F.; Zhang, Z.X.; Tringe, S.G.; Wang, H. Insight into the bacterial endophytic communities of peach cultivars related to crown gall disease resistance. Appl. Environ. Microbiol. 2019, 85, e02931-18. [Google Scholar] [CrossRef] [Green Version]
- Pan, K.; Yao, Y. Influence of different cucumber varieties’ root exudates on the rhizosphere soil microorganisms and soil nutrient. Northern Hortic. 2008, 8, 18–20. [Google Scholar]
- Ofek, M.; Hadar, Y.; Minz, D. Colonization of cucumber seeds by bacteria during germination. Environ. Microbiol. 2011, 13, 2794–2807. [Google Scholar] [CrossRef]
- Ofek, M.; Hadar, Y.; Minz, D. Ecology of root colonizing Massilia (Oxalobacteraceae). PLoS ONE 2012, 7, e40117. [Google Scholar] [CrossRef]
- Upreti, R.; Thomas, P. Root-associated bacterial endophytes from Ralstonia solanacearum resistant and susceptible tomato cultivars and their pathogen antagonistic effects. Front. Microbiol. 2015, 6, 255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazcano, C.; Boyd, E.; Holmes, G.; Hewavitharana, S.; Pasulka, A.; Ivors, K. The rhizosphere microbiome plays a role in the resistance to soil-borne pathogens and nutrient uptake of strawberry cultivars under field conditions. Sci. Rep. 2021, 11, 3188. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Fang, X.; Wu, H.; Cai, X.; and Xiao, H. Effects of plant cultivars on the structure of bacterial and fungal communities associated with ginseng. Plant Soil 2021, 465, 143–156. [Google Scholar] [CrossRef]
- Xu, L.; Nicolaisen, M.; Larsen, J.; Zeng, R.; Gao, S.; Dai, F. Pathogen infection and host-resistance interactively affect root-associated fungal communities in watermelon. Front. Microbiol. 2020, 11, 605–622. [Google Scholar] [CrossRef] [PubMed]
- Baltrus, D.A. Adaptation, specialization, and coevolution within phytobiomes. Curr. Opin. Plant Biol. 2017, 38, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Podolich, O.; Ardanov, P.; Zaets, I.; Pirttila, A.; Kozyrovska, N. Reviving of the endophytic bacterial community as a putative mechanism of plant resistance. Plant Soil 2015, 388, 367–377. [Google Scholar] [CrossRef]
- Yu, P.; He, X.; Baer, M.; Beirinckx, S.; Tian, T.; Moya, Y.A.T.; Zhang, X.; Deichmann, M.; Frey, F.P.; Bresgen, V.; et al. Plant flavones enrich rhizosphere Oxalobacteraceae to improve maize performance under nitrogen deprivation. Nat. Plants 2021, 7, 481–499. [Google Scholar] [CrossRef]
- Zhang, Y.; Xun, W.; Wang, X.; Tian, S.; Zhang, Y.; Li, D.; Zhou, Y.; Qin, Y.; Zhang, B.; Zhao, G.; et al. Root-secreted bitter triterpene modulates the rhizosphere microbiota to improve plant fitness. Nat. Plants 2022, 8, 887–896. [Google Scholar] [CrossRef]
- Kim, D.R.; Jeon, C.W.; Cho, G.; Thomashow, L.S.; Weller, D.M.; Paik, M.J.; Lee, Y.B.; Kwak, Y.S. Glutamic acid reshapes the plant microbiota to protect plants against pathogens. Microbiome 2021, 9, 244. [Google Scholar] [CrossRef]
- Berendsen, R.L.; Vismans, G.; Yu, K.; Song, Y.; de Jonge, R.; Burgman, W.P.; Burmølle, M.; Herschend, J.; Bakker, P.A.H.M.; Pieterse, C.M.J. Disease-induced assemblage of a plant-beneficial bacterial consortium. ISME J. 2018, 12, 1496–1507. [Google Scholar] [CrossRef] [Green Version]
- Kidd, B.N.; Edgar, C.I.; Kumar, K.K.; Aitken, E.A.; Schenk, P.M.; Manners, J.M.; Kazan, K. The mediator complex subunit PFT1 is a key regulator of jasmonate-dependent defense in Arabidopsis. Plant Cell 2009, 21, 2237–2252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romdhane, S.; Spor, A.; Aubert, J.; Bru, D.; Breuil, M.C.; Hallin, S.; Mounier, A.; Ouadah, S.; Tsiknia, M.; Philippot, L. Unraveling negative biotic interactions determining soil microbial community assembly and functioning. ISME J. 2022, 16, 296–306. [Google Scholar] [CrossRef] [PubMed]
- Hultberg, M.; Alsberg, T.; Khalil, S.; Alsanius, B. Suppression of disease in tomato infected by Pythium ultimum with a biosurfactant produced by Pseudomonas koreensis. Biocontrol. 2010, 55, 435–444. [Google Scholar] [CrossRef]
- Hultberg, M.; Bengtsson, T.; Liljeroth, E. Late blight on potato is suppressed by the biosurfactant-producing strain Pseudomonas koreensis 2.74 and its biosurfactant. Biocontrol. 2010, 55, 543–550. [Google Scholar] [CrossRef]
- Maisuria, V.B.; Gohel, V.; Mehta, A.N.; Patel, R.R.; Chhatpar, H.S. Biological control of Fusarium wilt of pigeonpea by Pantoea dispersa, a field assessment. Ann. Microbiol. 2008, 58, 411–419. [Google Scholar] [CrossRef]
- Jiang, L.M.; Jeong, J.C.; Lee, J.S.; Park, J.M.; Yang, J.W.; Lee, M.H.; Choi, S.H.; Kim, C.Y.; Kim, D.H.; Kim, S.W.; et al. Potential of Pantoea dispersa as an effective biocontrol agent for black rot in sweet potato. Sci. Rep. 2019, 9, 16354. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Fan, J.B.; Du, L.; Xu, H.; Zhang, Q.H.; He, Y.Q. The application of phosphate solubilizing endophyte Pantoea dispersa triggers the microbial community in red acidic soil. Appl. Soil Ecol. 2014, 84, 235–244. [Google Scholar] [CrossRef]
- Altankhuu, K.; Kim, J. Massilia solisilvae sp. nov., Massilia terrae sp. nov. and Massilia agilis sp. nov., isolated from forest soil in South Korea by using a newly developed culture method. Int. J. Syst. Evol. Microbiol. 2017, 67, 3026–3032. [Google Scholar] [CrossRef] [PubMed]
- Bai, H.; Zhao, F.; Li, M.; Qin, L.; Yu, H.; Lu, L.; Zhang, T. Citric acid can force Staphylococcus aureus into viable but nonculturable state and its characteristics. Int. J. Food Microbiol. 2019, 305, 108254. [Google Scholar] [CrossRef]
- Zhang, J.J.; Liu, Y.X.; Guo, X.X.; Qin, Y.; Garrido-Oter, R.; Schulze-Lefert, P.; Bai, Y. High-throughput cultivation and identification of bacteria from the plant root microbiota. Nat. Protoc. 2021, 16, 988–1012. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
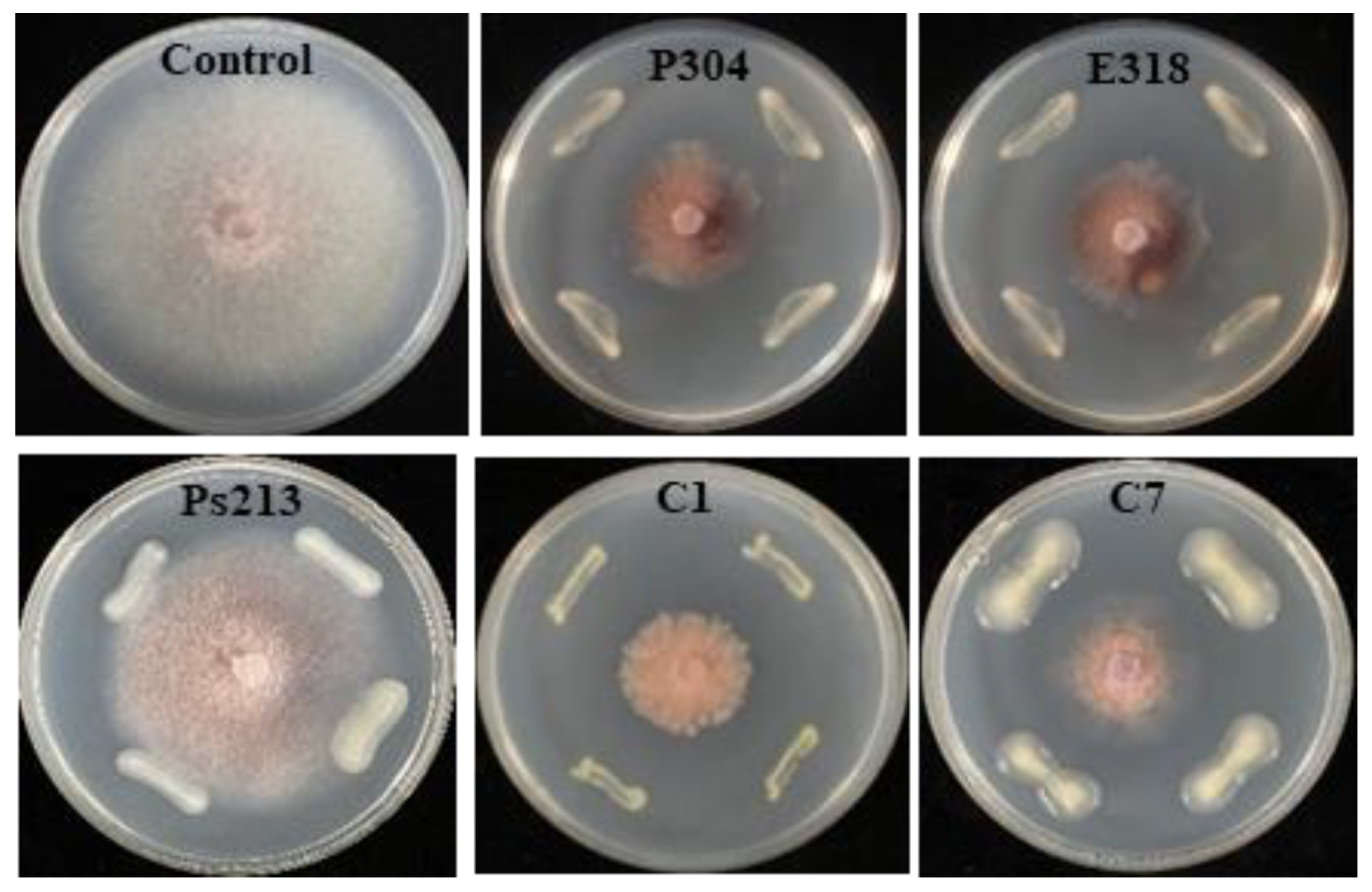{kind=link}
| Treatment | Cucumber Cultivar | Good’s Coverage | Chao | Shannon | Simpson |
|---|---|---|---|---|---|
| Mock | CL11 | 1.00 ± 0.00 | 43.00 ± 12.49 | 2.40 ± 0.18 | 0.18 ± 0.02 |
| EZZ | 1.00 ± 0.00 | 44.33 ± 4.04 | 2.29 ± 0.31 | 0.21 ± 0.08 | |
| JY409 | 1.00 ± 0.00 | 43.33 ± 4.62 | 2.87 ± 0.16 | 0.10 ± 0.02 | |
| JY35 | 1.00 ± 0.00 | 48.67 ± 11.02 | 2.64 ± 0.46 | 0.17 ± 0.11 | |
| ZN106 | 1.00 ± 0.00 | 42.00 ± 11.53 | 2.27 ± 0.16 | 0.04 ± 0.20 | |
| ZN6 | 1.00 ± 0.00 | 53.33 ± 10.50 | 2.44 ± 0.10 | 0.20 ± 0.00 | |
| Foc inoculation | CL11 | 1.00 ± 0.00 | 41.33 ± 4.04 | 2.05 ± 0.23 | 0.30 ± 0.08 |
| EZZ | 1.00 ± 0.00 | 44.33 ± 8.51 | 2.38 ± 0.37 | 0.21 ± 0.09 | |
| JY409 | 1.00 ± 0.00 | 47.67 ± 16.17 | 2.68 ± 0.37 | 0.15 ± 0.05 | |
| JY35 | 1.00 ± 0.00 | 50.33 ± 6.66 | 2.83 ± 0.16 | 0.12 ± 0.03 | |
| ZN106 | 1.00 ± 0.00 | 41.67 ± 5.13 | 2.31 ± 0.04 | 0.21 ± 0.01 | |
| ZN6 | 1.00 ± 0.00 | 54.67 ± 12.90 | 2.65 ± 0.43 | 0.15 ± 0.07 |
| Cucumber Cultivar | Isolate | Inhibition Rate (%) | Taxa Based on the 16S rRNA Gene Sequence | Taxa and ASVs Based on the Illumina High-Throughput Analysis | Homology (%) | |
|---|---|---|---|---|---|---|
| Genus | ASV | |||||
| CL11 | E11 | 20.40 ± 1.37 | Enterobacter | Unclassified_f__Enterobacteriaceae | ASV341 | 100.0 |
| Pa301 | 28.57 ± 3.80 | Paenibacillus | Paenibacillus | ASV62 | 93.7 | |
| P300 | 29.89 ± 2.88 | Pantoea | Unclassified_f__Enterobacteriaceae | ASV478 | 96.8 | |
| S305 | 8.41 ± 2.44 | Sphingomonas | Sphingomonas | ASV463 | 99.7 | |
| Ag302 | 18.82 ± 0.77 | Rhizobium | Unclassified_c__Gammaproteobacteria | ASV53 | 94.0 | |
| Br303 | 27.11 ± 1.37 | Brucella | Unclassified_f__Rhizobiaceae | ASV298 | 95.8 | |
| P304 | 41.84 ± 0.31 | Pantoea | Enterobacter | ASV343 | 98.4 | |
| EZZ | B312 | 16.39 ± 4.04 | Bordetella | Unclassified_f__Alcaligenaceae | ASV156 | 99.7 |
| A308 | 12.88 ± 1.96 | Agrobacterium | Allorhizobium-Neorhizobium-Pararhizobium-Rhizobium | ASV53 | 94.0 | |
| E307 | 19.83 ± 2.93 | Enterobacter | Enterobacter | ASV343 | 100.0 | |
| B309 | 18.75 ± 2.58 | Brucella/Ochrobactrum | Ochrobactrum | ASV298 | 95.8 | |
| B311 | 12.16 ± 4.17 | Bordetella | Unclassified_f__Alcaligenaceae | ASV5 | 100.0 | |
| B310 | 11.15 ± 1.35 | Bordetella | Unclassified_f__Alcaligenaceae410 | ASV5 | 100.0 | |
| JY409 | E12 | 28.40 ± 0.89 | Enterobacter | Unclassified_o__Enterobacterales | ASV410 | 99.7 |
| E318 | 41.14 ± 1.92 | Pantoea | Enterobacter | ASV343 | 98.4 | |
| E314 | 21.68 ± 6.29 | Enterobacter | Unclassified_f__Enterobacteriaceae | ASV55 | 94.0 | |
| E315 | 21.22 ± 5.39 | Enterobacter | Unclassified_f__Enterobacteriaceae | ASV55 | 95.0 | |
| E316 | 17.20 ± 0.96 | Enterobacter | Unclassified_f__Enterobacteriaceae | ASV55 | 95.0 | |
| JY35 | B13 | 21.30 ± 1.35 | Bordetella | Unclassified_f__Alcaligenaceae | ASV5 | 100.0 |
| E317 | 31.85 ± 2.71 | Enterobacter | Enterobacter | ASV343 | 100.0 | |
| S313 | 22.75 ± 2.58 | Stenotrophomonas | Stenotrophomonas | ASV46 | 99.7 | |
| Br319 | 27.61 ± 0.07 | Brucella/Ochrobactrum | Ochrobactrum | ASV298 | 95.8 | |
| ZN106 | C1-C5, C7-C9 | 35.50 ± 5.00 ~ 55.71 ± 2.84 | Cronobacter | Cronobacter | ASV162 | 100.0 |
| ZN6 | Ar10 | 15.20 ± 3.13 | Arachidicoccus | Arachidicoccus | ASV439 | 99.5 |
| E14 | 37.20 ± 2.65 | Enterobacter | Unclassified_f__Enterobacteriaceae | ASV55 | 99.7 | |
| Ps20 -Ps24 | 17.10 ± 1.10 | Pseudomonas | Pseudomonas | ASV263 | 99.7 | |
| Bacteria a | Species | Corresponding ASV | Disease Incidence (%) b | Disease Index | Control Efficacy (%) |
|---|---|---|---|---|---|
| P304 | Pantoea dispersa | ASV343 | 43.3 ± 8.17 ab | 19.6 ± 5.20 a | 46.3 |
| E318 | P. dispersa | ASV343 | 50.0 ± 0.00 ac | 20.8 ± 12.50 a | 42.9 |
| Ps213 | Pseudomonas koreensis | ASV466 | 41.7 ± 16.67 ab | 15.6 ± 3.99 a | 57.1 |
| BCX1 | P. dispersa + P. koreensis | ASV343+ASV466 | 29.2 ± 4.81 b | 8.3 ± 2.95 b | 77.2 |
| BCX2 | Cronobacter | ASV162 | 29.2 ± 15.96 b | 14.6 ± 2.41 ab | 60.0 |
| Control | - | - | 66.7 ± 13.61 c | 36.5 ± 6.25 c | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.-L.; Guo, X.-J.; Huang, X.; Guo, R.-J.; Lu, X.-H.; Li, S.-D.; Zhang, H. The Co-Association of Enterobacteriaceae and Pseudomonas with Specific Resistant Cucumber against Fusarium Wilt Disease. Biology 2023, 12, 143. https://doi.org/10.3390/biology12020143
Zhang Y-L, Guo X-J, Huang X, Guo R-J, Lu X-H, Li S-D, Zhang H. The Co-Association of Enterobacteriaceae and Pseudomonas with Specific Resistant Cucumber against Fusarium Wilt Disease. Biology. 2023; 12(2):143. https://doi.org/10.3390/biology12020143
Chicago/Turabian StyleZhang, Yu-Lu, Xiao-Jing Guo, Xin Huang, Rong-Jun Guo, Xiao-Hong Lu, Shi-Dong Li, and Hao Zhang. 2023. "The Co-Association of Enterobacteriaceae and Pseudomonas with Specific Resistant Cucumber against Fusarium Wilt Disease" Biology 12, no. 2: 143. https://doi.org/10.3390/biology12020143
APA StyleZhang, Y. -L., Guo, X. -J., Huang, X., Guo, R. -J., Lu, X. -H., Li, S. -D., & Zhang, H. (2023). The Co-Association of Enterobacteriaceae and Pseudomonas with Specific Resistant Cucumber against Fusarium Wilt Disease. Biology, 12(2), 143. https://doi.org/10.3390/biology12020143